

Abstract
Antibodies to neuraminidase (NA), the second most abundant surface protein on influenza virus, contribute toward protection against influenza. Traditional methods to measure NA inhibiting (NI) antibody titers are not practical for routine serology. This protocol describes the enzyme-linked lectin assay (ELLA), a practical alternative method to measure NI titers that is performed in 96 well plates coated with a large glycoprotein substrate, fetuin. NA cleaves terminal sialic acids from fetuin, exposing the penultimate sugar, galactose. Peanut agglutinin (PNA) is a lectin with specificity for galactose and therefore the extent of desialylation can be quantified using a PNA-horseradish peroxidase conjugate, followed by addition of a chromogenic peroxidase substrate. The optical density that is measured is proportional to NA activity. To measure NI antibody titers, serial dilutions of sera are incubated at 37 °C O/N on fetuin-coated plates with a fixed amount of NA. The reciprocal of the highest serum dilution that results in ≥50% inhibition of NA activity is designated as the NI antibody titer. The ELLA provides a practical format for routine evaluation of human antibody responses following influenza infection or vaccination.
Keywords: Immunology, Issue 115, Influenza, vaccine, serology, neuraminidase, antibody, inhibition
Introduction
Neuraminidase (NA) is a glycoprotein expressed on the surface of influenza virions. Its enzyme activity is essential for release of newly formed virus particles from infected cells 1,2. Antibodies that inhibit NA activity reduce virus titers and disease symptoms in animal models 3 and correlate with resistance against disease in humans 4. An increase in NA inhibition (NI) titers following vaccination may therefore serve an indicator of the vaccine's efficacy, however many past influenza immunogenicity studies did not include this end-point because the traditional assay to measure NI antibody titers is impractical for routine serology.
The traditional method to measure NI titers is based on quantifying the amount of sialic acid that is cleaved from glycoconjugates by NA 1. This method, often referred to as the thiobarbituric acid (TBA) method, uses hazardous chemicals to convert sialic acid to a chromophore that can be quantified by spectrometry. The assay is not suitable for testing large numbers of samples because individual glass tubes are used, making the assay cumbersome. Miniaturization of the assay to a 96 well plates provides a more practical format 5, however this assay still requires the use of toxic chemicals and therefore is not ideal.
An alternate assay to measure NI antibody titers was developed by Lambré et al. 6. This assay quantifies enzyme activity by measuring the amount of the penultimate sugar of glycoproteins, galactose, which becomes exposed when sialic acid is released by NA. Since peanut-agglutinin (PNA) binds specifically to galactose, a PNA-horseradish peroxidase (HRPO) conjugate can be used to obtain a colorimetric read-out. The optical density that is measured is therefore proportional to the NA activity in the sample. NI titers are measured by determining the highest dilution of serum that inhibits at least 50% of the NA activity. This enzyme-linked lectin assay (ELLA) is performed in 96-well plates coated with fetuin, a highly glycosylated serum protein, as the substrate for NA.
An important consideration for assays that measure NI titers, is the source of NA. This is because sera from vaccinated or infected individuals contain antibodies to hemagglutinin (HA) as well as antibodies to NA. Antibodies that bind to HA can interfere with NA activity and therefore, to avoid non-specific inhibition by HA-specific antibodies, purified NA or whole virus containing an antigenically-mismatched HA should be used in the assay. These viruses can be generated by classic reassortment or by reverse genetics; the objective is to rescue a virus that contains HA of a subtype that is not related to the target strain, and NA of the virus that is being studied. The assay described in this article employs influenza A viruses which are generated by reverse genetics to contain HA of subtype H6 and the targeted NA from influenza A viruses, subtypes H1N1 and H3N2 5.
Results generated by ELLA and miniaturized TBA assays show these methods are comparable, with similar subtype specificity and sensitivity 7. ELLA does not require the use of hazardous chemicals and therefore is the preferred method to measure NI titers. It is easy to perform, with only a few steps (Figure 1): samples are diluted and transferred to a fetuin-coated plate to which virus (the source of NA) is added. The plate is incubated O/N and washed before adding PNA-HRPO. After a 2 hr incubation, the plate is washed and peroxidase substrate is added; the color reaction is stopped and finally the optical density is measured and the reciprocal of the sample dilution that resulted in at least 50% inhibition of NA activity is reported as the 50% end-point titer. An intra-laboratory study to assess reproducibility of the ELLA, showed that plate-to-plate variability is minimal and operator-to-operator repeatability resulted in no more than 2-fold differences in titer 7. A subsequent inter-laboratory study of ELLA variability showed that the assay has good reproducibility when performed in different laboratories and that inclusion of a standard can further reduce variability in results 8. The ELLA is suitable for measuring NI antibody titers in serum panels from preclinical and clinical influenza vaccine studies 7 and can also be used to evaluate antigenic differences between the NAs of influenza viruses 9.
Protocol
All live reassortant viruses with avian components must be handled using biosafety level 2 (BSL2)-enhanced practices in a laboratory approved for use by the United States Department of Agriculture (USDA) and the institutional biosafety committee.
1. Preparation of Reagents and Starting Material
Note: Refer to the Materials Table to obtain the source of all reagents.
- Fetuin-coated Plates
- Prepare coating buffer by mixing 10 ml 10x coating buffer with 90 ml deionized H2O.
- Prepare a stock solution of fetuin at 25 mg/ml in 1x coating buffer and store in 500 µl aliquots at -20 °C.
- Prepare a working solution of fetuin (25 µg/ml) immediately before coating plates by diluting the stock solution 1,000-fold in 1x coating buffer.
- Use a multichannel pipette to dispense 100 µl of the working solution of fetuin into all wells of a 96-well plate that has high protein-binding capacity.
- Cover each plate with a plate sealer then stack the plates in groups of 10 and wrap them in foil.
- Place the plates in a refrigerator (2-8 °C) for at least 18 hr before performing an assay. Note: Plates may be coated in advance and stored with the coating solution at 2-8 °C for up to 2 months.
- Diluents
- To prepare the diluent to make virus and sample dilutions (2-(N-morpholino)ethanesulfonic acid (MES), pH 6.5, 20 mM CaCl2, 1% bovine serum albumin (BSA) and 0.5% Tween 20), mix 94.2 ml 1X MES, pH 6.5 with 2 ml CaCl2 (10 mg/ml), 3.3 ml 30% BSA and 0.5 ml Tween 20.
- To prepare the diluent for PNA-HRPO (MES, pH 6.5 with 20 mM CaCl2 and 1% BSA), mix 94.7 ml 1x MES, pH 6.5 with 2 ml CaCl2 (10 mg/ml), and 3.3 ml 30% BSA.
- Neuraminidase (NA) Note: H6Nx virus reassortants (these have HA of H6 subtype and the NA of interest), are generated following published methods 5,10. Request vials of inactivated reassortant virus from a collaborator if they are not available in-house. When using inactivated virus, confirm that the preparation is suitable for use in the ELLA through demonstration that the inactivation process did not have a significant impact on NA activity.
- Culture a stock of H6Nx virus in embryonated chicken eggs 11 and store 0.5 ml aliquots of neat allantoic fluid at -80 °C.
- Use a new aliquot of virus for each assay. Do not freeze and thaw the aliquots.
- Serum Samples and Controls
- Heat-inactivate all sera (test samples e.g., pre- and post-vaccination sera, as well as controls, e.g., sample with known NI titer) in a water bath at 56 °C for 45-60 min. Store the sera at -20 to -70 °C before or after the heat treatment. If the samples are to be tested repeatedly, make several aliquots before freezing so that the sample is not repeatedly frozen and thawed.
- Use the ELLA to measure the NI titers of human sera that are available in reasonable quantity (>5 ml) to identify at least one with low titer and another with a high titer. Store 100 µl aliquots as ≤-20 °C so that the same sample can be used as a control in many assays in order to track assay performance.
- Peanut Agglutinin (PNA)-Horseradish Peroxidase (HRPO)
- Prepare the PNA-HRPO diluent (MES, pH 6.5 with CaCl2 and 1% BSA).
- Prepare a PNA-HRPO stock solution by dissolving 1 mg of PNA-HRPO in 1 ml of diluent. Store 20-200 µl aliquots at -20 °C.
- Before using a new PNA-HRPO lot, test 1:500, 1:750, 1:1,000 and 1:2000 dilutions of this reagent to identify the amount that results in maximal OD with the positive control (virus only) and a background (no virus) that is <10% of the positive signal.
- Make quadruplicate virus dilutions as described in section 2.1.
- Transfer the dilutions to a fetuin-coated plate as described in section 2.2 and incubate the plate at 37 °C.
- Wash the plate 16-18 hr later and add 1:500, 1:750, 1:1,000 and 1:2,000 dilutions of PNA-HRPO (100 µl/well) to duplicate rows of the plate.
- Complete the assay as described in steps 2.3.4 to 2.3.9.
- Review the data to select the dilution that resulted in a maximum signal that is >10 times the background.
- Immediately before use, prepare the optimal dilution of PNA-HRPO identified in 1.5.3.
Prepare a large volume of Wash buffer (0.01 M phosphate buffered saline (PBS), pH 7.4, 0.05% Tween 20 (PBS-T)). Store at RT.
- Prepare the peroxidase substrate (o-Phenylenediamine dihydrochloride (OPD)): Note: alternative peroxidase substrates such as 3,3′,5,5′-Tetramethylbenzidine (TMB), may be used
- Prepare phosphate-citrate buffer by dissolving 1 capsule in 100 ml dH2O on the day of assay.
- Dissolve 1 OPD tablet (10 mg) in 20 ml of phosphate-citrate buffer immediately before use.
Prepare the stop solution (1 N H2SO4): add 27.2 ml stock 98% H2SO4 to 973 ml dH2O. Mix and then store at RT.
2. Determination of the Amount of NA to Use in ELLA
- Prepare Virus Dilutions in a 96-well Plate
- Dispense 120 µl sample diluent (section 1.2) into columns 1-11 of the dilution plate.
- To column 1, add an additional 96 µl sample diluent.
- Thaw and then vortex the vial of virus before adding 24 µl to duplicate wells in column 1. This gives a 1:10 dilution of virus.
- Make serial 2-fold dilutions of virus in sample diluent, by transferring 120 µl from one well to the next using clean pipette tips for each dilution.
- Transfer Virus Dilutions to a Fetuin-coated Plate
- Wash the fetuin-coated plate 3 times with PBS-T (section 1.6) and then blot each plate onto absorbent paper towel to remove any excess wash buffer.
- Add 50 µl of sample diluent (section 1.2.1) to each well in columns 1 to 11 of the fetuin-coated plate.
- Add 100 µl of sample diluent to column 12 (these wells are the negative control).
- Transfer 50 µl of diluted virus from columns 1-11 of the dilution plate to the corresponding wells of the fetuin-coated plate.
- Cover the plate with a plate sealer and then place it in a humidified incubator at 37 °C.
- Make note of the time at which remaining steps will be performed (16-18 hr later).
- Complete the NA Determination
- When the incubation is complete (16-18 hr), transfer the plate to the bench and remove the plate sealer.
- Wash the plate 6 times with PBS-T (section 1.7) and then invert and pat on absorbent paper towels to ensure all liquid has been removed from the wells.
- Add 100 µl/well PNA-HRPO solution (at the dilution determined in 1.5.3) to all wells and incubate the plate for 2 hr at RT.
- Less than 15 min before the incubation is to end, prepare the OPD solution as described in section 1.7.
- Wash the test plates 3 times to remove the PNA-HRPO and blot dry before adding 100 µl of the OPD substrate to each well.
- Incubate the plate for exactly 10 min at RT.
- Stop the reaction by adding 100 µl/well of 1N sulfuric acid.
- Use a plate reader to measure the Optical Density (OD) at 490 nm for 0.1 sec.
- Save and export all data files.
- Select the Virus Dilution that will be Used for Serology
- Draw a graph that plots OD490nm values at each virus dilution. Inspect the titration curve to identify the maximum signal (this is usually the signal that corresponds to a plateau at the beginning of the titration curve), the minimum signal (wells that contain no virus in column 12 of the plate provide the background control), and the dilutions of virus that result in OD that is proportional to the input dilution (i.e., linear region of the curve).
- Select the virus dilution that gives approximately 90% of the maximum signal and is within the linear range. Confirm that the OD at the selected dilution is at least 10-fold greater than the background signal. Use the selected virus dilution for all assays that employ this particular virus stock. Note: Alternatively, measure the NA activity of the virus and use ~15-20 µU NA activity/ml in the ELLA.
3. Enzyme-linked Lectin Assay
Note: Figure 2 shows the setup of the dilution and assay plates.
- Make Sample Dilutions
- Place the heat-inactivated samples on ice. Note: 8 sera can be diluted in each plate.
- For a set up using dilutions starting at 1:10 across the plate, add 120 µl sample diluent to all wells in columns 3-11.
- To column 2, add 216 µl of sample diluent and 24 µl of each sample. Mix the sample in the well by pipetting up and down 3 times and then transfer 120 µl to the next column.
- After changing pipette tips, mix the contents of the well by pipetting up and down and then transfer 120 µl to the next column.
- Repeat step 3.1.4 until the sample has been transferred to column 11 and the remaining 120 µl discarded.
- Add Samples and Virus to the Fetuin-coated Plate
- Thaw a vial of virus, vortex and resuspend the virus in the diluent (section 1.2) at the dilution that was selected in step 2.4.2. Prepare at least 5 ml of virus for each assay plate. Keep the diluted virus on ice until the plates are washed and serum samples have been added to the plate.
- Decide on the number of fetuin-coated plates that are needed for the assay (generally apply 4 sera per plate). Wash the fetuin-coated plate 3 times with PBS-T and then invert each plate and blot onto absorbent paper towel to remove excess wash buffer.
- Use a multichannel pipette to transfer 50 µl of each serum control or sample dilution from the dilution plate into duplicate wells in columns 2-11.
- Add 50 µl of diluted virus to all wells except for the negative control (column 12).
- Add 50 µl of sample diluent to wells in column 1 and add 100 µl of sample diluent to column 12.
- Cover the wells with a plate sealer and then mix by gently tapping sides of plate or placing on a plate shaker at moderate speed for 10 sec.
- Place the plate in a humidified incubator at 37 °C for 16-18 hr.
Add PNA-HRPO and complete the assay as described in section 2.3
4. Data Analysis
- Determine the Validity of the Assay Results
- Confirm that the background values (no virus) are less than 10% of the positive control (virus and no serum).
- Confirm that the titers of control sera run in different assays using the same conditions are within 2-fold of the median titer.
- Confirm that OD measurements of control wells are consistent (≤20% different) and that OD measurements of duplicate sample wells are consistent (≤10% different).
- Determine the root cause of invalid results and repeat the assay if the criteria listed in 4.1.1, 4.1.2 or 4.1.3, are not met. Consider the factors presented in Table 1 when trying to troubleshoot.
- Assign a 50% End-point Titer
- For each assay plate, subtract the average background (no antigen added to wells) from all readings.
- Calculate the percent inhibition at each serum dilution using the formula: 100 x (ODvirus only control - ODtest sample)/ ODvirus only control.
- Identify the highest dilution that resulted in at least 50% inhibition of the maximum signal.
- Report the reciprocal of this dilution as the 50% end-point titer. Note: If 50% inhibition was not achieved at any dilution, the titer is less than the first dilution tested, e.g., <10 when 1:10 is the first dilution tested.
- Calculate the 50% Inhibitory Concentration (IC50)
- Use four parameter logistic regressions to determine the IC50 titer as follows: subtract the average background from all readings and then transfer the results to a program that performs regression analysis.
- Use non-linear regression, with the maximum set to the virus only control and the minimum set to zero, to determine the dilution that corresponds with exactly 50% inhibition.
- Report the reciprocal of this dilution as the IC50 titer.
Representative Results
The assay presented in this manuscript is shown diagrammatically in Figure 1. Figure 2 shows the 96 well plate layout, and indicates that serial dilutions of the serum samples are prepared in a dilution plate before transferring them to the fetuin-coated plate. A critical parameter of the assay is the amount of neuraminidase that is used in the assay; this is determined through titration of the reassortant virus. Examples of H6N1 and H6N2 virus titrations are shown in Figure 3. At 1:10, 1:20 and 1:40 dilutions of H6N1, the optical density was similar, indicating that the conditions were such that a maximum reading was obtained. At a 1:80 dilution, the read-out was approximately 90% of the maximum and therefore this dilution of virus was used in subsequent assays. Examples of serum titrations are shown in Figure 4. The figure shows a human serum sample that was titrated against H6N1 and H6N2 viruses. Each data point shows the percent inhibition of NA activity that was achieved by serial dilutions of serum; the first serum dilution in the H6N1 assay was 1:80; the first dilution of serum in the H6N2 assay was 5. Since the highest dilution that results in ≥50% inhibition is the NI antibody titer, the titer of the serum shown in this example is 640 against the NA of NC/99 (N1) and 160 against the NA of WI/05 (N2).
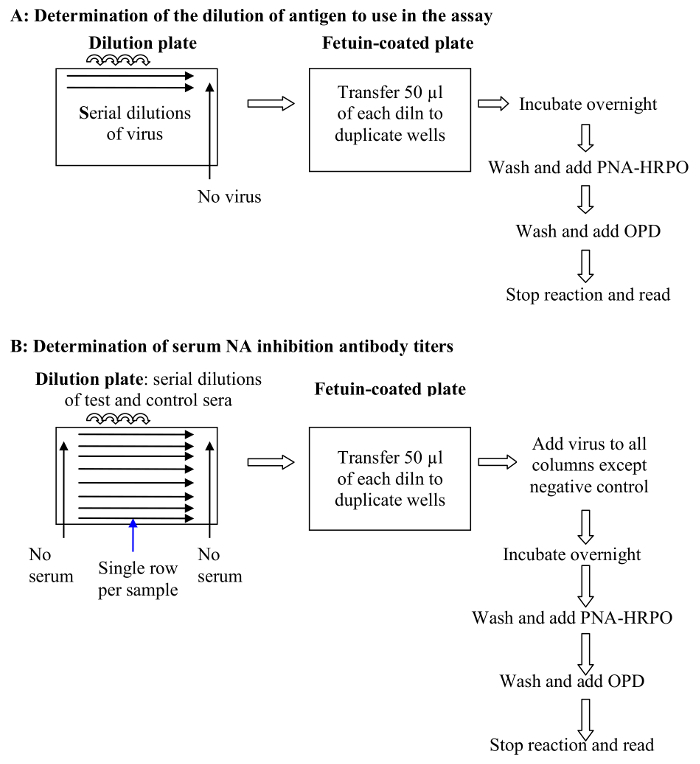 Figure 1.A schematic of the ELLA protocol. (A) Determination of the dilution of antigen (virus) to use in the assay (step 2 of the protocol): Virus dilutions are added to a fetuin coated plate and the NA activity measured by quantifying terminal galactose residues through binding of PNA-HRPO as described in step 2.3 of the protocol; (B) Determination of serum NI antibody titers (step 3 of the protocol). Samples are diluted and then transferred to a fetuin-coated plate where virus is added. The plate is incubated O/N before adding PNA-HRPO and completing the steps needed to generate a color reaction. Finally, the color reaction is stopped and the optical density is measured. The reciprocal of the sample dilution that results in at least 50% inhibition of NA activity is reported as the 50% end-point titer. Please click here to view a larger version of this figure.
Figure 1.A schematic of the ELLA protocol. (A) Determination of the dilution of antigen (virus) to use in the assay (step 2 of the protocol): Virus dilutions are added to a fetuin coated plate and the NA activity measured by quantifying terminal galactose residues through binding of PNA-HRPO as described in step 2.3 of the protocol; (B) Determination of serum NI antibody titers (step 3 of the protocol). Samples are diluted and then transferred to a fetuin-coated plate where virus is added. The plate is incubated O/N before adding PNA-HRPO and completing the steps needed to generate a color reaction. Finally, the color reaction is stopped and the optical density is measured. The reciprocal of the sample dilution that results in at least 50% inhibition of NA activity is reported as the 50% end-point titer. Please click here to view a larger version of this figure.
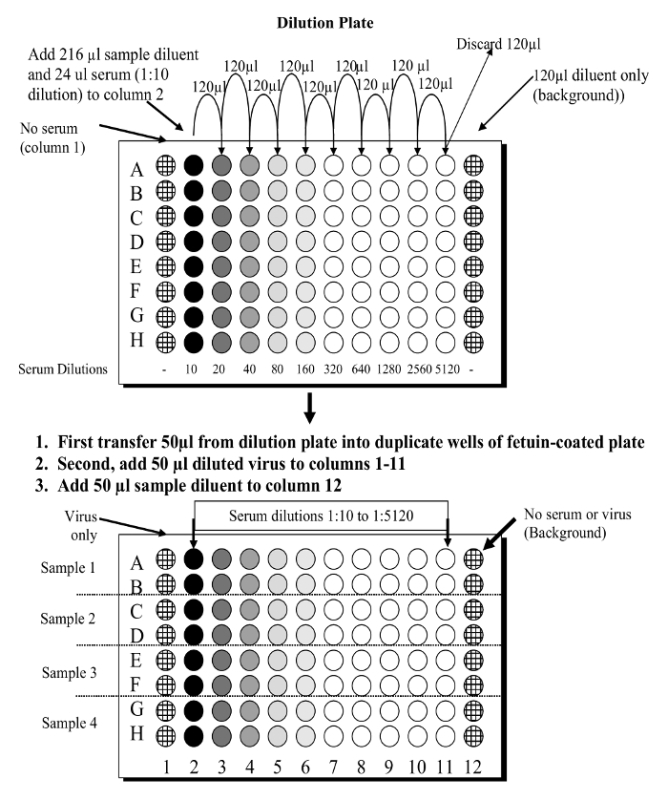 Figure 2. Diagram to show the ELLA plate set up. Serial dilutions of serum samples are made in a dilution plate and then transferred to a fetuin-coated plate. Please click here to view a larger version of this figure.
Figure 2. Diagram to show the ELLA plate set up. Serial dilutions of serum samples are made in a dilution plate and then transferred to a fetuin-coated plate. Please click here to view a larger version of this figure.
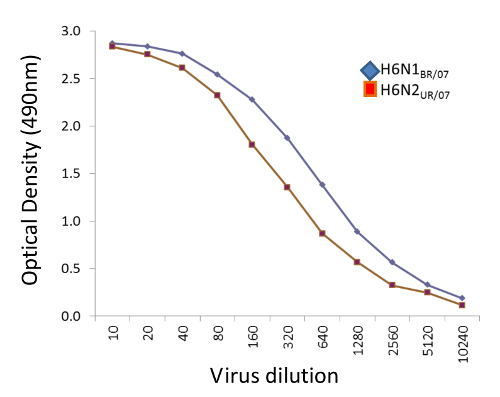 Figure 3.Examples of H6N1 and H6N2 virus titrations. Serial dilutions of H6N1BR/07 (NA of A/Brisbane/59/2007 (H1N1) shown in blue symbols) and H6N2UR/07 (NA of A/Uruguay/716/2007 (H3N2) shown in red symbols)were incubated for 18 hr in fetuin-coated plates and the reactivity with PNA-HRPO determined as described. AverageOD490nm of 2 wells is plotted against the reciprocal of the virus dilution. The dilution of H6N1 selected for use in the ELLA was 1:80 and the dilution of H6N2 selected was 1:40 because these dilutions resulted in approximately 90% of the maximum optical density and were within the linear range. Please click here to view a larger version of this figure.
Figure 3.Examples of H6N1 and H6N2 virus titrations. Serial dilutions of H6N1BR/07 (NA of A/Brisbane/59/2007 (H1N1) shown in blue symbols) and H6N2UR/07 (NA of A/Uruguay/716/2007 (H3N2) shown in red symbols)were incubated for 18 hr in fetuin-coated plates and the reactivity with PNA-HRPO determined as described. AverageOD490nm of 2 wells is plotted against the reciprocal of the virus dilution. The dilution of H6N1 selected for use in the ELLA was 1:80 and the dilution of H6N2 selected was 1:40 because these dilutions resulted in approximately 90% of the maximum optical density and were within the linear range. Please click here to view a larger version of this figure.
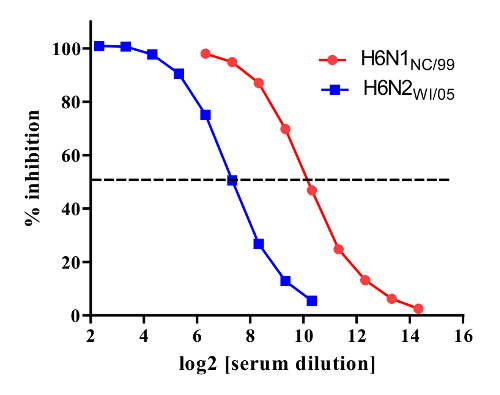 Figure 4.Titration of serum against NA of N1 (red symbols) and N2 (blue symbols) subtypes. NA inhibition titers were measured against reassortant viruses H6N1NC/99 (contains the NA of A/New Caledonia/20/1999 (H1N1)) and H6N2WI/05 (contains the NA of A/Wisconsin/67/2005 (H3N2)). Percent enzyme inhibition for serial dilutions of serum is shown with the dashed horizontal line indicating 50% inhibition. The NA inhibition titers against the NAs of A/New Caledonia/20/1999 (H1N1) (NC/99) and A/Wisconsin/67/2005 (H3N2) (WI/05) were 29.3 (640) and 27.3(160), respectively. Please click here to view a larger version of this figure.
Figure 4.Titration of serum against NA of N1 (red symbols) and N2 (blue symbols) subtypes. NA inhibition titers were measured against reassortant viruses H6N1NC/99 (contains the NA of A/New Caledonia/20/1999 (H1N1)) and H6N2WI/05 (contains the NA of A/Wisconsin/67/2005 (H3N2)). Percent enzyme inhibition for serial dilutions of serum is shown with the dashed horizontal line indicating 50% inhibition. The NA inhibition titers against the NAs of A/New Caledonia/20/1999 (H1N1) (NC/99) and A/Wisconsin/67/2005 (H3N2) (WI/05) were 29.3 (640) and 27.3(160), respectively. Please click here to view a larger version of this figure.
| Problem | Possible cause(s) | Solution |
| Weak signal or no plateau reached in virus titration | i) low NA enzyme activity ii) virus stock not stored under optimal conditions | i) confirm that the diluent has pH that is optimal for NA activity; if pH is optimal, prepare a new virus stock or concentrate virus ii) Regrow and aliquot stock; snap-freeze vials on dry ice before storing at -80 °C |
| Weak or no color in positive cell control wells | i) Virus dilution incorrectly assigned ii) Vial-to-vial variability in frozen virus aliquots iii) PNA- HRPO denatured or diluted too much iv) OPD incorrectly prepared | i) Repeat virus titration ii) Titrate several vials from the same batch to ensure there is no variability. If significant variability, prepare fresh aliquots iii) Use optimum virus dilution to retitrate PNA- HRPO iv) Repeat with fresh preparation of OPD |
| Weak or no inhibition by positive control sera | i) Too much virus used in assay ii) Serum deteriorated | i) Repeat virus titration ii) Obtain new antisera Check storage conditions |
| Inhibition by negative control sera | i) Inadequate heat-treatment of serum ii) Too little virus used in assay | i) Repeat heat-inactivation of serum ii) Repeat virus titration |
| High background | i) Possible contamination of plates ii) PNA- HRPO concentration too high/too low | i) Repeat using freshly coated plates ii) Titrate PNA-HRPO to identify correct dilution to use |
| Virus titration shows apparent inhibition of NA activity at low dilutions | i) Allantoic fluid may contain substrate for NA | ii) Use virus that has been pelleted through a sucrose cushion |
Table 1. Root cause analysis of assays that do not meet performance criteria. This table provides possible causes and solutions for problems that may occur when performing the ELLA.
Discussion
The enzyme-linked lectin assay is a practical method to measure NI antibody titers in sera. Although ELLA was described by Lambre et al., in 1990, its acceptance as a standard serologic assay has been more recent, with numerous laboratories performing the assay to measure NI antibody titers of clinical samples 12-16. Some modifications can be made to protocol steps without a large impact to the NI titers that are measured. For example, PBS can be used as diluent, however, it is very helpful to compare the virus titration curves in the recommended buffer (MES, pH 6.5) and PBS before a decision is made, as some NA subtypes have significantly reduced enzyme activity at pH >7.0. When the optimal pH is not used, the maximum signal of virus may be reduced, resulting in the use of an excessive amount of antigen (i.e., virus) in the assay; under these conditions, the assay may have reduced sensitivity.
Some non-specific inhibition is observed when untreated sera are tested in the ELLA. This is removed by heat-treatment (56 °C for 45 min), indicating the presence of thermolabile β-inhibitors. Other non-specific inhibitors (α and γ-class) of influenza HA have been described, particularly in relation to H3N2 virus hemagglutination and infectivity. Indeed, non-specific inhibition is also observed in ELLA when H3N2 viruses are used as the source of NA; this inhibition is removed by treatment of the serum samples with a small amount of sialidase 9.
As for other serologic assays, negative and positive controls should be included in each assay to provide a means to evaluate assay performance. When the assay has not met acceptance criteria, the reason should be identified. Table 1 provides a list of potential steps in the assay that can be addressed to troubleshoot problems.
The ELLA protocol provided in this report uses reassortant viruses that contain the HA originating from an avian virus, as the source of NA. This presents a limitation to performing the assay because a permit is required to work with low pathogenic avian viruses. H6Nx reassortant viruses can be shared between laboratories after they have been inactivated. Therefore the assay can be conducted through collaborations once the laboratory generating the reassortant virus has demonstrated that the inactivation is complete and the preparation has retained its NA activity. The constraint of using H6Nx reassortant viruses can be overcome by using non-infectious sources of NA. For example, some laboratories have used purified recombinant NA 17, while others have used virus-like particles (VLPs) 15. However, neither recombinant NA nor VLPs are readily available and therefore we continue to use influenza viruses with an antigenically-mismatched avian HA.
The traditional method to measure NI antibody titers is not practical for several reasons; of most concern are the harmful chemicals that are needed to produce a color reaction. In addition, large volumes of sample are needed and the assay is cumbersome to perform. In contrast, the ELLA is easy to perform and is in a format that allows titration of a reasonable number of samples. Data from studies that examined ELLA variability show that the assay yields reproducible results 7,8. The ELLA has consequently made routine measurement of NI antibody titers possible, and it is anticipated that it will be used more frequently by laboratories conducting serologic studies.
There are several critical steps that need to be considered when performing the ELLA. First, non-specific inhibitors of NA activity must be removed. These inhibitors are generally thermolabile and are destroyed by heat treatment at 56 °C for 45 min. Heat treatment is sufficient to remove non-specific inhibitors from samples when H6 reassortant viruses are used as the source of NA, however, when H3N2 viruses are used in the ELLA, serum factors that bind to hemagglutinin also interfere with the ELLA and should be removed by treatment with a small amount of sialidase prior to heat treatment 9. Second, an excessive amount of antigen, i.e., reassortant H6Nx virus, should not be used as this reduces the sensitivity of the assay. Careful titration of virus is therefore an essential step; the amount of antigen used in each assay must provide a signal that is well above the background, and must be within the linear range of the titration curve, i.e., the signal (optical density) must be proportional to the virus dilution.
In addition to routine serology, the ELLA can be used to evaluate antigenic differences between NAs of seasonal influenza viruses. This information may be very helpful when selecting virus strains for inclusion as vaccine candidates and will facilitate a greater understanding of immune pressures that result in antigenic drift of NA. In addition, antigenic analysis of NAs from swine or avian influenza viruses may provide critical information in determining the pandemic potential of emerging strains.
Disclosures
The authors have no conflicts of interest.
Acknowledgments
This project was funded by intramural PanFlu funds from the Center for Biologics Evaluation and Research (CBER). Plasmids used to prepare reassortant viruses were kindly provided by Robert Webster, St Jude Children's Research Hosptial, Memphis, TN. We are indebted to Ewan Plant and Haruhiko Murata for critical review of the manuscript.
References
- Kilbourne ED, Laver WG, Schulman JL, Webster RG. Antiviral activity of antiserum specific for an influenza virus neuraminidase. J Virol. 1968;2(4):281–288. doi: 10.1128/jvi.2.4.281-288.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compans RW, Dimmock NJ, Meier-Ewert H. Effect of antibody to neuraminidase on the maturation and hemagglutinating activity of an influenza A2 virus. J Virol. 1969;4(4):528–534. doi: 10.1128/jvi.4.4.528-534.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman JL, Khakpour M, Kilbourne ED. Protective effects of specific immunity to viral neuraminidase on influenza virus infection of mice. J Virol. 1968;2(8):778–786. doi: 10.1128/jvi.2.8.778-786.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BR, Kasel JA, Chanock RM. Association of serum anti-neuraminidase antibody with resistance to influenza in man. N Engl J Med. 1972;286(25):1329–1332. doi: 10.1056/NEJM197206222862502. [DOI] [PubMed] [Google Scholar]
- Sandbulte MR, Gao J, Straight TM, Eichelberger MC. A miniaturized assay for influenza neuraminidase-inhibiting antibodies utilizing reverse genetics-derived antigens. Influenza Other Respi Viruses. 2009;3(5):233–240. doi: 10.1111/j.1750-2659.2009.00094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambre CR, Terzidis H, Greffard A, Webster RG. Measurement of anti-influenza neuraminidase antibody using a peroxidase-linked lectin and microtitre plates coated with natural substrates. J Immunol Methods. 1990;135(1-2):49–57. doi: 10.1016/0022-1759(90)90255-t. [DOI] [PubMed] [Google Scholar]
- Couzens L, et al. An optimized enzyme-linked lectin assay to measure influenza A virus neuraminidase inhibition antibody titers in human sera. J Virol Methods. 2014;210C:7–14. doi: 10.1016/j.jviromet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Eichelberger MC, et al. Comparability of neuraminidase inhibition antibody titers measured by enzyme-linked lectin assay (ELLA) for the analysis of influenza vaccine immunogenicity. Vaccine. 2016;34(4):458–465. doi: 10.1016/j.vaccine.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westgeest KB, et al. Optimization of an enzyme-linked lectin assay suitable for rapid antigenic characterization of the neuraminidase of human influenza A(H3N2) viruses. J Virol Methods. 2015;217:55–63. doi: 10.1016/j.jviromet.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A. 2000;97(11):6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Manual of Animal Influenza Diagnosis and Surveillance. 2002.
- Cate TR, et al. A high dosage influenza vaccine induced significantly more neuraminidase antibody than standard vaccine among elderly subjects. Vaccine. 2010;28(9):2076–2079. doi: 10.1016/j.vaccine.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch RB, et al. Antibody correlates and predictors of immunity to naturally occurring influenza in humans and the importance of antibody to the neuraminidase. J Infect Dis. 2013;207(6):974–981. doi: 10.1093/infdis/jis935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz R, et al. Neuraminidase-Inhibiting Antibody Response to H5N1 Virus Vaccination in Chronically Ill and Immunocompromised Patients. Open Forum Infect Dis. 2014;1(2) doi: 10.1093/ofid/ofu072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries LF, Smith GE, Glenn GM. A recombinant viruslike particle influenza A (H7N9) vaccine. N Engl J Med. 2013;369(26):2564–2566. doi: 10.1056/NEJMc1313186. [DOI] [PubMed] [Google Scholar]
- Monto AS, et al. Antibody to Influenza Virus Neuraminidase: An Independent Correlate of Protection. J Infect Dis. 2015;212(8):1191–1199. doi: 10.1093/infdis/jiv195. [DOI] [PubMed] [Google Scholar]
- Fritz R, et al. A vero cell-derived whole-virus H5N1 vaccine effectively induces neuraminidase-inhibiting antibodies. J Infect Dis. 2012;205(1):28–34. doi: 10.1093/infdis/jir711. [DOI] [PubMed] [Google Scholar]