

Abstract
The etiology and impacts of human exposure to environmental pathogens are of major concern worldwide and, thus, the ability to assess exposure and infections using cost effective, high-throughput approaches would be indispensable. This manuscript describes the development and analysis of a bead-based multiplex immunoassay capable of measuring the presence of antibodies in human saliva to multiple pathogens simultaneously. Saliva is particularly attractive in this application because it is noninvasive, cheaper and easier to collect than serum. Antigens from environmental pathogens were coupled to carboxylated microspheres (beads) and used to measure antibodies in very small volumes of human saliva samples using a bead-based, solution-phase assay. Beads were coupled with antigens from Campylobacter jejuni, Helicobacter pylori, Toxoplasma gondii, noroviruses (G I.1 and G II.4) and hepatitis A virus. To ensure that the antigens were sufficiently coupled to the beads, coupling was confirmed using species-specific, animal-derived primary capture antibodies, followed by incubation with biotinylated anti-species secondary detection antibodies and streptavidin-R-phycoerythrin reporter (SAPE). As a control to measure non-specific binding, one bead set was treated identically to the others except it was not coupled to any antigen. The antigen-coupled and control beads were then incubated with prospectively-collected human saliva samples, measured on a high throughput analyzer based on the principles of flow cytometry, and the presence of antibodies to each antigen was measured in Median Fluorescence Intensity units (MFI). This multiplex immunoassay has a number of advantages, including more data with less sample; reduced costs and labor; and the ability to customize the assay to many targets of interest. Results indicate that the salivary multiplex immunoassay may be capable of identifying previous exposures and infections, which can be especially useful in surveillance studies involving large human populations.
Keywords: Immunology, Issue 115, Multiplex, immunoassay, salivary antibody, saliva, exposure, bead-based multiplexing, carboxylated microspheres, bead coupling, coupling confirmation
Introduction
Eighty-eight percent of diarrhea-related illness worldwide is associated with human exposure to contaminated water, unsafe food, and poor sanitation/hygiene, causing approximately 1.5 million deaths, the majority of whom are children1. This is a major cause of concern for public health officials and policy makers. In an effort to investigate exposures and illnesses associated with waterborne and other environmental pathogens, we developed a multiplex immunoassay to measure antibodies in human samples2-4. This method can be applied to epidemiological studies to determine human exposure to these pathogens and to better define immunoprevalence and incident infections.
Saliva holds considerable promise as an alternative to serum for human biomarker research. Among the advantages of using saliva are the non-invasiveness and ease of sample collection, low cost, and samples can easily be collected from children5-7. Serum and saliva samples have been studied extensively for antibodies against H. pylori2,3,8, Plasmodium falciparum9, Entamoeba histolytica10, Cryptosporidium parvum3,11, Streptococcus pneumonia12, hepatitis viruses A and C13-14, noroviruses2-4,15, T. gondii2-4, dengue virus16, human immunodeficiency virus (HIV)17, and Escherichia coli O157:H718.
A multiplex immunoassay allows for the analysis of multiple analytes simultaneously within a single sample volume and within a single cycle or run. Multiplexed antigens from C. jejuni, T. gondii, H. pylori, hepatitis A virus, and two noroviruses were used to measure human salivary IgG2-4 and IgA3,4 and plasma IgG2,3 antibody responses to these pathogens using a bead-based multiplexing immunoassay. When used in conjunction with epidemiological studies of exposure to microbes in water, soil and food, the type of assay described in this study may provide valuable information to enhance the understanding of infections caused by environmental pathogens. Moreover, salivary antibody data obtained from such studies can be used to improve risk assessment models19-22.
Protocol
Approval was obtained from the Institutional Review Board (IRB # 08-1844, University of North Carolina, Chapel Hill, NC, USA) for the collection of stimulated crevicular saliva samples from beachgoers at Boquerón Beach, Puerto Rico, as part of the United States Environmental Protection Agency (USEPA) National Epidemiological and Environmental Assessment of Recreational (NEEAR) Water Study23 to assess swimming associated exposures and illnesses. Study subjects provided informed consent and were instructed on the use of the saliva collection device by trained USEPA contractors. The saliva samples were shipped on ice and, upon receipt, they were centrifuged and stored at -80 °C as described4.
1. Bead Activation
Resuspend bead set stocks by vortexing and sonicating for 20 sec and transfer approximately 5.0 x 106 of the stock beads (400 µl) to microcentrifuge tubes. Note: The beads are supplied at a concentration of 12.5 x 106 beads/ml.
Pellet the stock beads by centrifuging at 10,000 x g for 2 min.
Remove the supernatant and resuspend the pelleted beads in 100 µl distilled water (dH2O) by vortex and sonication for 20 sec. Repeat step 1.2.
Remove the supernatant and resuspend the washed beads in 80 µl of 100 mM sodium phosphate monobasic, pH 6.2 by vortex and sonication for 20 sec.
Immediately before use, make a 50 mg/ml N-hydroxysulfosuccinimide (Sulfo-NHS) solution by adding 200 µl dH2O to the 10 mg Sulfo-NHS aliquot. Mix by vortex.
Add 10 µl of the 50 mg/ml Sulfo-NHS to the beads. Mix by vortex.
Immediately before use, make a 50 mg/ml 1-ethyl-[3dimethylaminopropyl] carbodiimide hydrochloride (EDC) solution by adding 200 µl distilled water (dH2O) to the 10 mg EDC aliquot. Mix by vortex.
Add 10 µl of 50 mg/ml EDC solution to the beads. Mix by vortex. Incubate the beads for 20 min at room temperature, in the dark, with mixing by vortex at 10 min intervals. Pellet the activated beads by microcentrifugation at 10,000 x g for 2 min.
Remove the supernatant and resuspend the beads in 250 µl of 50 mM 2-[N-Morpholino]ethanesulfonic acid (MES), pH 5.0 by vortex and sonication for 20 sec.
Pellet the activated beads by microcentrifugation at 10,000 x g for 2 min.
Repeat steps 1.9 and 1.10. Note: This provides a total of two washes with 50 mM MES, pH 5.0.
Resuspend the beads in 100 µl of 50 mM MES, pH 5.0 by vortex and sonication for 20 sec.
2. Bead Coupling
Couple the antigens to the bead sets using the concentrations shown in Table 1.
Add each antigen to the activated beads and bring the total volume to 500 µl in 50 mM MES, pH 5.0. Mix the antigens and beads by vortex.
Incubate the antigens and beads for 2 hr with mixing by rotation (~15 rpm) at room temperature in the dark. Pellet the coupled beads by microcentrifugation at 10,000 x g for 2 min.
Remove the supernatant and resuspend the beads in 500 µl of phosphate buffered saline (PBS)-bovine serum albumin (BSA)-polyoxyethylenesorbitan monolaurate (Tween-20)-sodium azide (PBS-TBN) pH 7.4 by vortex and sonication. Pellet the beads by microcentrifugation at 10,000 x g for 2 min and remove the supernatant. Caution: Sodium azide is an acutely toxic chemical. It is fatal if swallowed or gets in contact with skin. Do not breathe dust/fume/gas/mist/vapors or spray. Wear appropriate personal protective equipment (PPE's) when handling and dispose of in accordance with appropriate laws.
Resuspend the beads in 1 ml of PBS-TBN by vortex and sonication for 20 sec.
Pellet the beads by microcentrifugation at 10,000 x g for 2 min.
Repeat steps 2.5 and 2.6. Note: This provides a total of two washes with PBS-TBN.
Resuspend the coupled and washed beads in 1 ml of PBS, 1% BSA, 0.05% Azide, pH 7.4. Store the coupled beads in a 2-8 °C refrigerator in the dark.
3. Bead Count
Prepare a 1:10 dilution of the coupled beads in water or PBS buffer.
Load 10 µl of the bead dilution onto a hemocytometer at the sample introduction point.
Count the beads seen in one of the 4 x 4 corner grids. Calculate the total number of coupled beads using the following formula: Count (1 corner of 4 x 4 grid) x (1 x 104) x (dilution factor) x resuspension volume in ml.
4. Confirmation of Antigen Coupling
Resuspend the stock mixture of beads coupled to antigens of interest by vortex and sonication for 20 sec.
Prepare a working bead mixture by diluting the coupled bead stocks to a final concentration of 100 beads /µl of each unique bead set in PBS-1% BSA buffer (PBS-1% BSA, pH 7.4).
Prepare at least 7 two-fold serial dilutions of anti-species IgG primary antibody according to manufacturer's recommendations in 96-well round bottom plate with PBS-1% BSA buffer.
Pre-wet a separate 8-well column (8 rows) of a 96-well filter bottom plate for each antigen coupling confirmation test with 100 µl of wash buffer and remove supernatant by vacuum. Add 50 µl of working bead mixture (antigen-coupled beads) to the pre-wet wells.
Add 50 µl of antibody dilutions to rows 1-7 of each column of the 96-well filter plate and 50 µl of PBS-1% BSA buffer to row 8 in lieu of diluted antibody to serve as background wells. Mix with a multi-channel pipettor by pipetting up and down 5 times. Perform the same procedure for every antigen-coupled bead set to be confirmed.
Cover and allow to incubate in the dark at room temperature for 1 hr on a microplate shaker at 500 rpm. Remove supernatant by vacuum.
Wash wells with 100 µl of wash buffer and remove supernatant by vacuum. Repeat 1x. Resuspend the beads in 50 µl of PBS-1% BSA with a multi-channel pipettor.
Dilute biotinylated anti-species specific IgG secondary detection antibody to 16 µg/ml in PBS-1% BSA.
Add 50 µl of diluted secondary antibody to each well by pipetting up and down 5 times.
Cover filter plate and allow to incubate in the dark at room temperature for 30 min on a plate shaker. Remove supernatant by vacuum. Wash wells with 100 µl of wash buffer and remove supernatant by vacuum. Repeat wash 1x.
Resuspend the beads in 50 µl of PBS-1% BSA with a multi-channel pipettor.
Dilute streptavidin-R-phycoerythrin reporter (SAPE) to 24 µg/ml in PBS-1% BSA.
Add 50 µl of reporter to each well and mix by pipetting up and down 5 times.
Cover filter plate and allow to incubate in the dark at room temperature for 30 min on a plate shaker. Remove supernatant by vacuum. Wash wells with 100 µl of wash buffer and remove supernatant by vacuum. Repeat wash 1x.
Resuspend beads in 100 µl of PBS-1% BSA and analyze 50 µl using the analyzer29. Note: Results of the bead-based multiplex immunoassay are measured in Median Fluorescence Intensity (MFI). Always refer to the latest version of the software manual, if available to avoid errors.
5. Salivary Multiplex Immunoassay
Remove saliva from the -80 °C freezer and allow to thaw at room temperature.
Resuspend antigen coupled bead stocks by vortex and sonication for 20 sec.
Prepare a working bead mixture by diluting the coupled bead stocks to a final concentration of 100 beads/µl of each unique bead set in PBS-1% BSA buffer.
Prepare a 1:4 dilution of saliva with PBS-1% BSA buffer in a 96 well, deep well plate.
Pre-wet filter plate with 100 µl of wash buffer and remove supernatant by vacuum.
Add 50 µl of a working bead mixture and an equal volume of diluted saliva to 95 wells of the 96 well filter plates for a 1:8 final dilution. Mix reactions with a multi-channel pipettor. To the one control well, add 50 µl antigen-coupled beads plus 50 µl of PBS-1% BSA buffer (as a replacement for saliva).
Cover and allow to incubate in the dark at room temperature for 1 hr on a microplate shaker at 500 rpm. Remove supernatant by vacuum. Wash wells with 100 µl of wash buffer and remove supernatant by vacuum. Repeat wash 1x.
Resuspend beads in 50 µl of PBS-1% BSA with a multi-channel pipettor.
Dilute biotinylated goat anti-human IgG secondary detection antibody to 16 µg/ml in PBS-1% BSA.
Add 50 µl diluted secondary antibody to each well and mix contents with a multi-channel pipettor.
Cover filter plate and allow to incubate in the dark at room temperature for 30 min on a plate shaker. Remove supernatant by vacuum. Wash wells with 100 µl of wash buffer and remove supernatant by vacuum. Repeat wash 1x.
Resuspend beads in 50 µl of PBS-1% BSA with a multi-channel pipettor.
Dilute streptavidin-R-phycoerythrin reporter (SAPE) to 24 µg/ml in PBS-1% BSA.
Add 50 µl reporter to each well and mix with a multi-channel pipettor.
Cover filter plate and allow to incubate in the dark at room temperature for 30 min on a plate shaker. Remove supernatant by vacuum. Wash wells with 100 µl of wash buffer and remove supernatant by vacuum. Repeat wash 1x.
Resuspend beads in 100 µl of PBS-1% BSA and analyze 50 µl using the analyzer29. Note: Results of the multiplex immunoassay are measured in Median Fluorescence Intensity (MFI) units. Always refer to the latest version of the software manual, if available to avoid errors.
Representative Results
One unique bead set was used as a control to measure non-specific binding and sample to sample variability. These beads were treated identically to the antigen coupled beads with the exception that they were not incubated with any antigen in the coupling step. MFI values >500 obtained from the control beads incubated with all saliva samples were removed from further analyses due to suspected contamination from serum and the remaining responses were log distributed. The saliva can be contaminated with serum if the gums are rubbed too vigorously with the sponge attached to the collection device or if the study participant has periodontal disease. The log transformed MFI data were used to calculate an immunopositive cut-off point of 505 MFI based on the mean plus 3 standard deviations of the log MFI values. Additionally, antigen-coupled beads were added to specific wells that were treated as test wells in every manner except diluted saliva was replaced with PBS-1% BSA to evaluate background fluorescence and cross-reactivity. The MFI values obtained in these background wells were subtracted from the MFI values from each antigen-coupled bead set for each saliva sample.
Bead coupling was confirmed using animal derived species-specific primary detection antibodies specific for each antigen. To ensure that the antigen coupled beads were capable of approaching the dynamic range of the assay, we defined proper coupling as an MFI ≥18,000, observation of dose response and cross-reactivity between primary antibodies and non-targets of <10% as described2. Representative coupling confirmations for C. jejuni and T. gondii of the antigens in the assay are shown in Figure 1.
Based on the cut-off point established (505 MFI), the immunoprevalence rates for the samples (n = 2,078) ranged from about 2% (n = 41) for T. gondii to nearly 50% (n = 1,009) for norovirus GI.1 (Figure 2). The data indicate that the immunoprevalence rate was highest for the noroviruses followed by hepatitis A virus and H. pylori.
While 32% (n = 672) of the samples were immunonegative for all of the pathogens in the assay, 68% (n = 1,406) were immunopositive to one or more pathogens. Figure 3 shows the breakdown of immunopositivity to one or more of the pathogens.
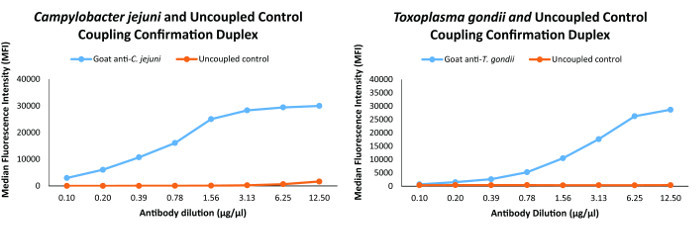 Figure 1: Coupling confirmation analyses for two of the organisms in the multiplex immunoassay. Coupling confirmation was performed for all of the antigens and the graphs for C. jejuni and T. gondii in this figure are representative of the coupling confirmations for the antigens in the multiplex immunoassay. Please click here to view a larger version of this figure.
Figure 1: Coupling confirmation analyses for two of the organisms in the multiplex immunoassay. Coupling confirmation was performed for all of the antigens and the graphs for C. jejuni and T. gondii in this figure are representative of the coupling confirmations for the antigens in the multiplex immunoassay. Please click here to view a larger version of this figure.
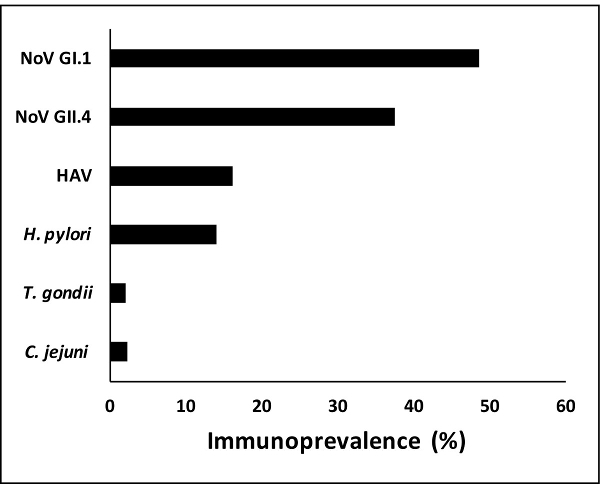 Figure 2:Immunoprevalence to specific pathogens. Immunoprevalence to specific pathogens in the multiplex immunoassay ranged from about 2% for T. gondii to nearly 50% for norovirus GI.1. Antibodies against the norovirus antigens were more commonly observed followed by HAV (hepatitis A virus) and H. pylori. T. gondii and C. jejuni antibodies appeared less frequently in the saliva samples analyzed. Please click here to view a larger version of this figure.
Figure 2:Immunoprevalence to specific pathogens. Immunoprevalence to specific pathogens in the multiplex immunoassay ranged from about 2% for T. gondii to nearly 50% for norovirus GI.1. Antibodies against the norovirus antigens were more commonly observed followed by HAV (hepatitis A virus) and H. pylori. T. gondii and C. jejuni antibodies appeared less frequently in the saliva samples analyzed. Please click here to view a larger version of this figure.
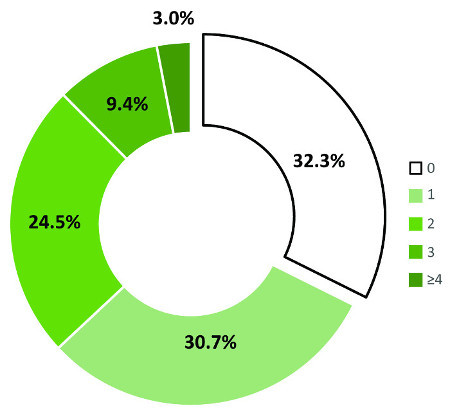 Figure 3: Breakdown of immunopositivity to multiple pathogens simultaneously. The multiplex immunoassay allows for the analysis of immunopositivity to multiple pathogens simultaneously in a 50 µl sample volume. Almost a third of the saliva samples assayed was immunonegative for antibodies against all of the antigens in the multiplex. Approximately 31% of the samples was immunopositive for one antigen while a quarter was positive to two antigens. 9.4% was immunopositive to three antigens. 3% was positive to four or more antigens simultaneously. Please click here to view a larger version of this figure.
Figure 3: Breakdown of immunopositivity to multiple pathogens simultaneously. The multiplex immunoassay allows for the analysis of immunopositivity to multiple pathogens simultaneously in a 50 µl sample volume. Almost a third of the saliva samples assayed was immunonegative for antibodies against all of the antigens in the multiplex. Approximately 31% of the samples was immunopositive for one antigen while a quarter was positive to two antigens. 9.4% was immunopositive to three antigens. 3% was positive to four or more antigens simultaneously. Please click here to view a larger version of this figure.
| Antigen | Bead Set | Coupling buffer | Ag Concentration (µg) | # of beads in 1 corner | # of beads/µl | Vol. for 100 B/uL for 4 ml (µl) |
| C. jejuni | 8 | MES 5.0 | 50 | 64 | 6,400 | 94 |
| H. pylori | 33 | MES 5.0 | 25 | 71 | 7,100 | 85 |
| Hepatitis A | 42 | MES 5.0 | 100 | 91 | 9,100 | 66 |
| T. gondii | 30 | MES 5.0 | 25 | 85 | 8,500 | 71 |
| Norovirus GII.4 | 55 | MES 5.0 | 5 | 72 | 7,200 | 83 |
| Norovirus GI.1 | 67 | MES 5.0 | 5 | 63 | 6,300 | 95 |
| Uncoupled | 80 | MES 5.0 | 60 | 6,000 | 100 | |
| Total volume of beads (µl) | 594 | |||||
| Total volume of buffer needed (µl) | 3,406 |
Table 1: Working bead mix for the multiplex immunoassay. After coupling and confirmation were completed, a master mix consisting of each bead set was prepared as shown. The master mix was distributed among all of the wells in the 96 well plate. All of the wells were incubated with saliva with the exception of one background control well which contained only beads and PBS-1% BSA buffer.
Discussion
These results indicate that the multiplex immunoassay method is useful for discriminating between saliva samples that are immunopositive or immunonegative. To determine immunopositivity, a single cut-off point was developed by calculating the mean plus three standard deviations of the log transformed MFI responses of the control uncoupled beads tested with all of the saliva samples. The cut-off point afforded the ability to assess exposure and immunoprevalence to either a single or multiple pathogens. This discriminative power may be useful in population studies to investigate health risks associated with exposure to environmental and other pathogens.
There are several other methods that can be used to determine a cut-off point depending on the distribution of the MFIs, uncoupled controls and what question the study is designed to answer. Some examples for determining cut-offs include finite mixed modeling24-26, mean plus 3 standard deviations of responses to controls, mean plus 2 standard deviations of responses to controls, and three times the mean of responses to controls27,28. For simple screenings, less stringent criteria may be used but for epidemiological studies, it may be more appropriate to employ a more stringent definition to reduce the probability of reporting false positives. Our initial application is to look at immunoconversion and immunoprevalence between swimmers and non-swimmers. In this study, the uncoupled control MFIs were not normally distributed and the results were log transformed.
Advantages to performing a bead-based multiplex immunoassay include the use of very low sample volumes, reduction in time and labor and the ability to measure responses for up to 500 analytes simultaneously while requiring a minimal amount of reagents. By contrast, traditional methods of assessing antibody levels indicative of exposure and/or infection (e.g., ELISA testing) can take several hours to complete, are labor intensive, and can only be used to measure one analyte at a time. These traditional methods also require larger volumes of patient/participant samples. For example, most ELISA's require at least 100 µl of sample while a multiplex immunoassay may require 50 µl or less.
Limitations of a multiplex immunoassay include the need for rigorous optimization to determine the optimal concentrations of primary capture and secondary detection antibodies, antigens and reporter. Cross-reactivity is also a major concern in immunoassays as antibodies against a particular antigen may cross-react with antigens from other organisms and thereby lead to false positive readings. Optimization, cross-reactivity and non-specific binding were addressed previously2-4. The success of any such assay is largely dependent on the ability to obtain highly immunogenic antigens as well as specific antibodies to these antigens for use in the bead coupling and coupling confirmation steps. Another limitation is the limited availability of characterized (diagnostically positive and negative) samples to validate the assays.
There are a number of critical steps within the protocol. These include: ensuring the antigens that will be coupled to the beads do not contain foreign proteins, azide, glycine, Tris or any primary amines. If any of these agents exist in the antigen preparation, they should be removed by dialysis or chromatography. It is recommended that one should start with 5 µg of antigen per 5 million beads and titrate to find the optimal concentration. Choose the optimal detection antibody concentration that gives the best sensitivity and dynamic range. Bear in mind that signals tend to decrease as antigen and detection antibody concentrations increase (hook effect). For example, if the signal decreases as antigen concentration increases then detection antibody may be limiting. Conversely, if signals decrease as detection antibody increases then reporter (SAPE) may be limiting. Check the multiplex for cross- reactivity by combining the optimal coupling for each protein (5,000 of each bead set per well). Test the multiplexed bead sets by combining with the optimal detection antibody amount. Optimize the reporter and make a standard curve of the antigen by testing each antigen individually.
Troubleshooting a multiplex immunoassay to improve sensitivity and to increase median fluorescent signal are critically important. To improve sensitivity, decrease either the amount of antigen coupled to the beads or the detection antibody concentration. The use of a higher affinity antibody for capture and/or detection may also improve sensitivity. Increasing the amount of antigen coupled to the beads may increase the median fluorescent signal. One study demonstrated that pre-incubating serum samples with polyvinylalcohol plus polyvinylpyrrolidone (PVX) buffer is able to suppress non-specific binding in serological assays using the bead-based multiplex assay30 such as the one that we are describing here. However, another study by our collaborators found no significant effect between PBS-BSA and PVX buffers. They concluded further that because of the higher viscosity of the PVX buffer, it created problems with bead acquisition in the analyzer and formed foam in the microplate wells3.
In conclusion, we have presented a method that is capable of measuring the presence of human salivary IgG antibodies to multiple pathogens simultaneously in a very small sample volume. In the future, this assay will be used to detect the presence of salivary antibodies associated with swimming related exposures or infections and to help inform risk assessment models. Additionally, the assay can be used to measure antibodies associated with airborne and foodborne exposures, determine incident infections or immunoconversions and provide critical immunological information to epidemiologists conducting questionnaire-type population studies.
Disclosures
The United States Environmental Protection Agency through its Office of Research and Development funded and managed the research described here. It has been subjected to Agency's administrative review and approved for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Acknowledgments
Clarissa Curioso was supported through an appointment to the Research Participation Program at the U.S. Environmental Protection Agency administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and U.S. EPA.
References
- Prüss-Üstün A, Gore F, Bartram J. Safer water, better health: costs, benefits and sustainability of interventions to protect and promote health. Geneva: World Health Organization; 2008. [Google Scholar]
- Augustine S, et al. Statistical approaches to developing a multiplex immunoassay for determining human exposure to environmental pathogens. J Immunol Methods. 2015;425:1–9. doi: 10.1016/j.jim.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Griffin S, et al. Application of salivary antibody immunoassays for the detection of incident infections with Norwalk virus in a group of volunteers. J Immunol Methods. 2015;424:53–63. doi: 10.1016/j.jim.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin S, Chen I, Fout G, Wade T, Egorov A. Development of a multiplex microsphere immunoassay for the quantitation of salivary antibody responses to selected waterborne pathogens. J Immunol Methods. 2011;364(1-2):83–93. doi: 10.1016/j.jim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Ferguson D. Current diagnostic uses of saliva. J Dent Res. 1987;66:420–424. doi: 10.1177/00220345870660020601. [DOI] [PubMed] [Google Scholar]
- Mandel I. The diagnostic uses of saliva. J Oral Pathol Med. 1990;19:119–125. doi: 10.1111/j.1600-0714.1990.tb00809.x. [DOI] [PubMed] [Google Scholar]
- Malamud D. Saliva as a Diagnostic Fluid. Brit Med J. 1992;305:207–208. doi: 10.1136/bmj.305.6847.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballam L, et al. Western blotting is useful in the salivary diagnosis of Helicobacter pylori infection. J Clin Pathol. 2000;53:314–317. doi: 10.1136/jcp.53.4.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estevez P, Satoguina J, Nwakanma D, West S, Conway D, Drakeley C. Human saliva as a source of anti-malarial antibodies to examine population exposure to Plasmodium falciparum. Malar J. 2011;10:104. doi: 10.1186/1475-2875-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd-Alla M, Jackson T, Reddy S, Ravdin J. Diagnosis of invasive amebiasis by enzyme-linked immunosorbent assay of saliva to detect amebic lectin antigen and anti-lectin immunoglobulin G antibodies. J Clin Microbiol. 2000;38(6):2344–2347. doi: 10.1128/jcm.38.6.2344-2347.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoguchi A, et al. Antibody reactivity to Cryptosporidium parvum in saliva of calves after experimental infection. J Vet Med Sci. 2000;62(11):1231–1234. doi: 10.1292/jvms.62.1231. [DOI] [PubMed] [Google Scholar]
- Choo S, Zhang Q, Seymour L, Akhtar S, Finn A. Primary and booster salivary antibody responses to a 7-valent pneumococcal conjugate vaccine in infants. J Infect Dis. 2000;182(4):1260–1263. doi: 10.1086/315834. [DOI] [PubMed] [Google Scholar]
- Tourinho R, et al. Importance of the cutoff ratio for detecting antibodies against hepatitis A virus in oral fluids by enzyme immunoassay. J Virol Methods. 2011;173(2):169–174. doi: 10.1016/j.jviromet.2011.01.014. [DOI] [PubMed] [Google Scholar]
- Moorthy M, Daniel H, Kurian G, Abraham P. An evaluation of saliva as an alternative to plasma for the detection of hepatitis C virus antibodies. Indian J Med Microbiol. 2008;26(4):327–332. doi: 10.4103/0255-0857.42116. [DOI] [PubMed] [Google Scholar]
- Moe C, Sair A, Lindesmith L, Estes M, Jaykus L. Diagnosis of norwalk virus infection by indirect enzyme immunoassay detection of salivary antibodies to recombinant norwalk virus antigen. Clin Diagn Lab Immunol. 2004;11(6):1028–1034. doi: 10.1128/CDLI.11.6.1028-1034.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap G, Sil B, Ng L. Use of saliva for early dengue diagnosis. PLoS Negl Trop Dis. 2011;5(5):e1046. doi: 10.1371/journal.pntd.0001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm W, Angulo G, Torres P, Burgess-Cassler A. A simple saliva-based test for detecting antibodies to human immunodeficiency virus. Clin Diagn Lab Immunol. 1999;6(4):577–580. doi: 10.1128/cdli.6.4.577-580.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chart H, Perry N, Willshaw G, Cheasty T. Analysis of saliva for antibodies to the LPS of Escherichia coli O157 in patients with serum antibodies to E. coli O157 LPS. J Med Microbiol. 2003;52(Pt 7):569–572. doi: 10.1099/jmm.0.05126-0. [DOI] [PubMed] [Google Scholar]
- Ashbolt N, Bruno M. Application and refinement of the WHO risk framework for recreational waters in Sydney, Australia. J Water Health. 2003;1(3):125–131. [PubMed] [Google Scholar]
- Westrell T, Schonning C, Stenstrom T, Ashbolt N. QMRA (quantitative microbial risk assessment) and HACCP (hazard analysis and critical control points) for management of pathogens in wastewater and sewage sludge treatment and reuse. Water Sci Technol. 2004;50(2):23–30. [PubMed] [Google Scholar]
- Ashbolt N. Microbial contamination of drinking water and disease outcomes in developing regions. Toxicology. 2004;198(1-3):229–238. doi: 10.1016/j.tox.2004.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg J, Soller J, Scott J, Eisenberg D, Colford J., Jr A dynamic model to assess microbial health risks associated with beneficial uses of biosolids. Risk Anal. 2004;24:221–236. doi: 10.1111/j.0272-4332.2004.00425.x. [DOI] [PubMed] [Google Scholar]
- Wade TJ, et al. Report on 2009 National Epidemiologic and Environmental Assessment of Recreational Water Epidemiology Studies, US EPA. US EPA Report Number: EPA/600/R-10/168: US EPA2011. 2011.
- Fujii Y, et al. Serological surveillance development for tropical infectious diseases using simultaneous microsphere-based multiplex assays and finite mixture models. PLoS Negl Trop Dis. 2014;8:e3040. doi: 10.1371/journal.pntd.0003040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota M, Massari M, Gabutti G, Guido M, De Donno A, Ciofi degli Atti M. Measles serological survey in the Italian population: interpretation of results using mixture model. Vaccine. 2008;26(34):4403–4409. doi: 10.1016/j.vaccine.2008.05.094. [DOI] [PubMed] [Google Scholar]
- Vyse A, Gay N, Hesketh L, Pebody R, Morgan-Capner P, Miller P. Interpreting serological surveys using mixture models: the seroepidemiology of measles, mumps and rubella in England and Wales at the beginning of the 21st century. Epidemiol Infect. 2006;134(6):1303–1312. doi: 10.1017/S0950268806006340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman A, et al. Establishment of diagnostic cutoff points for levels of serum antibodies to pertussis toxin, filamentous hemagglutinin, and fimbriae in adolescents and adults in the United States. Clin Diagn Lab Immunol. 2004;11(6):1045–1053. doi: 10.1128/CDLI.11.6.1045-1053.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clotilde L, Bernard C, IV, Hartman G, Lau D, Carter J. Microbead-based immunoassay for simultaneous detection of Shiga toxins and isolation of Escherichia coli O157 in foods. J Food Prot. 2011;74(3):373–379. doi: 10.4315/0362-028X.JFP-10-344. [DOI] [PubMed] [Google Scholar]
- Luminex User Software Manual (RUO) xPONENT 3.1 Rev. 2. 2014;3:6–102. [Google Scholar]
- Waterboer T, Sehr P, Pawlita M. Suppression of non-specific binding in serological Luminex assays. J Immunol Methods. 2006;309(1-2):200–204. doi: 10.1016/j.jim.2005.11.008. [DOI] [PubMed] [Google Scholar]