

Abstract
There is an increasing need for efficient phenotyping and histopathology of a variety of tissues. This phenotyping need is evident with the ambitious projects to disrupt every gene in the mouse genome. The research community needs rapid and inexpensive means to phenotype tissues via histology. Histological analyses of skeletal tissues are often time consuming and semi-quantitative at best, regularly requiring subjective interpretation of slides from trained individuals. Here, we present a cryohistological paradigm for efficient and inexpensive phenotyping of mineralized tissues. First, we present a novel method of tape-stabilized cryosectioning that preserves the morphology of mineralized tissues. These sections are then adhered rigidly to glass slides and imaged repeatedly over several rounds of staining. The resultant images are then aligned either manually or via computer software to yield composite stacks of several layered images. The protocol allows for co-localization of numerous molecular signals to specific cells within a given section. In addition, these fluorescent signals can be quantified objectively via computer software. This protocol overcomes many of the shortcomings associated with histology of mineralized tissues and can serve as a platform for high-throughput, high-content phenotyping of musculoskeletal tissues moving forward.
Keywords: Cellular Biology, Issue 115, high-throughput, cryosectioning, cryotape, fluorescent imaging, mineralization labels, fluorescent proteins, multiphoton imaging
Introduction
Biological research often requires efficient phenotyping, which is frequently associated with some sort of histological analysis1-3. This need is even more evident with the ambitious projects to disrupt each gene in the mouse genome4. These histological analyses can range from assessing cell morphology and/or anatomical features to mapping expression of specific genes or proteins to individual cells. In fact, one of the fundamental contributions of histology to the field of genomics is the ability to associate a specific molecular signal to a specific region or cell type.
Traditional methods of histology, especially for musculoskeletal tissues, are often time consuming and laborious, requiring sometimes weeks to fix, decalcify, section, stain, and image the specimen then analyze the images via human interpretation. Analyzing multiple molecular signals, whether via immunohistochemistry, in situ hybridization, or special stains, requires multiple sections and even multiple specimens to perform appropriately. In addition, these multiple responses cannot be co-localized to the same cell and sometimes cannot be co-localized to a specific region within a given specimen. As the genomics and epigenomics field moves into the digital age, the histological field must also follow suit to provide efficient, high-throughput, and automated analysis of a variety of molecular signals within a single histological section.
Indeed, there is a demand for improved histological techniques that can associate multiple molecular signals to specific cells within a given specimen. Recently, we have published a new high-throughput cryohistological method for assessing several response measures within a given section from mineralized tissue5-14. The process involves stabilizing the cryosection with frozen cryotape, adhering the taped section rigidly to a microscope slide, and conducting several rounds of staining and imaging on each section. These rounds of images are then aligned manually or via computer automation prior to image analysis (Figure 1). Here, we present detailed protocols of this process and provide examples where these techniques have improved our understanding of different biological processes.
Protocol
The University of Connecticut Health Center institutional animal care and use committee approved all animal procedures.
1. Fixation and Embedding
Euthanize the animal via CO2 asphyxiation or other approved methods.
Harvest the tissue of interest (e.g., limb, vertebrae, etc.) and place in 10% neutral buffered formalin at 4 °C until properly fixed. Take special care to maintain consistent anatomical placement prior to fixation. For instance, fix limbs with joints at consistent flexion, internal rotation, and external rotation angles11. Typically, fix whole mouse limbs for 1 - 3 days. Caution: Formalin is toxic and should be handled in a fume hood while wearing appropriate personal protective equipment. Note: The timeframe of fixation depends on the size of the specimen. If the experiment allows, cutting open the bone will improve fixation of the marrow compartment. Some specimens may require perfusion fixation15 to minimize fluorescent background (e.g., faint fluorescent signals within bone marrow).
Transfer specimens from formalin to 30% sucrose made in 1x PBS and incubate for 12 - 24 hr at 4 °C. Note: Once incubated for 12 - 24 hr, specimens can then be placed at -80 °C for long-term storage. This method of storage is recommended for experiments in which researchers intend to embed multiple samples in the same block but may not harvest all of these samples at the same time (e.g., experiments with multiple time points).
Remove specimens from the sucrose and dissect away any excess tissue.
Fill a cryomold (size of mold is dependent on specimen size) part of the way with cryo embedding medium. Place sample in mold such that cutting plane for the region of interest is parallel with the bottom of the mold. Note: An example of specific tissue placement and orientation can be found in Figure 2A - B.
Place the cryomold onto a piece of dry ice until a thin layer of cryo embedding medium freezes and subsequently fixes the specimen in place. Fill the remaining volume of the cryomold with cryo embedding medium while maintaining the cryomold on the dry ice pellet.
Place the cryomold in a container containing 2-methyl-butane pre-chilled by dry ice. Once specimens are completely frozen, remove cryomolds and shake off excess 2-methyl-butane. Wrap cryomolds in cellophane and place in a -20 °C or -80 °C freezer for storage. Note: Samples can dry out over time when stored in cryo embedding medium, especially at -20 °C. We recommend storage of samples for longer than 1 - 2 months in cryo embedding medium at -80 °C or in 30% sucrose at -80 °C (see step 1.3).
2. Tape-stabilized Tissue Sectioning
Remove specimen blocks from freezer and place in cryostat with the temperature set between -20 and -25 °C.
Remove the block from the cryomold and trim away excess cryo embedding medium with a razor blade.
Place some cryo embedding medium onto the specimen disc and then align the block such that the surface of the specimen disc is parallel to the bottom surface of the block thereby establishing the proper cutting plane for the region of interest.
Place the specimen disc in the cryostat and allow for the cryo embedding medium to freeze.
Place the specimen disc onto the specimen head and adjust the head such that the blade cuts parallel to the surface of the specimen block.
Trim the block down to a level within the region of interest and brush off any shavings from the surface of the block.
Cut a piece of the cryotape large enough to cover the region of interest and pre-chill in the cryostat. Note: Multiple pieces can be cut of consistent sizes and stored within the cryostat during sectioning.
Remove non-adherent backing from the cryotape by grasping the tape by the non-sticky silver/gold tabs using forceps then place the tape onto the block sticky-side down.
Apply pressure to the cryotape using the roller (Figure 2C).
Make a section (5 - 8 μm) using either the automatic motor or manual cutting wheel of the cryostat. Note: It is good practice to hold the edge of the cryotape as it comes onto the stage such that the section does not fall off the stage during sectioning (Figure 2D).
Place the section tissue side up on a plastic microscope slide within the cryostat.
Remove the plastic slide from the cryostat and allow the cryo embedding medium to melt such that the section adheres to the surface of the slide.
Repeat sectioning for serial sections or other regions of interest.
Once finished, store the sections in a slide box at 4, -20 or -80 °C depending on the length of storage required and the downstream response measures. For instance, use slides that are going to be stained for enzymatic activity (see 5.2 - 5.3) within a month if stored at 4 °C.
3. Adhesion of Sections to Microscope Slides
- UV-curable adhesive method.
- Label the microscope slide. Note: For increased automation, samples and slides can be labeled with barcodes.
- Apply a drop of UV-activated optical adhesive to two glass slides and use the edge of a plastic slide to spread a thin layer of adhesive across the surface of the glass slides.
- Cut off the silver/gold tab and lay the taped section tissue side up on the adhesive of the first slide. Apply the section in a rolling motion from one edge to the other in order to minimize the formation of entrapped bubbles underneath the tape. NOTE: The first slide is used to pre-wet the underside of the tape prior to placing it on the second (permanent) slide (step 3.1.4).
- Remove the taped section from the first slide and place it on the adhesive layer of the second slide (Figure 3A). Again, take care to avoid entrapment of bubbles. NOTE: This step takes practice to master.
- Once the appropriate number of sections for the given experiment is placed on each slide, wipe off the excess adhesive from the surrounding areas.
- Inspect each section closely for bubbles or fibers underneath the tape. Perform this inspection under a microscope or magnifying glass. If there are obstructions, remove the individual section, remove the obstructions, and then reapply it to the glass slide.
- Once it is determined that there are no obstructions under the taped sections, place the microscope slides under the UV black light for 5 - 10 min to crosslink the adhesive.
- Chitosan adhesive method.
- Prepare the 1% chitosan adhesive. Prepare acetic acid solution by dissolving 0.25 ml of concentrated acetic acid in 100 ml DI water (0.25% v/v). Dissolve 1 g of chitosan powder in 100 ml of acetic acid solution and stir the solution O/N or until all the chitosan powder is dissolved. NOTE: The solution is stable at RT.
- Deposit a drop of chitosan solution for each section that will be placed on the slide.
- Cut off the silver/gold tab of the tape and place each taped section tissue side up onto the adhesive (Figure 3A). Use great care to avoid entrapment of bubbles.
- Once all sections are placed on the slide, tilt the slide up on one of its long edges and drag excessive chitosan to the bottom edge using forceps.
- Place slides in this orientation on top of a paper towel in a slide box. Gravity will then cause the excess chitosan to fall down onto the towel, yielding a flat and uniform layer of chitosan.
- Place the slide box with its lid propped open in the refrigerator O/N to allow the chitosan to dry. NOTE: See Table 1 for a comparison between the two adhesive methods.
4. Application of Reference Markers to Slides
Prepare reference marker solution by dissolving 50 μl of green and 50 μl of red microspheres in 100 μl of water. Store the solution in the dark at 4 °C.
Place a 10 μl or 20 μl pipet tip into the microsphere solution. Add a small drop of the microsphere solution to each section adjacent to the region of interest (Figure 3C). Be careful to not get the microspheres within the region of interest.
Dry the slides at RT (protected from light) for 30 min if processing the slides on that day. If not, place the slides in a slide box at 4 °C. Note: As long as the microsphere solution dries prior to hydrating the slides, the microspheres will remain adhered to the slides during multiple rounds of staining/imaging.
5. Multiple Rounds of Staining
NOTE: By choosing a compatible sequence of imaging, staining, and reimaging steps, it is possible to detect and co-localize many biological signals on the same tissue section. Each round of imaging/staining/reimaging has to be developed for the particular histological question. The imaging/staining/reimaging sequence typically involves acquiring the endogenous fluorescent signals (e.g., cellular GFP, mineralization dyes, in vivo imaging probes) on the first round of imaging followed by fluorescent multiplexed immunostaining and multiple rounds of enzymatic activity stains. Lastly, the section can be stained using chromogenic dyes (e.g., H&E, toluidine blue, safranin O, etc.) to highlight the tissue architecture. Presented in this section are custom methods adapted from commercially available protocols.
- Calcein blue staining for accumulated mineral NOTE: Calcein blue staining yields a consistent mineral surface that is amenable to automated image analysis unlike darkfield or differential interference contrast (DIC) imaging. The problem with calcein blue staining is that the fluorescent spectrum overlaps with tetracycline and demeclocycline labeling. Therefore, if the sample contains these mineral labels, image the labels in the first round of imaging prior to staining with calcein blue.
- If the endogenous signals within the tissue are imaged prior to this staining step, submerge the slides from the previous imaging round in a coplin jar filled with 1xPBS until the cover slips fall away from the slides. Remove and dry the cover slips.
- Prepare 30 mg/ml of calcein blue solution in 2% NaHCO3 (made by dissolving 2 g NaHCO3 in 100 ml H2O and adjusting to pH of 7.4 with HCl). Aliquot and store the solution at -20°C.
- Immerse the slides in a coplin jar containing 1x PBS for 15 min to hydrate the sections, if not already hydrated from the previous round.
- Remove the slides and dry the back and edges with a paper towel.
- Apply 100 - 500 μl of calcein blue solution per slide and incubate for 10 min in a humidity chamber at RT.
- Rinse the slides in 1x PBS for 10 min with 3 changes.
- Apply ~ 200 μl of 50% glycerol in 1x PBS to each slide and mount the cover slip.
- Remove excess glycerol and dry the bottom of the slides.
- Tartrate-resistant acid phosphatase (TRAP) enzymatic activity staining NOTE: TRAP is expressed by multiple cell types in the hematopoietic lineage including osteoclasts. The TRAP staining presented here uses a yellow fluorescent acid phosphatase substrate. Certain fluorescent signals will overlap with the custom yellow filter set including tetracycline/demeclocycline mineralization labels, calcein blue stain, and DAPI counterstain.
- Submerge slides from the previous imaging round in a coplin jar filled with 1x PBS until the cover slips fall away from the slides. Remove and dry the cover slips.
- Prepare Buffer 1 by dissolving 9.2 g of sodium acetate anhydrous and 11.4 g of sodium tartrate dibasic dihydrate in water and bringing the final volume to 1,000 ml. Adjust the pH to 4.2 with concentrated glacial acetic acid (~ 1.5 - 2 ml). Store the solution at 4°C for up to 12 months.
- Prepare Buffer 2 by dissolving 40 mg of sodium nitrite in water and bringing the final volume to 1 ml. Store the solution at 4 °C for up to 1 week.
- On the day of staining, prepare the reaction buffer by mixing 7.5 ml of buffer 1 and 150 µl of buffer 2.
- Apply the reaction buffer without the substrate to the slides for 10 - 15 min to equilibrate the tissue at the lower pH. NOTE: The typical volume is ~ 200 μl but needs to be large enough to cover all sections.
- Dilute the yellow fluorescent acid phosphatase substrate between 1:50 and 1:100 in the reaction buffer. NOTE: Dilution will be dictated by TRAP activity within the sample.
- Pour the reaction buffer off of the slides and apply the reaction buffer with the yellow fluorescent substrate to the slides.
- Place the slides under the UV black light for ~ 5 min to activate the substrate. NOTE: Incubation time may vary depending on TRAP activity within the sample.
- Rinse the slides in 1x PBS for 10 min with 3 changes.
- Apply ~ 200 μl of 50% glycerol in 1x PBS to each slide and mount the cover slip.
- Remove excess glycerol and dry the bottom of the slides. NOTE: The acidic TRAP buffer will decalcify the sections. The added benefit of the decalcification is that certain colors will be available again for downstream staining (e.g., mineralization labels). For instance, the calcein blue staining will be removed during the TRAP staining. Therefore, DAPI counterstain can be imaged in this channel during a subsequent round.
- Alkaline phosphatase (AP) enzymatic activity staining NOTE: Multiple cell types involved with mineralization including osteoblasts and mineralizing chondrocytes express AP. The Fast Red substrate used in this protocol elicits both a fluorescent red and chromogenic red signal. Because of this, the chromogenic substrate will quench fluorescent signals in the regions where the substrate precipitates. Therefore, it is best to do this stain after imaging the fluorescent signals that may be quenched by the AP substrate.
- Submerge slides from the previous imaging round in a coplin jar filled with 1x PBS until the cover slips fall away from the slides. Remove and dry the cover slips.
- Prepare 1 M Tris by dissolving 12.1 g of Tris in 100 ml of water. Adjust pH to 9.5 with 1 M NaOH.
- Prepare 1 M MgCl2 hexahydrate by dissolving 20.33 g of MgCl2 in 100 ml of deionized water.
- Prepare 2 M NaCl by dissolving 11.68 g NaCl in 100 ml of deionized water.
- Prepare 100x (20 mg/ml) stock solution of Fast Red TR Salt by dissolving 1 g powder in 50 ml of deionized water. Make 100 μl aliquots and store at -20°C.
- Prepare 100x (10 mg/ml) Naphthol AS-MX phosphate stock solution by dissolving 500 mg powder in 50 ml of N,N dimethylformamide. Make 100 μl aliquots and store at -20 °C.
- On the day of staining, prepare the AP buffer by adding 1 ml of 1 M Tris, 0.5 ml of 1 M MgCl2, 0.5 ml of 2 M NaCl and bring the final volume to 10 ml using deionized water (final concentrations: 100 mM Tris, 50 mM MgCl2, 100 mM NaCl).
- Place AP buffer on each slide and incubate in a humidity chamber for 10 min at RT.
- Prepare AP substrate buffer by diluting 100x stocks of Fast Red TR Salt and Naphthol AS-MX to 1x in the AP buffer.
- Pour AP buffer off of slides and replace with AP substrate buffer. Incubate slides for 5 - 10 min in a humidity chamber at RT. NOTE: Incubation time will be dictated by AP activity of the sample.
- Wash slides 3x in 1x PBS for 5 min each.
- Mount cover slips with DAPI counterstaining solution (1:1,000 dilution of DAPI in 50% glycerol in 1x PBS).
- Toluidine blue O (TB) chromogenic staining NOTE: Because all previous steps are imaged under temporary aqueous mounting in 50% glycerol/PBS solution, the chromogenic step is also imaged under aqueous conditions. However, the staining solution may tend to leach out over time. Therefore, it is suggested to image the toluidine blue as soon as possible after staining.
- Submerge slides from previous imaging round in a coplin jar filled with deionized water until the cover slips fall away from the slides. Remove and dry the cover slips.
- After removing the cover slips, replace with fresh water and incubate the slides for at least 10 min so that salts from PBS are sufficiently removed from the tissue.
- Prepare 0.025% TB solution in deionized water.
- Place the slides in the TB solution for 1 - 5 min. NOTE: Incubation time is dependent on user preference but should be kept consistent across all samples.
- Place slides into coplin jar with deionized water and wash slides 3x for 5 min each.
- Mount cover slip with 30% glycerol dissolved in deionized water (NOT 1x PBS as this will cause the stain to leach out from the tissue). NOTE: Glycerol mounting medium can be substituted with fructose syrup, which helps prevent diffusion of the stain from the tissue. To prepare the fructose syrup, dissolve 30 g of fructose in 10 ml of deionized water and heat to 60 °C until dissolved.
6. Multiple Rounds of Imaging
Load the slides into the microscope trays (Figure 3D). Note: The trays are spring loaded.
Insert the trays into the tray stack of the slide-scanning microscope (Figure 3E).
Click on the profile list to load a profile for each slide with appropriate exposure times for each fluorophore. Click on the slide name to name each slide manually or have the software read the barcode provided on the slide label. Click the start preview scan button to take a preview image of the slide.
Set the regions of interest in the tissue detection wizard and save them for subsequent rounds of imaging. Once each slide is setup, start the scan process by clicking the start scan button. Note: The system can hold up to 100 slides at a time.
Once the imaging has finished, export the images from each individual channel and imaging round as .tif or .jpg files for image assembly and analysis.
7. Image Assembly
- Manual method using Photoshop (or similar image editing software capable of producing layered images).
- Open each individual channel image from each imaging round.
- Assemble the individual images as layers within one composite image. To do so, click on the layer within the layer palette from one image. Then drag the layer onto another image. This drag and drop operation will create a new layer in the image. Do this for all other images. Alternatively, use a script (File>Scripts>Load Files into Stack…) to automate this process by loading multiple image files as layers in an image stack.
- Once each image is listed as an individual layer in the composite image, double click on the layer name to rename the layers as needed to discern the different channels.
- In order to see through each layer and create a truly composite image, change the blend mode for each layer from "Normal" to "Screen" within the layer palette. Note: To speed up this step, change a single layer to screen, then right-click on the layer, select "copy layer style", then highlight all other layers and select "paste layer style" to apply the screen style to the other layers.
- If desired, decrease the opacity (change value to 10 - 40%) of the chromogenic layer (i.e., toluidine blue) to better visualize the fluorescent signals through the chromogenic layer. Note: The tiled scanning microscope used in this protocol holds the slides on 3 edges, thereby minimizing the amount of rotation that can occur to the slide during each round of imaging. Therefore, it is much easier for the user to manually align the images without having to rotate images derived from different rounds of imaging.
Representative Results
A General Workflow for the High-Throughput, Multi-Image Cryohistology
Figure 1 represents the general workflow used for this technique. It includes several steps from fixation through several rounds of imaging and finally image alignment/analysis. The process can take as little as a week to go from sample fixation through 4 rounds of imaging, which is less time than it takes to decalcify these type of samples. The order of the imaging typically begins with the endogenous signals that are already in the specimen (e.g., GFPs, mineralization labels, etc.), then multiplexed fluorescent immunostaining followed by fluorescent enzymatic activity assays (e.g., TRAP, AP, etc.) and cell cycle analysis assays (e.g., EdU) and finally finishes with a chromogenic stain (e.g., toluidine blue O, hematoxylin, safranin O, etc.).
Representative Example from a Juvenile Ankle Joint 6
The purpose of this particular study was to demonstrate the correlation between expression of different types of collagen (i.e., Col1a1, Col2a1, and Col10a1) with mineralization of the fibrocartilage of the Achilles tendon-to-bone insertion site (i.e., enthesis). Therefore, a triple transgenic fluorescent reporter mouse including Col1a1-GFPTpz, Col2a1-CFP, and Col10a1-mcherry was used to identify cells expressing each transgene. The first round of imaging was of the endogenous transgene expression from a two-week-old mouse, which corresponds to when the enthesis mineralizes in the Achilles tendon (Figure 4B). The second round of imaging was then conducted on the multiphoton microscope to acquire images of the collagen architecture via two photon second harmonic generation (SHG, Figure 4C). This step was used to identify cells at the base of the collagen fibers within the enthesis. The third round of imaging was an immunostaining step for Indian hedgehog (IHH), which is one of the main signaling ligands that promotes mineralization of the enthesis (Figure 4D). The fourth round of imaging was AP staining using a blue alkaline phosphatase substrate kit, which elicits both Cy5 fluorescence and blue chromogenic signals, to visualize areas of active mineral deposition (Figure 4E). Finally, the fifth round of imaging was TB staining to visualize the anatomical features including the proteoglycan content within the fibrocartilage (Figure 4F). All images were manually aligned within image editing software. The five rounds of imaging were conducted over a 4-day period, which followed 3 days of sample processing and sectioning (7 days total).
Representative Example from an Adult Knee Joint 11
The purpose of this experiment was to determine the mineralization changes that occur in the enthesis of the medial collateral ligament (MCL) of the knee following joint destabilization via transection of the anterior cruciate ligament (ACL). Mineralization changes can be seen in the MCL enthesis as early as two weeks post-surgery in these 3-month-old mice. To monitor mineral apposition of fibrocartilage within the enthesis, a mineral label was given to the mice on the day of surgery (demeclocycline) and the day before sacrifice (calcein) at 2 weeks post-surgery. The mice also included Col1a1-CFP and Col10a1-mcherry fluorescent reporters to monitor collagen expression of unmineralized and mineralized fibrochondrocytes, respectively. The first round of imaging was of the endogenous signals, which in this case corresponded to the fluorescent proteins and fluorescent mineralization labels (Figure 5A). The second round included TRAP staining to demonstrate expression of this enzyme in mineralizing fibrochondrocytes in the enthesis as well as osteoclasts in the underlying bone marrow (Figure 5B). The third round was AP staining to demonstrate regions of active mineralization of fibrochondrocytes as well as osteoblasts of the underlying bone (Figure 5C). Finally, TB staining was conducted for the fourth round (Figure 5D). All images were manually aligned within image editing software. Once again the total time taken from tissue harvest to image alignment was 7 days.
Representative Example of Trabecular Bone from Distal Femur
The purpose of this study is to phenotype the skeleton of mice with varying genetic backgrounds by conducting automated dynamic histomorphometry (www.bonebase.org). Three-month-old mice were given two mineralization labels (calcein 7 days prior to sacrifice and alizarin complexone 2 days prior to sacrifice). Distal femurs from four separate mice were embedded within the same frozen block and then cut together. The section containing four bones was glued down to the slide. Next, another section containing 4 additional bones was glued down adjacent to the first section. Therefore, a total of 8 bones were applied to each slide (Figure 3B). This process was conducted on 8 female and 8 male mice at 3 different levels within the bone marrow, yielding a total number of 12 slides. The reference markers (microspheres) were applied within the epiphysis and the mid-diaphysis on the dry sections (Figure 3C). All 12 slides were stained for calcein blue and imaged for accumulated mineral (calcein blue) and mineralization labels (calcein and alizarin complexone) during the first round of imaging (Figure 6A). The slides were then imaged for TRAP activity (Figure 6B) in the second round and AP activity (Figure 6C) in the third round of imaging. Finally, the slides were stained with toluidine blue in the fourth round (Figure 6D). The reference markers were imaged during every imaging round, including the chromogenic round, and were aligned using the custom software. To provide an idea of the throughput for this procedure, 32 bones (16 femurs and 16 vertebrae) were embedded in 8 blocks, 3 sections were taken from each block, the sections were distributed across 12 slides, and the 12 slides were imaged 4 times producing 96 composite image stacks. The total time to perform this experiment was 8 days.
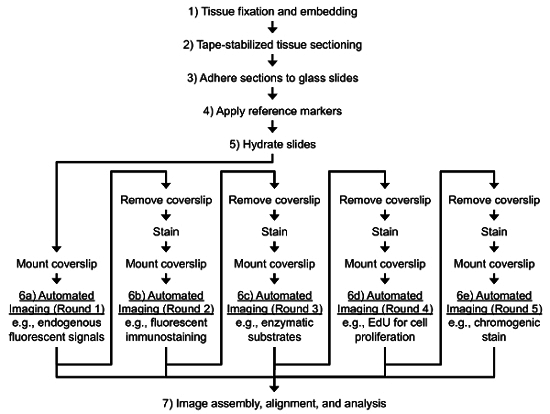 Figure 1. Typical Workflow for the Protocol. General steps include 1) tissue fixation, 2) tape-stabilized cryosectioning, 3) adherence of taped sections to glass slides, 4) application of reference markers, 5) multiple rounds of staining and 6) imaging, and 7) image assembly, alignment, and analysis. Please click here to view a larger version of this figure.
Figure 1. Typical Workflow for the Protocol. General steps include 1) tissue fixation, 2) tape-stabilized cryosectioning, 3) adherence of taped sections to glass slides, 4) application of reference markers, 5) multiple rounds of staining and 6) imaging, and 7) image assembly, alignment, and analysis. Please click here to view a larger version of this figure.
 Figure 2.High-throughput Embedding and Tape-stabilized Cryosectioning. Because of the stability the cryotape provides, multiple bones can be embedded adjacent to each other (A-B) and sectioned simultaneously (C-D). A piece of dry ice is used during the embedding process to rigidly fix the bones in place prior to freezing the entire cryo-block (B). The cryotape is rolled onto the block (C) and the section remains stuck to the tape during sectioning (D). Please click here to view a larger version of this figure.
Figure 2.High-throughput Embedding and Tape-stabilized Cryosectioning. Because of the stability the cryotape provides, multiple bones can be embedded adjacent to each other (A-B) and sectioned simultaneously (C-D). A piece of dry ice is used during the embedding process to rigidly fix the bones in place prior to freezing the entire cryo-block (B). The cryotape is rolled onto the block (C) and the section remains stuck to the tape during sectioning (D). Please click here to view a larger version of this figure.
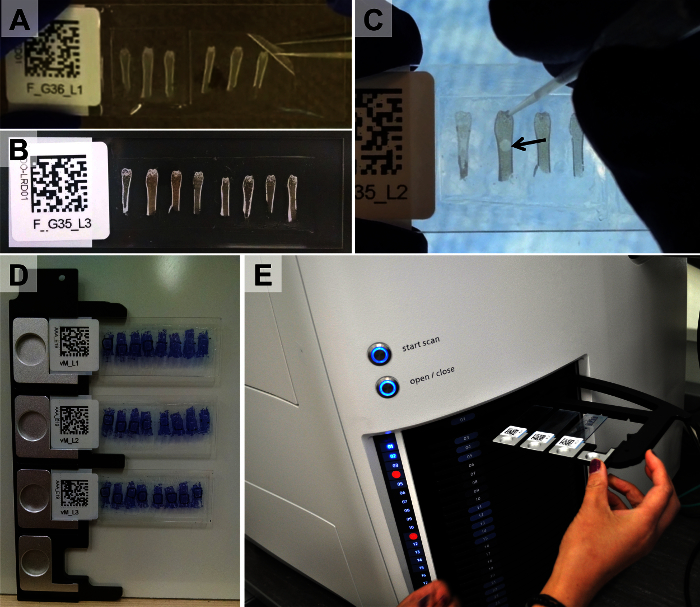 Figure 3.Adherence of Taped Sections to Glass Slides, Application of Reference Markers, and Loading of Slides into Tray of Slide-scanning Microscope. Taped sections are glued to the surface of glass slides with either UV-curing or chitosan-based adhesive (A). Following curing or drying, only a thin, flat layer of adhesive remains between the cryotape and glass surface (B). Reference markers are applied to dry slides (C, arrow points to drop of microsphere solution). Slides are then mounted in microscope trays (D) and loaded into the microscope (E). Please click here to view a larger version of this figure.
Figure 3.Adherence of Taped Sections to Glass Slides, Application of Reference Markers, and Loading of Slides into Tray of Slide-scanning Microscope. Taped sections are glued to the surface of glass slides with either UV-curing or chitosan-based adhesive (A). Following curing or drying, only a thin, flat layer of adhesive remains between the cryotape and glass surface (B). Reference markers are applied to dry slides (C, arrow points to drop of microsphere solution). Slides are then mounted in microscope trays (D) and loaded into the microscope (E). Please click here to view a larger version of this figure.
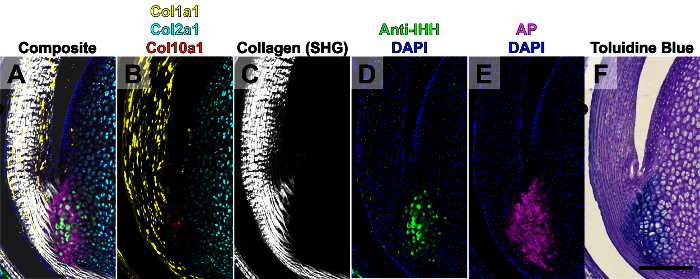 Figure 4. Five Rounds of Imaging of the Achilles Tendon from a Two-week-old Mouse. The composite image stack (A) was created from five rounds of imaging. Round 1 (B): endogenous Col1a1-GFPTpz, Col2a1-CFP, and Col10a1-mcherry transgene expression. Round 2 (C): collagen second harmonic generation (SHG) on the two photon microscope. Round 3 (D): immunostaining for IHH. Round 4 (E): AP enzymatic activity. Round 5: toluidine blue O (F). This figure has been modified from Dyment et al., 20156. Scale bar = 200 μm. Please click here to view a larger version of this figure.
Figure 4. Five Rounds of Imaging of the Achilles Tendon from a Two-week-old Mouse. The composite image stack (A) was created from five rounds of imaging. Round 1 (B): endogenous Col1a1-GFPTpz, Col2a1-CFP, and Col10a1-mcherry transgene expression. Round 2 (C): collagen second harmonic generation (SHG) on the two photon microscope. Round 3 (D): immunostaining for IHH. Round 4 (E): AP enzymatic activity. Round 5: toluidine blue O (F). This figure has been modified from Dyment et al., 20156. Scale bar = 200 μm. Please click here to view a larger version of this figure.
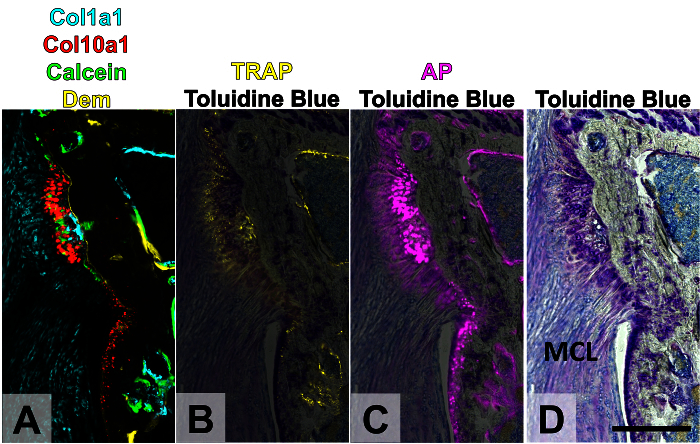 Figure 5. Four Rounds of Imaging of MCL Enthesis Following Joint Destabilization. Knees were destabilized in three-month-old mice, leading to increased mineralization of the MCL enthesis. A demeclocycline mineral label was given the day of surgery and a calcein mineral label was given the day before sacrifice. Round 1 (A): endogenous Col1a1-CFP, Col10a1-mcherry transgene expression with demeclocycline and calcein mineral labels. Round 2 (B): TRAP enzymatic activity. Round 3 (C): AP enzymatic activity. Round 4 (D): toluidine blue O. The TRAP and AP signal was overlaid on top of the TB signal in panels B-C. The yellow channel can be used again for TRAP because the TRAP buffer decalcifies the tissue, removing the demeclocycline label. This figure has been modified from Dyment et al., 201511. Scale bar = 200 μm. Dem: demeclocycline, TRAP: tartrate-resistant acid phosphatase, AP: alkaline phosphatase, MCL: medial collateral ligament. Please click here to view a larger version of this figure.
Figure 5. Four Rounds of Imaging of MCL Enthesis Following Joint Destabilization. Knees were destabilized in three-month-old mice, leading to increased mineralization of the MCL enthesis. A demeclocycline mineral label was given the day of surgery and a calcein mineral label was given the day before sacrifice. Round 1 (A): endogenous Col1a1-CFP, Col10a1-mcherry transgene expression with demeclocycline and calcein mineral labels. Round 2 (B): TRAP enzymatic activity. Round 3 (C): AP enzymatic activity. Round 4 (D): toluidine blue O. The TRAP and AP signal was overlaid on top of the TB signal in panels B-C. The yellow channel can be used again for TRAP because the TRAP buffer decalcifies the tissue, removing the demeclocycline label. This figure has been modified from Dyment et al., 201511. Scale bar = 200 μm. Dem: demeclocycline, TRAP: tartrate-resistant acid phosphatase, AP: alkaline phosphatase, MCL: medial collateral ligament. Please click here to view a larger version of this figure.
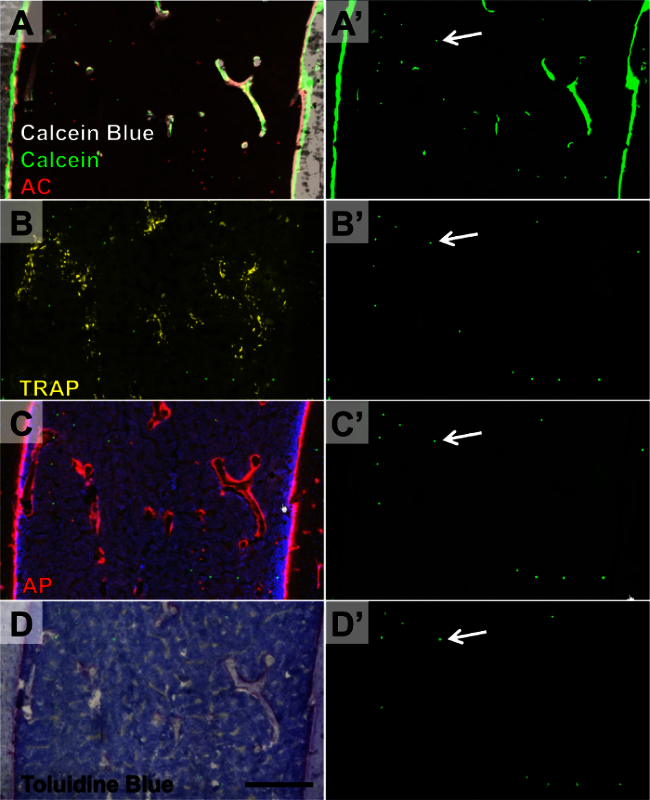 Figure 6.Four Rounds of Imaging within Distal Femur Containing Microsphere Reference Markers. Three-month-old mice were given calcein and alizarin complexone mineral labels. Round 1 (A-A'): endogenous calcein and alizarain complexone labels in addition to calcein blue staining of accumulated mineral. Round 2(B-B'): TRAP enzymatic activity. Round 3(C-C'): AP enzymatic activity. Round 4(D-D'): toluidine blue O. The green microspheres were imaged during each round (A'-D', arrow denotes the same microsphere in all images). Scale bar = 200 μm. Please click here to view a larger version of this figure.
Figure 6.Four Rounds of Imaging within Distal Femur Containing Microsphere Reference Markers. Three-month-old mice were given calcein and alizarin complexone mineral labels. Round 1 (A-A'): endogenous calcein and alizarain complexone labels in addition to calcein blue staining of accumulated mineral. Round 2(B-B'): TRAP enzymatic activity. Round 3(C-C'): AP enzymatic activity. Round 4(D-D'): toluidine blue O. The green microspheres were imaged during each round (A'-D', arrow denotes the same microsphere in all images). Scale bar = 200 μm. Please click here to view a larger version of this figure.
| Chitosan adhesive | UV-curing adhesive | |
| Adhesive mechanism | Evaporation | UV Polymerization |
| Curing time | > 24 hr | < 20 min |
| Can sections be removed after adhesive cures? | Yes | No |
| Is cured adhesive dissolvable? | Yes, in acidic solutions with low pH | No |
| Does adhesive withstand heat antigen retrieval? | No | Yes |
| Is adhesive auto-fluorescent? | No | Minimal in UV range |
Table 1. Comparison Between Chitosan Adhesive and UV-curing Adhesive
| Cryotape | Tape-Transfer System | |
| Possible to section mineralized bone using this system? | Yes | Yes, but pieces of mineralized bone may not transfer completely to the slide |
| Possible to section mineralized joints using this system? | Yes | Yes |
| Possible to section brain using this system? | Yes, but tissue may fall off of tape after multiple rounds of imaging | Yes |
| Possible to cut multiple samples embedded in the same block? | Yes | Yes |
| Possible to conduct multiple rounds of imaging on same section? | Yes | Yes |
Table 2. Comparison Between Cryotape System and Tape-transfer System
Discussion
Here we have presented a detailed cryohistology protocol to co-localize and quantify several biological measures by aligning images from multiple rounds of staining/imaging on a single section. The method outlined using the cryotape is especially useful as it maintains the morphology of difficult to section tissue (e.g., mineralized bone and cartilage). In addition, the sectioned tissue is adhered firmly to the glass slide, allowing for multiple rounds of staining/imaging of the same section; unlike traditional methods where serial sections are each stained with a different protocol. Using serial sections can pose an issue when attempting to co-localize signals between the sections, something that is not a problem with single sections that have been stained and imaged with multiple rounds.
The first tape-stabilized tissue sectioning product on the market was a tape transfer system16, 17. It uses plastic tape coated with a cold temperature adhesive that is placed on the surface of the tissue cryoblock. When the cryostat blade cuts beneath the tape, the section is removed intact and adherent to the tape. Subsequently the tape is placed sample side down onto slides that are coated with UV-curing glue such that the tissue becomes rigidly attached to the slide. Once the tape is pealed away from the section, the slide can be processed for either fluorescence or chromogenic histology. The background fluorescence of the adhesives is low, and multiple rounds of staining and imaging works well with most soft tissues. However with mineralized sections, there can be loss of mineral fragments when transferring the tissue from the adhesive tape to the glass slides. In addition, it is a relatively expensive system to install in the cryostat (> $ 8,000) and consumable costs are high (> $ 2/slide).
Another tape stabilization strategy developed by Dr. Tadafumi Kawamoto uses an adhesive coated polyvinylidene chloride film to capture the tissue section. In their protocol, a fresh frozen tissue is sectioned onto the tape and immediately fixed in PFA or ethanol18. Subsequently, the tape is stained and then mounted on a glass slide with the sample side down for microscopic examination. Thus each staining protocol is performed on a different taped section.
We have modified and added to the Kawamoto protocol in a number of ways including fixing the sample in formalin prior to embedding. This step is critical to fix the soluble cytoplasmic GFP of transgenic animals within the limits of the cell. We have utilized the cryotape for a wide range of tissue types5-13. Laboratory personnel with limited histological experience can produce high-quality sections with this method. The primary advantages of this protocol are the relative low cost (no special instrumentation, < $ 1.00 per slide), no loss of mineral fragments, and the ability to perform multiple rounds of staining and imaging on the same section.
Because imaging the same regions of a slide multiple times in repetition would be unreasonable for a large number of slides even using a motorized stage, another critical step of this protocol is the utilization of a slide-scanning microscope to increase consistency and throughput. The microscope is a digital slide scanner that operates under both epifluorescence and transmitted light. It is equipped with a 10-position motorized turret and both B&W and color cameras. The true novelty of the system, in our hands, is that specific regions of interest can be quickly and easily defined by the user or detected automatically by the imaging software. These regions of interest can then be saved from the first round of imaging and then reloaded quickly during the subsequent rounds. Therefore, the same regions of interest are imaged during each round of imaging. Based on user-defined parameters in the software, the microscope will autofocus and scan tiled images that are stitched during acquisition within the regions of interest.
The order by which the user performs the multiple rounds of staining is important. The general order is outlined in Figure 1. The number of fluorophores that can be sufficiently separated via fluorescent microscopy is the primary limiting factor to the number of response measures that can be acquired on a given section. However, fluorescent channels can be reused in certain cases. For instance, calcein blue and demeclocycline both emit a strong signal in the yellow channel used to measure TRAP activity. This bleed-through is avoided though because the TRAP buffer decalcifies the section, thereby removing the mineral prior to imaging the TRAP activity. In addition, DAPI counterstain will emit in the yellow channel as well. However, the user can replace DAPI with another counterstain in the red or far-red range. In addition, antibodies can be stripped and re-stained with different antibodies using the same fluorescent channels. Therefore, this method is quite adaptable to increasing the number of response measures that can be recorded on a given section.
There are certain limitations with the presented protocol. 1) The cryotape may not adhere sufficiently to certain tissues. For instance, formalin-fixed brain sections tend to fall off the tape after multiple rounds of staining. For tissues such as this, the tape transfer system may be more appropriate as the tissue is adhered to the glass surface via UV-activated adhesive (see Table 2 for comparison between these two methods). 2) The chitosan adhesive, while providing transparent optical properties, will not survive harsh processing steps that include high temperatures or strong acids. Therefore, the UV-activated adhesive is more suitable for these applications (see Table 1 for comparison between these two adhesives).
The cryohistological protocols presented here also lend themselves to higher throughput and automation. For instance, the tape stabilization provided by the cryotape allows for relatively novice users to cut multiple bones or joints at once in the same block, significantly increasing throughput. Using an automated scanner significantly reduces technician time and cost related to imaging, which has typically been the rate-limiting step in our experience. Being able to consistently and reliably image the same region of interest several times in a short period of time provides a platform for automated, objective analysis of tissues. In fact, our group has developed a platform for computer-automated alignment and histomorphometry of bone. First, the software aligns the multiple rounds of images based on the fluorescent reference markers. Then, the aligned images are fed into a histomorphometry pipeline where the computer software defines the region of interest and objectively measures several static and dynamic measurements (see more at www.bonebase.org)13. This platform was made possible because of the increased throughput and consistency provided by the cryotape sectioning, digital slide-scanning microscope, and custom analysis software. This protocol represents a significant advancement in high throughput cryohistology and should be of use to those imaging difficult to section tissues such as bone as well as those inexperienced with tissue sectioning.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors would like to acknowledge the following funding sources: NIH R01-AR063702, R21-AR064941, K99-AR067283, and T90-DE021989.
References
- Schofield PN, Vogel P, Gkoutos GV, Sundberg JP. Exploring the elephant: histopathology in high-throughput phenotyping of mutant mice. Dis Model Mech. 2012;5(1):19–25. doi: 10.1242/dmm.008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adissu HA, et al. Histopathology reveals correlative and unique phenotypes in a high-throughput mouse phenotyping screen. Disease Models and Mechanisms. 2014;7(5):515–524. doi: 10.1242/dmm.015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JT, et al. Virtual histology of transgenic mouse embryos for high-throughput phenotyping. PLoS Genet. 2006;2(4):e61. doi: 10.1371/journal.pgen.0020061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayadi A, et al. Mouse large-scale phenotyping initiatives: overview of the European Mouse Disease Clinic (EUMODIC) and of the Wellcome Trust Sanger Institute Mouse Genetics Project. Mamm.Genome. 2012;23(9-10):600–610. doi: 10.1007/s00335-012-9418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utreja A, et al. Cell and matrix response of temporomandibular cartilage to mechanical loading. Osteoarthr Cartil. 2016;24(2):335–344. doi: 10.1016/j.joca.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyment NA, et al. Gdf5 progenitors give rise to fibrocartilage cells that mineralize via hedgehog signaling to form the zonal enthesis. Dev.Biol. 2015;405(1):96–107. doi: 10.1016/j.ydbio.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalley AL, et al. Improved biomechanical and biological outcomes in the MRL/MpJ murine strain following a full-length patellar tendon injury. J.Orthop.Res. 2015;33(11):1693–1703. doi: 10.1002/jor.22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenbach AP, et al. Ablating hedgehog signaling in tenocytes during development impairs biomechanics and matrix organization of the adult murine patellar tendon enthesis. J.Orthop.Res. 2015;33(8):1142–1151. doi: 10.1002/jor.22899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushiku C, Adams D, Jiang X, Wang L, Rowe D. Long Bone Fracture Repair in Mice Harboring GFP Reporters for Cells within the Osteoblastic Lineage. J.Orthop.Res. 2010;28(10):1338–1347. doi: 10.1002/jor.21105. [DOI] [PubMed] [Google Scholar]
- Matthews BG, et al. Analysis of asMA-labeled progenitor cell commitment identifies notch signaling as an important pathway in fracture healing. J.Bone Miner.Res. 2014;29(5):1283–1294. doi: 10.1002/jbmr.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyment NA, Hagiwara Y, Jiang X, Huang J, Adams DJ, Rowe DW. Response of knee fibrocartilage to joint destabilization. Osteoarthritis Cartilage. 2015;23(6):996–1006. doi: 10.1016/j.joca.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grcevic D, et al. In vivo fate mapping identifies mesenchymal progenitor cells. Stem Cells. 2012;30(2):187–196. doi: 10.1002/stem.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Jiang X, Chen L, Josh P, Shin DG, Rowe D. Computer-Automated Static, Dynamic and Cellular Bone Histomorphometry. J Tissue Sci Eng. 2012;Suppl 1:004. doi: 10.4172/2157-7552.S1-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BG, Torreggiani E, Roeder E, Matic I, Grcevic D, Kalajzic I. Osteogenic potential of alpha smooth muscle actin expressing muscle resident progenitor cells. Bone. 2016;84:69–77. doi: 10.1016/j.bone.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara Y, et al. Fixation stability dictates the differentiation pathway of periosteal progenitor cells in fracture repair. J.Orthop.Res. 2015;33(7):948–956. doi: 10.1002/jor.22816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, et al. Histological analysis of GFP expression in murine bone. J.Histochem.Cytochem. 2005;53(5):593–602. doi: 10.1369/jhc.4A6401.2005. [DOI] [PubMed] [Google Scholar]
- Nissanov J, Bertrand L, Tretiak O. Cryosectioning distortion reduction using tape support. Microsc.Res.Tech. 2001;53(3):239–240. doi: 10.1002/jemt.1089. [DOI] [PubMed] [Google Scholar]
- Kawamoto T. Use of a new adhesive film for the preparation of multi-purpose fresh-frozen sections from hard tissues, whole-animals, insects and plants. Arch.Histol.Cytol. 2003;66(2):123–143. doi: 10.1679/aohc.66.123. [DOI] [PubMed] [Google Scholar]