

Abstract
Yarrowia lipolytica is a non-pathogenic, dimorphic and strictly aerobic yeast species. Owing to its distinctive physiological features and metabolic characteristics, this unconventional yeast is not only a good model for the study of the fundamental nature of fungal differentiation but is also a promising microbial platform for biochemical production and various biotechnological applications, which require extensive genetic manipulations. However, genetic manipulations of Y. lipolytica have been limited due to the lack of an efficient and stable genetic transformation system as well as very high rates of non-homologous recombination that can be mainly attributed to the KU70 gene. Here, we report an easy and rapid protocol for the efficient genetic transformation and for gene deletion in Y. lipolytica Po1g. First, a protocol for the efficient transformation of exogenous DNA into Y. lipolytica Po1g was established. Second, to achieve the enhanced double-crossover homologous recombination rate for further deletion of target genes, the KU70 gene was deleted by transforming a disruption cassette carrying 1 kb homology arms. Third, to demonstrate the enhanced gene deletion efficiency after deletion of the KU70 gene, we individually deleted 11 target genes encoding alcohol dehydrogenase and alcohol oxidase using the same procedures on the KU70 knockout platform strain. It was observed that the rate of precise homologous recombination increased substantially from less than 0.5% for deletionof the KU70 gene in Po1g to 33%-71% for the single gene deletion of the 11 target genes in Po1g KU70Δ. A replicative plasmid carrying the hygromycin B resistance marker and the Cre/LoxP system was constructed, and the selection marker gene in the yeast knockout strains was eventually removed by expression of Cre recombinase to facilitate multiple rounds of targeted genetic manipulations. The resulting single-gene deletion mutants have potential applications in biofuel and biochemical production.
Keywords: Genetics, Issue 115, Bioengineering, Unconventional yeast, Yarrowia lipolytica, KU70 gene, genetic transformation, gene deletion, genetic manipulation, homologous recombination, marker rescue
Introduction
Unlike Saccharomyces cerevisiae, Yarrowia lipolytica, an unconventional yeast, can grow in the form of yeast or mycelium in response to changes in environmental conditions 1,2. Thus, this dimorphic yeast can be used as a good model for the study of fungal differentiation, morphogenesis and taxonomy 3,4,5. It is generally regarded as a safe (GRAS) yeast species, which is widely used to produce a variety of food additives such as organic acids, polyalcohols, aroma compounds, emulsifiers and surfactants 6,7,8,9. It is an obligate aerobe and a well-known oleaginous yeast capable of naturally accumulating lipids at high amounts, i.e., up to 70% of cell dry weight 10. It can also utilize a wide spectrum of carbon sources for growth, including different kinds of residues in waste resources as nutrients 11,12,13. All of these unique features make Y. lipolytica very attractive for various biotechnological applications.
Although the whole genome sequence of the Y. lipolytica has been published 14,15, genetic manipulation of this unconventional yeast is more complex than other yeast species. First, transformation of this yeast species is much less efficient due to the absence of a stable and efficient genetic transformation system 16,17. Second, laborious genomic integration of linear expression cassettes is commonly used for the expression of genes of interest as no natural episomal plasmid system has been found in this yeast 18. Third, generation of genetic knock-outs and knock-ins are limited because the gene targeting efficiency via accurate homologous recombination in this yeast is low and most integration events occur through non-homologous end joining (NHEJ) 19.
In this study, we report an optimized transformation protocol for the Y. lipolytica Po1g strain, which is easy, rapid, efficient and reproducible. To enhance the frequency of precise homologous recombination, we deleted the KU70 gene, which encodes a key enzyme in the NHEJ pathway. By using the optimized transformation protocol and transforming a linear knockout cassette containing flanking homology regions of 1 kb, the KU70 gene of the Y. lipolytica Po1g was successfully deleted. The robustness of this gene deletion methodology was then demonstrated by targeting alcohol dehydrogenase and alcohol oxidase genes in the Po1g KU70Δ strain. It was observed that the KU70 deletion strain exhibited a considerably higher efficiency of homologous recombination-mediated gene targeting than that of the wild-type Po1g strain. In addition, a replicative Cre expression plasmid carrying the hygromycin B resistance marker was constructed to perform marker rescue. The marker rescue facilitates multiple rounds of gene targeting in the obtained gene deletion mutants. Besides gene deletion, our protocol for genetic transformation and gene deletion described here can be applied to insert genes to specific loci, and to introduce site-specific mutations into the Y. lipolytica genome.
Protocol
1. Generation of the Y. lipolytica KU70 Deletion Strain
- Construction of the disruption cassette Note: See Table 1 for all primers used in polymerase chain reaction (PCR) amplifications.
- Design primers 20 to PCR amplify the LEU2 expression cassette (see Table 1) from a Y. lipolytica expression vector and introduce LoxP sites into the 5' and 3' ends of the LEU2 cassette with a long forward primer (# 1; Table 1) and a long reverse primer (# 2; Table 1), respectively. To introduce an additional restriction site for subsequent steps of the cloning process, add a BamHI restriction site in primer # 1.
- Perform PCR using a high-fidelity DNA polymerase as detailed in Table 2.
- Purify the PCR product LoxP-promoter-LEU2-terminator-LoxP cassette using a PCR purification kit according to the manufacturer's instructions. Add 3'-A overhangs to the purified PCR product according to the manufacturer's instructions 21. Use T4 DNA ligase to ligate the A-tailed PCR product into a TA cloning vector to yield the plasmid T-LEU2 as per Table 3.
- PCR amplify the 1 kb 5' upstream sequence of the KU70 gene using primers # 3/# 4 from the Y. lipolytica Po1g genomic DNA 22 as per Table 2. Purify the PCR product using a PCR purification kit according to the manufacturer's instructions.
- Double digest both the purified PCR product and T-LEU2 plasmid with SacII and BamHI enzymes as per Table 4. Purify the digestion mixture using a PCR purification kit as per manufacturer's instructions. Use T4 DNA ligase to ligate the purified and digested PCR product to the SacII/BamHI sites of T-LEU2 to yield the plasmid T-LEU2-5E as per Table 3.
- PCR amplify the 1 kb 3' downstream sequence of the KU70 gene using primers # 5/# 6 from the Y. lipolytica Po1g genomic DNA 22 as per Table 2. Purify the PCR product using a PCR purification kit according to the manufacturer's instructions. Double digest both the purified PCR product and T-LEU2-5E plasmid with NotI and NdeI enzymes as per Table 4.
- Purify the digestion mixture using a PCR purification kit as per manufacturer's instructions. Then, ligate the purified and digested PCR product to the NotI/NdeI sites of T-LEU2-5E to yield the plasmid T-KO using T4 DNA ligase as per Table 3.
- Digest the plasmid T-KO with SacII and NdeI enzymes as per Table 4 to produce the disruption cassette (Figure 1). Purify the digested DNA fragments using a PCR purification kit as per manufacturer's instructions.
- Competent cell preparation and transformation of Y. lipolytica Po1g strain
- Competent cell preparation
- Inoculate a colony of Y. lipolytica Po1g strain from a fresh yeast extract-peptone-dextrose (YPD) plate in 10 ml of YPD medium (1% yeast extract, 2% peptone, 2% dextrose and 50 mM citrate buffer pH 4.0) in a 100 ml flask. Incubate in a 30 °C shaking incubator at 225 rpm for 20 hr until saturation (an OD600 of about 15, measured using a spectrophotometer).
- Pellet the cells by centrifuging for 5 min at 5,000 x g at room temperature. Wash the cells with 20 ml Tris-EDTA (TE) buffer (10 mM Tris, 1 mM EDTA, pH 7.5), and pellet the cells as described in step 1.2.1.2. Resuspend the cells in 1 ml of 0.1 M lithium acetate (pH 6.0, adjusted with acetic acid), and incubate for 10 min at room temperature.
- Aliquot the competent cells (100 µl) into sterile 1.5 ml tubes. Proceed to the transformation steps below immediately, or add glycerol to a final concentration of 25% (v/v) and store at -80 °C for long-term storage.
- Transformation
- Gently mix 10 µl of denatured salmon sperm DNA (10 mg/ml) and 1-5 µg of the purified disruption cassette together with 100 µl of competent cells, and incubate at 30 °C for 15 min.
- Add 700 µl of 40% polyethylene glycol (PEG)-4000 (dissolved in 0.1 M lithium acetate pH 6.0), mix well and incubate in a 30 °C shaking incubator at 225 rpm for 60 min. Note: It is important to use PEG with average molecular weight of 4000, instead of PEG-3350 (typically used in the transformation of conventional yeast).
- Heat shock the transformation mixture by placing the tube in a 39 °C water bath for 60 min.
- Add 1 ml YPD medium and recover for 2 hr at 30 °C and 225 rpm. Centrifuge at 9,000 x g for 1 min at room temperature, remove the supernatant and resuspend the pellet in 1 ml of TE buffer.
- Repellet the cells by centrifugation at 9,000 x g for 1 min at room temperature and discard the supernatant. Resuspend the pellet in 100 µl of TE buffer and plate onto selective plates (e.g., leucine-deficient plates), and incubate at 30 °C for 2-3 days.
- Pick 4 to 10 single colonies and inoculate separately into 2 ml of YPD medium. Incubate overnight in a 30 °C shaking incubator at 225 rpm. Identify positive colonies by PCR analysis of genomic DNA from the transformants 22 as per Table 5 using two sets of primers # 55/# 56, and # 56/# 57, respectively. Note: The NotI linearized pYLEX1 vector is transformed into Y. lipolytica Po1g competent cells in order to determine the transformation efficiency by counting the number of colony forming units (cfu) per µg plasmid DNA used 23.
2. Marker Rescue
- Construction of the Cre expression plasmid
- Construction of pYLEX1-CRE and pYLEX1-HPH
- Design primers 20 to PCR amplify the open reading frames of cre and hph. Add an extra adenine nucleotide upstream of the start codons in the forward primers # 82 and # 84, and insert KpnI restriction sites downstream of the stop codons in the reverse primers # 83 and # 85. PCR amplify the open reading frames of cre and hph from pSH69 (accession number HQ412578) 24 as per Table 2.
- Purify both cre and hph fragments using a PCR purification kit according to the manufacturer's instructions. Digest both the purified cre and hph fragments with KpnI enzyme as per Table 4. Double digest the pYLEX1 plasmid with PmlI and KpnI enzymes as per Table 4. Purify the digestion mixture using a PCR purification kit as per manufacturer's instructions.
- Use T4 DNA ligase as per Table 3 to ligate the purified and digested cre and hph fragments to PmlI/KpnI sites of the Y. lipolytica expression vector, yielding pYLEX1-CRE and pYLEX1-HPH, respectively.
- Construction of pSL16-CRE-HPH
- PCR amplify the promoter-gene-terminator cassette of cre from pYLEX1-CRE using primers # 86/# 87 flanking the SalI/PstI restriction sites as per Table 2. Purify the PCR product using a PCR purification kit according to the manufacturer's instructions.
- Double digest both the purified PCR product and pSL16-CEN1-1 (227) plasmid 25 with SalI and PstI enzymes as per Table 4. Purify the digestion mixture using a PCR purification kit as per manufacturer's instructions. Then, ligate the purified and digested PCR product into pSL16-CEN1-1 (227) at the corresponding sites to create pSL16-CRE using T4 DNA ligase as per Table 3.
- PCR amplify the promoter-gene-terminator cassette of hph from pYLEX1-HPH using primers # 88/# 89 flanking the XhoI/BglII restriction sites as per Table 2. Purify the PCR product using a PCR purification kit according to the manufacturer's instructions.
- Double digest both the purified PCR product and pSL16-CRE plasmid with XhoI and BglII enzymes as per Table 4. Purify the digestion mixture using a PCR purification kit as per manufacturer's instructions. Then, ligate the purified and digested PCR product into pSL16-CRE at the corresponding sites to generate pSL16-CRE-HPH using T4 DNA ligase as per Table 3. Note: The resulting Cre expression plasmid, pSL16-CRE-HPH, harbors a selectable hygromycin B marker and is a centromeric and episomally replicating vector (Figure 2).
- Verify all the constructs by digestion with appropriate restriction enzymes (e.g., SalI/PstI, to excise the insert from the vector) as per Table 4.
- Cre recombinase-mediated marker rescue
- Transform pSL16-CRE-HPH into competent cells of the KU70 knockout strain following the transformation protocol described in section 1.2 above. Plate transformed cells onto YPD plus hygromycin B (YPDH) plates (containing 400 µg/ml hygromycin B), and incubate at 30 °C for 2-3 days.
- Perform colony PCR as per Table 5 using primers # 84/# 85 to identify positive colonies containing pSL16-CRE-HPH plasmid after transformation. Note: The forward primer # 84 and the reverse primer # 85 are located inside the coding region of hph gene (Table 1). Only positive clones generate a specific PCR product in the PCR reaction.
- Pick a positive clone and inoculate into 2 ml of YPDH medium, and incubate in a 30 °C shaking incubator at 225 rpm overnight to saturation. Measure the cell OD600 with a spectrophotometer. Harvest the cells at an OD600 of ~15. Centrifuge at 5,000 x g for 5 min at room temperature, and wash once with 2 ml of sterile water.
- Re-inoculate cells into 2 ml of YPD medium with an initial OD600 of 0.1 and allow the cells to grow in a 30 °C shaking incubator at 225 rpm overnight. Streak the overnight cell culture onto YPD plates to isolate single colonies 23. Replica plate colonies onto YPD, YPDH, and leucine-deficient plates 23. Note: Colonies that have lost both LEU2 marker and pSL16-CRE-HPH plasmid can only grow on YPD plates (Figure 3).
3. Deletion of Alcohol Dehydrogenase and Alcohol Oxidase Genes
Perform a search on the Y. lipolytica genome database using the Basic Local Alignment Search Tool (BLAST) to identify the gene candidates for deletion 26. Input the protein sequence of S. cerevisiae alcohol dehydrogenase I into BLAST search tool (http://www.genome.jp/tools-bin/search_sequence?prog=blast&db=yli&seqid=aa). Note: The BLAST search tool returns the most similar protein sequences in the Y. lipolytica genome database.
Construct the deletion cassettes by PCR with primers # 7 to # 56 (Table 1), and perform restriction enzyme digestion and ligation reactions using the same methods as described in section 1.1 above.
Prepare competent cells of the KU70 knockout strain, carry out transformations and perform PCR to identify positive colonies with primers # 55, # 58 to # 81 (Table 1) by following the protocol described in section 1.2 above. Note: As an example, the procedure for the YALI0E17787g gene deletion is described in Figures 4 and 5. The effects of 1 kb and 2 kb long homologous region on homologous recombination rate are reported.
Representative Results
The linearized Y. lipolytica expression vector was inserted into the pBR docking platform in the genome of Y. lipolytica Po1g strain by performing a single crossover recombination 27. By using the rapid chemical transformation procedure established in this study, the linearized Y. lipolytica expression vector was successfully transformed into the wild-type Po1g strain at a transformation efficiency of >100 cfu/µg DNA. A knockout cassette flanked by 1 kb homologous sequences was transformed into the Po1g strain and the KU70 gene was successfully deleted (Figure 1). Here, the forward primer # 56 anneals to the sequence located within the 5' homology arm of the KU70 gene. The reverse primer # 55 is located inside the coding region of the LEU2 marker. The reverse primer # 57 is located inside the coding region of the KU70 gene. However, out of over 200 colonies, only one positive colony with the deleted KU70 gene was observed, suggesting a very low gene deletion rate (<0.5%). Subsequently, a replicative plasmid carrying the hygromycin B resistance marker and Cre/LoxP system was constructed (Figure 2), and the selection marker gene in the KU70 knockout strain was eventually removed by expression of Cre recombinase (Figure 3). Using the BLAST search tool, eleven putative alcohol dehydrogenase genes and one alcohol oxidase gene were identified in Y. lipolytica genome. The putative alcohol dehydrogenase genes are YALI0E17787g, YALI0D25630g, YALI0A16379g, YALI0E15818g, YALI0D02167g, YALI0A15147g, YALI0E07766g, YALI0F09603g, YALI0B10175g, YALI0F08129g, YALI0E07810g. The putative alcohol oxidase gene is YALI0B14014g. To verify the increase of gene deletion rate after deletion of the KU70 gene, we individually deleted 11 genes in parallel (except forYALI0A16379g) by using the disruption cassettes with long homologous sequences in the Y. lipolytica KU70 knockout strain (Figures 4 and 5). Here, the forward primers (P1-F and P2-F) anneal to the sequence located within the 5' homology arm of target genes. The reverse primers (P1-R) are located inside the coding region of LEU2 marker. The reverse primers (P2-R) are located inside the coding region of target genes (Figure 4). The knock-out cassettes differ only slightly in the lengths of the homologous sequences and the restriction sites (Table 1). Gene deletion rates [homologous recombination / (homologous recombination + non-homologous recombination)] of 33%-71% were achieved, suggesting a dramatic increase in the targeted homologous recombination frequency over the KU70 gene deletion. For instance, the deletion rate of YALI0E17787g was 40% (~100 cfu/µg DNA). Furthermore, the possibility of further increase in gene deletion rate was investigated when longer homologous sequences were used. It showed that the YALI0E17787g gene disruption cassette with 2 kb homologous sequences gave a deletion rate of 87.5% (~400 cfu/µg DNA). In addition, the introduced selection marker gene LEU2 in the obtained deletion mutants was removed by transforming the vector pSL16-CRE-HPH (hp4d-cre, hph), thus enabling multiple rounds of targeted gene manipulations.
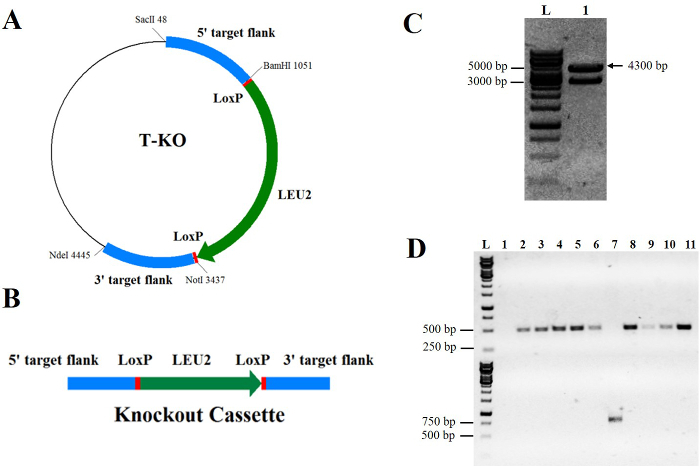 Figure 1. Construction of the KU70 knockout cassette and the KU70 deficient mutant.A. The KU70 knockout plasmid T-KO. B. The schematic diagram of the KU70 knockout cassette. Upstream and downstream regions (1 kb) of the KU70 gene were cloned to 5'- and 3'-end of the disruption cassette for homologous recombination.The LEU2 expression cassette was inserted between two homologous flanking sequences. C. Gel electrophoresis of the KU70 deletion cassette after SacII and NdeI digestion from T-KO. L: 1 kb DNA Ladder; 1: The KU70 deletion cassette (the expected band size is 4.3 kb). D. PCR confirmation of the KU70 gene deletion in Y. lipolytica Po1g. PCR products were amplified from genomic DNA of transformants (lanes 1-10) and wild-type strain (lane 11). Lane 7 illustrates a positive knockout, while lane 1 shows a false positive. In the top gel, a fragment of 500 bp is obtained with the primer set # 56/# 57 and is only absent in lanes 1 and 7. In the bottom gel, the successful knockouts generate a fragment of 750 bp, which is obtained with the primer set # 55/# 56. The 750 bp band is only seen in lane 7. L: 1 kb DNA Ladder. Please click here to view a larger version of this figure.
Figure 1. Construction of the KU70 knockout cassette and the KU70 deficient mutant.A. The KU70 knockout plasmid T-KO. B. The schematic diagram of the KU70 knockout cassette. Upstream and downstream regions (1 kb) of the KU70 gene were cloned to 5'- and 3'-end of the disruption cassette for homologous recombination.The LEU2 expression cassette was inserted between two homologous flanking sequences. C. Gel electrophoresis of the KU70 deletion cassette after SacII and NdeI digestion from T-KO. L: 1 kb DNA Ladder; 1: The KU70 deletion cassette (the expected band size is 4.3 kb). D. PCR confirmation of the KU70 gene deletion in Y. lipolytica Po1g. PCR products were amplified from genomic DNA of transformants (lanes 1-10) and wild-type strain (lane 11). Lane 7 illustrates a positive knockout, while lane 1 shows a false positive. In the top gel, a fragment of 500 bp is obtained with the primer set # 56/# 57 and is only absent in lanes 1 and 7. In the bottom gel, the successful knockouts generate a fragment of 750 bp, which is obtained with the primer set # 55/# 56. The 750 bp band is only seen in lane 7. L: 1 kb DNA Ladder. Please click here to view a larger version of this figure.
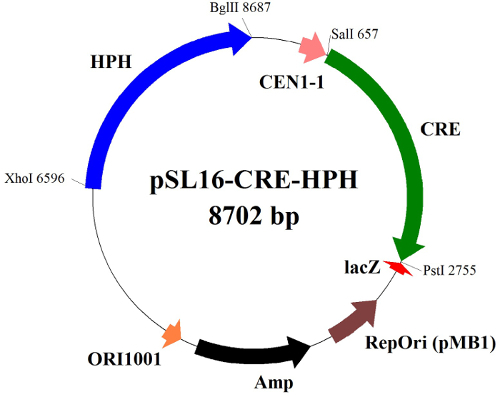 Figure 2.Schematic diagram of the Cre recombinase expression plasmid for marker rescue. The episomally replicating plasmid pSL16-CRE-HPH bears a Cre recombinase (CRE), a hygromycin B resistance marker (HPH), an ampicillin resistance gene (Amp), an origin of replication of Escherichia coli (pMB1), a replication origin of Y. lipolytica (ORI1001), a centromere DNA sequence of Y. lipolytica (CEN1-1) and a lacZ gene encoding the enzyme β-galactosidase. Please click here to view a larger version of this figure.
Figure 2.Schematic diagram of the Cre recombinase expression plasmid for marker rescue. The episomally replicating plasmid pSL16-CRE-HPH bears a Cre recombinase (CRE), a hygromycin B resistance marker (HPH), an ampicillin resistance gene (Amp), an origin of replication of Escherichia coli (pMB1), a replication origin of Y. lipolytica (ORI1001), a centromere DNA sequence of Y. lipolytica (CEN1-1) and a lacZ gene encoding the enzyme β-galactosidase. Please click here to view a larger version of this figure.
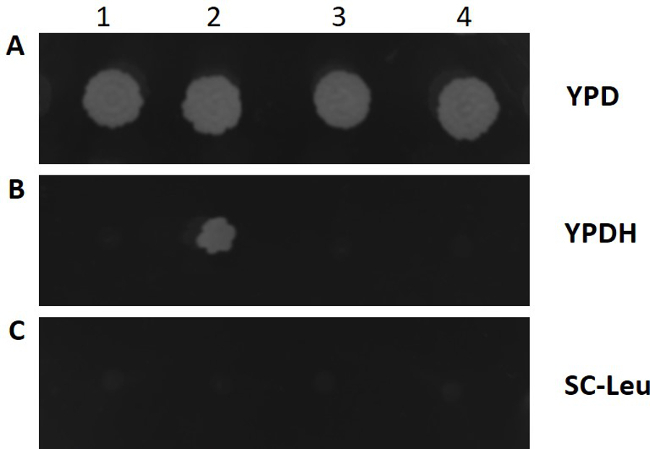 Figure 3.Growth screening of positive marker-lessY. lipolytica strains after marker rescue. Transformants (1-4) were diluted and spotted onto YPD plates, YPDH plates and leucine-deficient plates, respectively. After 3 days of growth at 30 °C, positive marker-free transformants (1, 3, 4) can only grow on YPD plate, which indicates the excision of LEU2 marker gene upon expression of the Cre recombinase and the loss of plasmid pSL16-CRE-HPH. A: YPD plate, B: YPDH plate, C: leucine-deficient plate. Please click here to view a larger version of this figure.
Figure 3.Growth screening of positive marker-lessY. lipolytica strains after marker rescue. Transformants (1-4) were diluted and spotted onto YPD plates, YPDH plates and leucine-deficient plates, respectively. After 3 days of growth at 30 °C, positive marker-free transformants (1, 3, 4) can only grow on YPD plate, which indicates the excision of LEU2 marker gene upon expression of the Cre recombinase and the loss of plasmid pSL16-CRE-HPH. A: YPD plate, B: YPDH plate, C: leucine-deficient plate. Please click here to view a larger version of this figure.
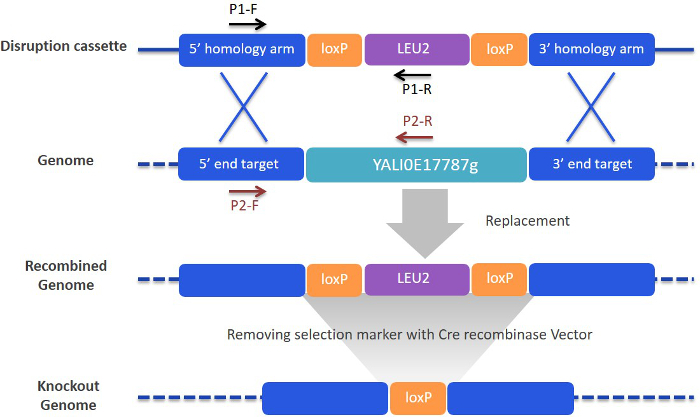 Figure 4.Schematic diagram of targeted gene deletion and marker rescue procedure. The gene disruption cassette consists of a selection marker gene (LEU2) flanked by two LoxP recognition sites and two targeting arms (upstream and downstream flanking sequences). After transformation of the disruption cassette into Y. lipolytica cells, a gene replacement event occurs via double-crossover homologous recombination within the two flanking homology arms at the targeted locus. Two sets of PCR primers were used to verify integration of the disruption cassette (P1-F and P1-R) and concurrent deletion of the targeted genomic regions (P2-F and P2-R). Finally, the LEU2 marker is removed by expression of Cre recombinase, leaving behind a single LoxP site at the chromosomal locus. Targeted gene deletion and replacement of YALI0E17787 gene was shown here as an example. Please click here to view a larger version of this figure.
Figure 4.Schematic diagram of targeted gene deletion and marker rescue procedure. The gene disruption cassette consists of a selection marker gene (LEU2) flanked by two LoxP recognition sites and two targeting arms (upstream and downstream flanking sequences). After transformation of the disruption cassette into Y. lipolytica cells, a gene replacement event occurs via double-crossover homologous recombination within the two flanking homology arms at the targeted locus. Two sets of PCR primers were used to verify integration of the disruption cassette (P1-F and P1-R) and concurrent deletion of the targeted genomic regions (P2-F and P2-R). Finally, the LEU2 marker is removed by expression of Cre recombinase, leaving behind a single LoxP site at the chromosomal locus. Targeted gene deletion and replacement of YALI0E17787 gene was shown here as an example. Please click here to view a larger version of this figure.
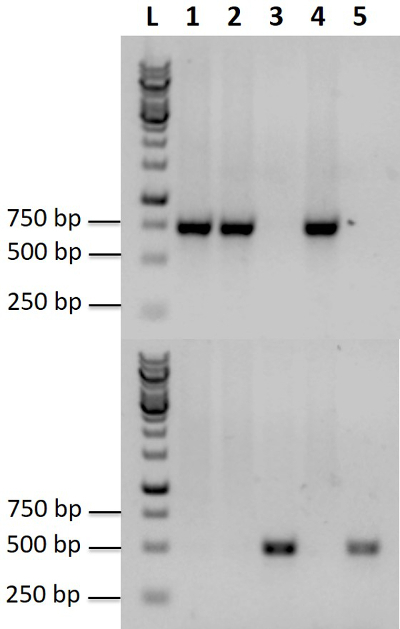 Figure 5.PCR confirmation of YALI0E17787 gene disruption inY. lipolytica Po1g. PCR products wereamplified from genomic DNA of transformants (lanes 1-4) and wild-type strain (lane 5). Lanes 1, 2, and 4 illustrate positive knockouts, while lane 3 shows a false positive. In the top gel, the successful knockouts generate a fragment of 750 bp, which is obtained with the primer set P1-F and P1-R. The 750 bp band is seen in lanes 1, 2, and 4, but not in lanes 3 and 5. In the bottom gel, a fragment of 500 bp is obtained with the primer set P2-F and P2-R and is only present in lanes 3 and 5. L: 1 kb DNA Ladder. Please click here to view a larger version of this figure.
Figure 5.PCR confirmation of YALI0E17787 gene disruption inY. lipolytica Po1g. PCR products wereamplified from genomic DNA of transformants (lanes 1-4) and wild-type strain (lane 5). Lanes 1, 2, and 4 illustrate positive knockouts, while lane 3 shows a false positive. In the top gel, the successful knockouts generate a fragment of 750 bp, which is obtained with the primer set P1-F and P1-R. The 750 bp band is seen in lanes 1, 2, and 4, but not in lanes 3 and 5. In the bottom gel, a fragment of 500 bp is obtained with the primer set P2-F and P2-R and is only present in lanes 3 and 5. L: 1 kb DNA Ladder. Please click here to view a larger version of this figure.
Table 1. Primers used in this study. Please click here to download this table.
| PCR component | ||
| PCR materials | Volumes | Final concentration |
| PCR buffer | 10 µl | 1x |
| dNTP mix | 1 µl | 200 µM |
| Forward primer | 2.5 µl | 500 nM |
| Reverse primer | 2.5 µl | 500 nM |
| DNA polymerase | 0.5 µl | 1 U |
| DNA template (diluted genomic or plasmid DNA) | 0.5-2 µl | 50 ng |
| Sterile water | up to 50 µl | |
| PCR conditions | ||
| Temperature | Time | Number of cycles |
| 98 °C | 30 sec | 1 |
| 98 °C | 10 sec | 30 |
| 55 °C | 30 sec | |
| 72 °C | 30 sec | |
| 72 °C | 10 min | 1 |
| Perform PCR amplification using a thermal cycler |
Table 2. PCR set-up and conditions for the high-fidelity DNA polymerase.
| Component | |
| Materials | Final concentration |
| T4 DNA ligase Buffer | 1x |
| Vector DNA | 0.03 pmol |
| Insert DNA | 0.15 pmol |
| T4 DNA ligase | 400 U |
| Sterile water | up to 20 µl |
| Conditions | |
| Temperature | Time |
| 16 °C | 16 hr |
Table 3. DNA ligation reaction conditions using T4 DNA ligase.
| Component | |
| Materials | Final concentration |
| Restriction digestion buffer | 1x |
| DNA (Plasmid or PCR product) | 1 µg |
| 1st restriction enzyme | 5 U |
| 2nd restriction enzyme (optional) | 5 U |
| Sterile water | up to 50 µl |
| Conditions | |
| Temperature | Time |
| 37 °C | 6 hr |
| Perform single digestion with a single restriction enzyme | |
| Perform double digestion with a pair of restriction enzymes |
Table 4. Conditions for restriction enzyme digestion of DNA.
| PCR component | ||
| PCR materials | Volumes | Final concentration |
| PCR buffer | 2.5 µl | 1x |
| dNTP mix | 0.5 µl | 200 µM |
| Forward primer | 0.5 µl | 200 nM |
| Reverse primer | 0.5 µl | 200 nM |
| DNA polymerase | 0.2 µl | 1 U |
| DNA template (diluted genomic or plasmid DNA) | 0.5-2 µl | 50 ng |
| Sterile water | up to 25 µl | |
| PCR conditions | ||
| Temperature | Time | Number of cycles |
| 95 °C | 5 min | 1 |
| 95 °C | 30 sec | 30 |
| 50 °C | 30 sec | |
| 72 °C | 30 sec | |
| 72 °C | 10 min | 1 |
| Perform PCR amplification using a thermal cycler | ||
| Perform colony PCR directly using a colony as template, rather than a DNA sample |
Table 5. PCR/Colony PCR set-up and conditions for Taq DNA polymerase.
Discussion
Our objective for this study is to enable quick and efficient generation of targeted gene knockouts in the Y. lipolytica Po1g strain. Several considerations need to be addressed to achieve this. First, a high transformation efficiency is required. Thus, an efficient and convenient chemical transformation protocol for the Y. lipolytica Po1g strain was described in this study. The use of PEG-4000 is a critical factor for the successful transformation of this strain. No transformants were obtained for plasmid transformation when using PEG-3350 (for the transformation of S. cerevisiae) instead of PEG-4000. In addition, preparation of competent cells using freshly prepared cells is required to obtain high transformation efficiency in this strain. Second, to reduce non-homologous recombination and random integration events via dominant NHEJ in this strain, an efficient transformation method was then employed to construct the KU70 knockout mutant. Third, to improve the efficiency of gene replacement by homologous recombination, long flanking arms (1 kb) upstream and downstream of the target genes were used in gene deletion cassettes. The long flanking homologous regions in the disruption cassettes are essential for efficient homologous recombination in this strain. No positive colonies were obtained when using disruption cassettes with 50 bp homologous regions (sufficient for efficient homologous recombination in S. cerevisiae). Using all these strategies, the KU70 gene of the Y. lipolytica Po1g strain was successfully deleted. Nonetheless, it is extremely difficult to obtain the KU70 knockout strain due to very high rate of non-homologous recombination in the Y. lipolytica Po1g strain. Care must be taken when handling the antibiotic hygromycin B containing media as hygromycin B is light sensitive. Using media with degraded hygromycin B will result in false positives in steps 2.2.1 and 2.2.2 and false negatives in step 2.2.7.
When the KU70 deletion strain was used to generate targeted deletion of individual genes encoding alcohol dehydrogenases and alcohol oxidase, a much higher deletion rate was achieved compared to the deletion of the KU70 gene. It demonstrates that the use of the KU70 deletion platform strain resulted in a dramatic increase in homologous recombination frequency. After the issue of very low homologous recombination rate was addressed, screening of Y. lipolytica single-gene knockout mutants was highly sped up. We also noted that the 1 kb length of homologous region was efficient enough for targeted gene deletion. An increase in homologous recombination frequency could be achieved when using longer homologous sequences (2 kb). Notably, these genes encode alcohol dehydrogenases and alcohol oxidase, which catalyze the reversible conversion between alcohols and aldehydes. The deletion of these genes can lead to increased accumulation of biofuel and biochemical compounds including short-chain fatty acids, medium-chain fatty acids, long-chain fatty acids, hydroxy fatty acids, alcohols, aldehydes and lipids. Further studies will be performed to verify this possibility.
A PCR-mediated gene disruption method has been reported in Y. lipolytica Po1d strain 28. However, this method is laborious and time-consuming. One major limitation of this method is the difficulty of obtaining sufficient quantities of the gene deletion cassettes for one transformation experiment. Another limitation is that this method is limited to the URA3 marker. The method described in this study specifically addressed these issues in the PCR-based technique by combining the digestion-ligation approach and the Cre/LoxP system. This method allows the usage of different antibiotic and auxotrophic markers and thus is more versatile. It can facilitate rapid gene deletion in the Y. lipolytica Po1g strain. We herein present a detailed description of the targeted single-gene deletion strategy through homologous recombination in Y. lipolytica Po1g cells. However, one LoxP site remains as a scar within the genome after the successful marker rescue (Figure 4). In multiple-gene deletion, multiple LoxP scars are introduced into the genome and this may result in genomic instability due to potential recombination events that can occur between the LoxP sites. Nonetheless, the knockout strains constructed in this study showed very good genetic stability, indicating that unintended recombination events did not occur between the LoxP sites.
In summary, an efficient genetic transformation protocol has been established for the Y. lipolytica Po1g strain. The targeted deletion of single genes in this strain was demonstrated as a proof of concept. The Y. lipolytica Po1g KU70 deletion platform strain constructed in this study showed very efficient homologous recombination frequency, which enables an easier and faster screening of the targeted gene deletions. Thus, this platform strain provides a more efficient system and a better choice for applications in pathway engineering and strain optimization when constructing other chromosomal deletion mutants. The described methodology and the constructed KU70 deletion strain could also be applied to the insertion of target genes into specific loci and the creation of site-specific mutations into the genome.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We gratefully acknowledge the funding support from the National Environment Agency of Singapore (ETRP 1201102), the Competitive Research Program of the National Research Foundation of Singapore (NRF-CRP5-2009-03), the Agency for Science, Technology and Research of Singapore (1324004108), Global R&D Project Program, the Ministry of Knowledge Economy, the Republic of Korea (N0000677), the Defense Threat Reduction Agency (DTRA, HDTRA1-13-1-0037) and the Synthetic Biology Initiative of the National University of Singapore (DPRT/943/09/14).
References
- Jiménez-Bremont JF, Rodrìguez-Hernández AA, Rodrìguez-Kessler M. J Ruiz-Herrera., editor. Development and dimorphism of the yeast Yarrowia lipolytica. Dimorphic fungi: Their importance as models for differentiation and fungal pathogenesis. 2012. pp. 58–66.
- Coelho M, Amaral P, Belo I. Méndez-Vilas A, editor. Yarrowia lipolytica: an industrial workhorse. Current research, technology and education topics in applied microbiology and microbial biotechnology. 2010. pp. 930–940.
- Martinez-Vazquez A, et al. Identification of the transcription factor Znc1p, which regulates the yeast-to-hypha transition in the dimorphic yeast Yarrowia lipolytica. Plos One. 2013;8(6):e66790. doi: 10.1371/journal.pone.0066790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard M, Quijano RR, Bezzate S, Bordon-Pallier F, Gaillardin C. Tagging morphogenetic genes by insertional mutagenesis in the yeast Yarrowia lipolytica. J. Bacteriol. 2001;183(10):3098–3107. doi: 10.1128/JB.183.10.3098-3107.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CL, Martìnez-Costa OH, Sánchez V, Gancedo C, Aragòn JJ. The dimorphic yeast Yarrowia lipolytica possesses an atypical phosphofructokinase: characterization of the enzyme and its encoding gene. Microbiology. 2005;151(5):1465–1474. doi: 10.1099/mic.0.27856-0. [DOI] [PubMed] [Google Scholar]
- Domìnguez Á, et al. Non-conventional yeasts as hosts for heterologous protein production. Int. Microbiol. 1998;1(2):131–142. [PubMed] [Google Scholar]
- Zinjarde SS. Food-related applications of Yarrowia lipolytica. Food Chem. 2014;152:1–10. doi: 10.1016/j.foodchem.2013.11.117. [DOI] [PubMed] [Google Scholar]
- Finogenova T, Morgunov I, Kamzolova S, Chernyavskaya O. Organic acid production by the yeast Yarrowia lipolytica: a review of prospects. Appl. Biochem. Micro+ 2005;41(5):418–425. [PubMed] [Google Scholar]
- Groguenin A, et al. Genetic engineering of the β-oxidation pathway in the yeast Yarrowia lipolytica to increase the production of aroma compounds. J. Mol. Catal. B-Enzym. 2004;28(2):75–79. [Google Scholar]
- Meng X, et al. Biodiesel production from oleaginous microorganisms. Renew. Energ. 2009;34(1):1–5. [Google Scholar]
- Papanikolaou S, Aggelis G. Lipid production by Yarrowia lipolytica growing on industrial glycerol in a single-stage continuous culture. Bioresource Technol. 2002;82(1):43–49. doi: 10.1016/s0960-8524(01)00149-3. [DOI] [PubMed] [Google Scholar]
- Tai M, Stephanopoulos G. Engineering the push and pull of lipid biosynthesis in oleaginous yeast Yarrowia lipolytica for biofuel production. Metab. Eng. 2013;15:1–9. doi: 10.1016/j.ymben.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Liu HH, Ji XJ, Huang H. Biotechnological applications of Yarrowia lipolytica: Past, present and future. Biotechnol. Adv. 2015;33(8):1522–1546. doi: 10.1016/j.biotechadv.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Liu L, Alper HS. Draft genome sequence of the oleaginous yeast Yarrowia lipolytica PO1f, a commonly used metabolic engineering host. Genome Announc. 2014;2(4):e00652–e00614. doi: 10.1128/genomeA.00652-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B, et al. Genome evolution in yeasts. Nature. 2004;430(6995):35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- Chen DC, Beckerich JM, Gaillardin C. One-step transformation of the dimorphic yeast Yarrowia lipolytica. Appl. Microbial. Biotechnol. 1997;48(2):232–235. doi: 10.1007/s002530051043. [DOI] [PubMed] [Google Scholar]
- Wang JH, Hung W, Tsai SH. High efficiency transformation by electroporation of Yarrowia lipolytica. J. Microbiol. 2011;49(3):469–472. doi: 10.1007/s12275-011-0433-6. [DOI] [PubMed] [Google Scholar]
- Matsuoka M, et al. Analysis of regions essential for the function of chromosomal replicator sequences from Yarrowia lipolytica. Mol. Gen. Genet. 1993;237(3):327–333. doi: 10.1007/BF00279435. [DOI] [PubMed] [Google Scholar]
- Kretzschmar A, et al. Increased homologous integration frequency in Yarrowia lipolytica strains defective in non-homologous end-joining. Curr. Genet. 2013;59(1-2):63–72. doi: 10.1007/s00294-013-0389-7. [DOI] [PubMed] [Google Scholar]
- Lorenz TC. Polymerase Chain Reaction: Basic Protocol Plus Troubleshooting and Optimization Strategies. J. Vis. Exp. 2012. p. e3998. [DOI] [PMC free article] [PubMed]
- Kobs G. Cloning blunt-end DNA fragments into the pGEM-T Vector Systems. Promega Notes. 1997;62:15–18. [Google Scholar]
- JoVE Science Education Database. Essentials of Biology 1: yeast, Drosophila and C. elegans. Cambridge, MA: JoVE; 2016. Isolating Nucleic Acids from Yeast. Available from: http://www.jove.com/science-education/5096/isolating-nucleic-acids-from-yeast. [Google Scholar]
- Sanders ER. Aseptic laboratory techniques: plating methods. J. Vis. Exp. 2012. p. e3064. [DOI] [PMC free article] [PubMed]
- Hegemann JH, Heick SB. Delete and repeat: a comprehensive toolkit for sequential gene knockout in the budding yeast Saccharomyces cerevisiae. Methods Mol. Biol. 2011;765:189–206. doi: 10.1007/978-1-61779-197-0_12. [DOI] [PubMed] [Google Scholar]
- Yamane T, Sakai H, Nagahama K, Ogawa T, Matsuoka M. Dissection of centromeric DNA from yeast Yarrowia lipolytica and identification of protein-binding site required for plasmid transmission. J. Biosci. Bioeng. 2008;105(6):571–578. doi: 10.1263/jbb.105.571. [DOI] [PubMed] [Google Scholar]
- McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32((Web Server issue)):W20–W25. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madzak C, Tréton B, Blanchin-Roland S. Strong hybrid promoters and integrative expression/secretion vectors for quasi-constitutive expression of heterologous proteins in the yeast Yarrowia lipolytica. J. Mol. Microbiol. Biotechnol. 2000;2(2):207–216. [PubMed] [Google Scholar]
- Nicaud JM, Le Clainche A, Le Dall MT, Wang H, Gaillardin C. Yarrowia lipolytica, a yeast model for the genetic studies of hydroxy fatty acids biotransformation into lactones. J. Mol. Catal. B-Enzym. 1998;5(1):175–181. [Google Scholar]