

Abstract
One of the limiting factors to the adoption and advancement of personalized medicine is the inability to develop diagnostic tools to probe individual nuances in expression from patient to patient. Current methodologies that try to separate cells to fill this niche result in disruption of physiological expression, making the separation technique useless as a diagnostic tool. In this protocol, we describe the functionalization and optimization of a surface for the cellular capture and release. This functionalized surface integrates biotinylated antibodies with a glass surface functionalized with an aminosilane (APTES), desthiobiotin and streptavidin. Cell release is facilitated through the introduction of biotin, allowing the recollection and purification of cells captured by the surface. This release is done through the targeting of the secondary moiety desthiobiotin, which results in a much more gentle release paradigm. This reduction in harsh reagents and shear forces reduces changes in cellular expression. The functionalized surface captures up to 80% of cells in a single cell mixture and has demonstrated 50% capture in a dual-cell mixture. Applications of this technology to xenografts and cancer separation studies are investigated. Quantification techniques for surface verification such as plate reader and ImageJ analyses are described as well.
Keywords: Cancer Research, Issue 115, Bioengineering, cell isolation, personalized medicine, desthiobiotin, SAv, biotin, glass functionalization, APTES, MCF7-GFP, RAW 264.7
Introduction
Current bench-top cell separation approaches (e.g., fluorescence activated cell sorting1, laser capture micro-dissection2, immuno-magnetic bead separation1) can take several hours of preparation and sorting. These large time scales can affect physiological response and expression levels, resulting in analyses that are not representative of the physiological response3. Systems are needed that can rapidly and efficiently isolate specific cell types without disrupting cell-surface receptor-levels in order to improve cell isolation and enrichment for biomedical applications. Therefore, the rationale for our approach is to develop a gentle approach for cell isolation.
The "lab on a chip" concept offers the promise of orders of magnitude quicker (hours-to-minutes) cell isolation, and most frequently involves capturing cells onto a surface and releasing cells or intracellular contents through physical4,5 or chemical methods6. Although these approaches offer a few advantages such as identifying protein7,8 expression, identifying RNA expression9-11, or even providing cells for in vitro culture12,13, many of these techniques cannot be translated to diagnostics such as cell receptor profiling due to their non-physiological environments. Enzymatic lifting agents such as collagenases can also affect these receptor quantities14,15, meaning cell receptor quantification techniques that use these lifting agents will not generate accurate physiological data. Cellular lysis prevents differentiation between the native surface receptors, and those which were previously internalized16. This protocol describes a fast and gentle approach for cell isolation.
Protocol
1. Cleaning the Glass Surface and Preparing Reagents
Place a glass surface in an oxygen plasma machine for 5 min at 50% power to clean it.
Prepare 2.5 ml 2% reconstituted (3-aminopropyl) triethoxysilane (APTES) solution, by adding 50 µl of APTES and 2.45 ml of ethanol in a conical tube.
2. APTES and DSB Functionalization
Add APTES solution to the surfaces. Pipette 150 µl per well for 8 well plates. Pipette 100 µl per well for 24 well plates. Pipette 1.1 ml for 60 x 15 mm glass dishes. Cover the surfaces to prevent evaporation and uneven distribution of APTES solution. Place the surfaces on a platform shaker for 50 min at room temperature, which creates an even distribution of the APTES layer. NOTE: APTES is an aminosilane that forms the first layer of the surface. If using a different surface, heuristically determine the volume of solution needed to cover the surface.
Select the temperature for the oven, while the surfaces are on the shaker: Heat to 55 °C for 2 hr for the 8 well plates and 24 well plates. Heat glass only dishes to 90 °C for 1 hr.
- Rinse the surfaces with ethanol.
- Administer the amount of ethanol required by referencing based on the surface used. Add 150 µl ethanol to each well for 8 well plates. Add 125 µl ethanol to each well for 24 well plates. Add 1.1 ml ethanol for glass dishes.
- Rinse the surface by discharging and drawing the liquid from a fixed point such as the corner of the well. Hold the pipette at approximately a 70° angle so the tip is not directly pointed into the surface. Rinse twice more using ethanol. NOTE: If any of the glass bottom well plates have any plastic in them, do not heat them above 65 °C, as the plastic will begin to melt and warp.
Dry with 100% nitrogen gas dispensed from a tank. Place the surfaces in the oven.
Prepare 2.5 ml of d-desthiobiotin (DSB) solution by combining 1.5 mg/ml DSB in 37.5 µl of dimethyl sulfoxide (DMSO), and 5 mg/ml 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) in 2462.5 µl of the 0.1 M 4-morpholinoethanesulfonic acid hydrate (MES) (pH 6) buffer. Then combine both solutions.
Add 1 µl of 2-β mercaptoethanol, after 15 min, into the solution to quench the reaction between DSB and EDC. Remove hot APTES functionalized glass surfaces from the oven. Wait 5-10 min for the glass surfaces to cool.
Add MES buffer the surface to rinse, using amounts based on the surface. Add 150 µl MES buffer to each well for 8 well plates. Add 125 µl MES buffer to each well for 24 well plates. Add 1.1 ml MES buffer for glass dishes.
Rinse surface by discharging and drawing the liquid from a fixed point such as the corner of the well. Hold the pipette at approximately a 70° angle so the tip is not directly pointed into the surface. Rinse twice more with MES buffer.
Apply the DSB solution to the surfaces to allow them to incubate. Add 150 µl DSB solution per well for 8 well plates. Add 100 µl DSB solution per well for 24 well plates. Add 1.1 ml for glass dishes DSB solution.
Place the DSB covered glass surfaces on a damp paper towel inside a Petri dish. Cover and incubate in a 4 °C fridge for 18-24 hr.
3. Streptavidin Functionalization
Rinse each glass surface three times with 1 ml of 1.0x Phosphate Buffered Saline (PBS). Rinse surface by discharging and drawing the liquid from a fixed point such as the corner of the well. Hold the pipette at approximately a 70° angle so the tip is not directly pointed into the surface. Dilute the streptavidin (SAv) stock solution to 0.4 mg/ml (recommended). NOTE: PBS is isotonic so it can be used as a medium for dilution and rinsing. PBS is used as a solvent for the SAv, and therefore, will not affect the ability of the SAv to bind to the surface.
Apply 0.4 mg/ml of SAv solution evenly to the surfaces so that a thin layer of forms at the bottom of the glass, select the amount of solution based on surface. For 8 well plates, add 150 µl 0.4 mg/ml SAv solution per well. For 24 well plates, add 100 µl 0.4 mg/ml SAv solution per well. For glass dishes, add 1.1 ml 0.4 mg/ml SAv solution.
Cover and move the plates to a 14 cm Petri dish to retain moisture. Incubate the Petri dish in refrigerator for 18-24 hr. NOTE: It is essential to utilize consistent volumes of APTES, DSB, and SAv on each surface.
Rinse each glass surface three times with 150 µl of PBS to remove SAv. Rinse surface by discharging and drawing the liquid from a fixed point such as the corner of the well. Hold the pipette at approximately a 70° angle so the tip is not directly pointed into the surface.
Wet a paper towel with de-ionized water and place the paper towel flat in 14 cm Petri dish surrounding the plates to retain moisture in the wells. Cover the Petri dish containing the wells. Incubate the Petri dish in a 4 °C Biosafety level 1 (BSL-1) refrigerator until needed.
4. Cell Capture and Release
Start with T175 flask(s) of cells intended for the experiment. Aspirate the media from flask(s). Wash out the remaining media with 5 ml of room temperature PBS. Aspirate PBS from the flask(s).
Add 10 ml of non-enzymatic lifting agent, such as Cell Dissociation Solution, to the T-175 flask of cells. Put the flask in the incubator for 6 min to allow for lifting of the cells from the flask.
After 6 min, add 10 ml of cold Hank's Balanced Salt Solution (HBSS; See List of Materials) to inactivate the lifting agent. Take out 20 µl of cells to count number of cells in solution using a hemacytometer. NOTE: Depending on the functionalized surfaces used in the capture experiment, the number of cells needed varies. For functionalized 8 well plates, use 300,000 cells per well. For functionalized glass dishes, use 1.1 million cells. For functionalized 24 well plates, 125,000 cells are recommended. More information on cell counting is found in the Supplement.
Repeat steps 4.1- 4.3 for another flask of cells, if fewer cells are present than needed for the experiment. Combine cell suspensions and recount cells to get total number of cells in solution.
Spin down cell suspension in a centrifuge at 500 x g for 5 min at 4 °C to get a pellet of concentrated cells. Find the appropriate amount of HBSS to add to the cell suspension to get a concentration of 1 x 106 cells per ml, then aspirate the supernatant from the spun down cells, and add the calculated volume. This volume can be calculated using the equations in the Supplement.
Pipette up and down (triturate) to resuspend the cells in solution and reduce cellular clumping in solution that may reduce antibody binding. Split the cellular solution into separate control and experimental solutions. View Supplemental File for components and calculations.
Add the biotinylated antibodies to the respective cell solutions as described in the Supplemental File. Incubate for 30 min at 4 °C on an end-over-end mixer. NOTE: For MCF7GFP cells, 0.5 mg/ml hIgG or 0.5 mg/ml HLA-ABC antibodies are recommended. For RAW macrophages, 1 mg/ml mCD11b are recommended at half the volume to account for the dilution differences. For Human Umbilical Vein Endothelial Cells (HUVECs), 0.5 mg/ml hCD31 is recommended.
- Wash the functionalized glass surface with HBSS. For this experiment, an 8 well plate is advised.
- Add 150 µl HBSS to each well for 8 well plates. Rinse twice more using HBSS. Rinse surface by discharging and drawing the liquid from a fixed point such as the corner of the well. Hold the pipette at approximately a 70° angle so the tip is not directly pointed into the surface. Keep cold at 4 °C.
Add cell solutions to the wells and wait 45 min for the cells to incubate on ice on the shaker. Make the solution of biotin in sterile HBSS by using calculated volumes as described in the Supplement.
- Remove the cell solution from the glass wells by using HBSS.
- Gently pipette 150 µl HBSS into each well. Then pipette out the HBSS. Rinse surface by discharging and drawing the liquid from a fixed point such as the corner of the well. Hold the pipette at approximately a 70° angle so the tip is not directly pointed into the surface.
- Repeat two more times. After washing, add HBSS to the wells to keep the cells wet.
Add 150 µl of 20 mM biotin solution to each respective release well, and then wait 20 min to allow for reaction.
Recollect non-specifically bound cells by washing with HBSS as stated above, and then if using fluorescently labeled cells, proceed to fluorescence image the cells. Also image live cells in a flask (as a control, to compare with the cells in the well). If using green fluorescent protein (GFP) transfected cells, the excitation will be 470 nm, and the emission will be 515 nm.
5. Antibody Optimization: Antibody Titration
Begin by lifting the cells from the T75 or T175 flask as explained in step 4.1- 4.3. NOTE: These volumes are calibrated for 24 well plates, but can be changed to suit any glass functionalized surfaces through heuristic testing. A representative antibody titration is shown in Figure 1.
Count cells using the hemacytometer, and then spin down the cell solution in a conical tube at 500 x g for 5 min at 4 °C. Reconstitute cells to 1 million cells per ml, using the calculations described in the Supplement.
- Split the 1 million cells per ml solution into six different solutions of 500 µl in different centrifuge tubes for the antibody (Ab) solutions.
- Dilute the antibody stock solution to 10 µg/ml, 1 µg/ml, 100 ng/ml, 10 ng/ml, 1 ng/ml. Create the controls by using 100 µl of Stain Buffer (PBS + 1% sodium azide + 1% BSA), and 500 µl of cells for the control with no Ab in it. For the blank control (No Ab and no cells), use a solution of 300 µl PBS. NOTE: CAUTION: Sodium azide is extremely toxic, explosive, and extreme care must be taken when using it. Please consult the material safety data sheet (MSDS) and use the appropriate safety equipment.
Combine and incubate the Ab solutions with the cell solutions in the end-over-end mixer at 4 °C for 30 min. Rinse the functionalized 24 well plates with HBSS three times. Rinse the surface by discharging and drawing the liquid from a fixed point such as the corner of the well. Hold the pipette at approximately a 70° angle so the tip is not directly pointed into the surface.
Aliquot 125 µl of each sample solution into the appropriate APTES, DSB, and SAv functionalized well. Incubate at 4 °C or on ice for 45 min on the glass surface. Rinse the surface with HBSS three times to remove non-specifically attached cells. Rinse surface by discharging and drawing the liquid from a fixed point such as the corner of the well. Hold the pipette at approximately a 70° angle so the tip is not directly pointed into the surface. Do NOT rinse the bottom row, as those are controls.
Add 150 µl of HBSS to each washed well, put the 24 well plates on ice, and then use a plate reader to measure fluorescence of GFP cells (Excitation 485 nm / Emission 528 nm) .
6. Cell Optimization: Cell Titration
Lift the cells as mentioned in steps 4.1 -4.3.
Count cells using a hemacytometer. Spin down cell solution in conical tube at 500 x g for 5 min at 4 °C. Reconstitute cells to 1 million cells per ml. NOTE: More information on the use of the hemacytometer can be found in the Supplement.
Pipette 1.6 ml from the cell solution for the 1.6 million cells stock and place in a conical tube. Pipette 800 µl of cells from the cell solution for the 800,000 cells stock and place in a conical tube. Pipette 80 µl from the cell stock for the 80,000 cells stock and place in a conical tube. Pipette 8 µl from the cell stock for 8,000 cells stock and place in a conical tube. NOTE: Concentrations are given per 8 well plate measurements, replicate as needed. An example of plate output is shown in Figure 2.
Take the four stock solutions and spin in a centrifuge at 500 x g for 5 min at 4 °C. Re-suspend all stock solutions in 400 µl of HBSS.
Add 1.6 µl of 100 ng/ml antibody to the 1.6 million cell stock, 0.8 µl to the 800,000 cell stock, and 0.5 µl to all other stocks. Incubate the antibody for 30 min in the end-over-end mixer for 30 min at 4 °C. Wash the functionalized glass surface with HBSS three times. NOTE: The functionalized 8 well plate is recommended for microscopy quantification, while the functionalized 24 well plate is recommended for plate reader quantification. Antibody concentration for 8,000 cell stock was not reduced to allow the limiting factor of the capture surface to not be the lack of antibodies in solution, but rather the capturing properties of the surface.
Apply 150 µl of the sample solutions to each well. Apply the 1.6 million cell stock to the first column of wells on the left. Apply the 800,000 cell stock to the second column from the left. Apply the 80,000 cell stock to the third column from the left. Finally apply the 8,000 cell stock to the last column. Incubate for 45 min at 4 °C on the shaker. NOTE: The 1.6 million cell stock serves as the highest concentration to be tested, and is double the amount of cells that is used typically in cell separation experiments (600,000 cells per well) The 800,000 cell stock serves as the control for the experiment as it is the typical cellular concentration that is used in cell separation experiments (3,000,000 cells per well). The 80,000 cell stock serves as a ten-fold dilution to test for lower concentrations for cellular capture (30,000 cells per well). The 8,000 cell stock serves as a hundred fold dilution to use as a test to check the lower limit of cellular separation (3,000 cells per well).
Rinse with HBSS three times. Rinse surface by discharging and drawing the liquid from a fixed point such as the corner of the well. Hold the pipette at approximately a 70° angle so the tip is not directly pointed into the surface. Collect all washes.
Image the cells on a fluorescence microscope, if using 8 well plates, take pictures of each surface, and apply 150 µl biotin to each surface for one hour at 4 °C on the shaker. After one hour, rinse biotin release wells with HBSS 3 times as before. Collect all washes from wells for counting with the hemocytometer and image the release wells.
Apply 150 µl of 20 mM excess biotin solution to appropriate biotin release wells for 1 hr, if using 24 well plates, and then rinse those wells 3 times with HBSS. Quantitate 24 well plates using a plate reader. Use a hemacytometer to count cells from all collected well washes.
7. Image Analysis
Note: The FIJI software package (http://fiji.sc/Fiji) is recommended for image analysis. Initially, the images were converted into grayscale image, and then the brightness/contrast was altered to bring out the cells.
Load up the image, and convert to grayscale by clicking the image tab then scrolling down to "Type" and then clicking "8 bit".
Increase the contrast of the image by clicking the image tab and then scrolling to "Adjust". Click on "Brightness and Contrast", and then use the scroll bars to make the cells stand out. If using fluorescence images, invert the images to make the cells more defined.
Load a plugin onto ImageJ called ITCN (http://rsb.info.nih.gov/ij/plugins/itcn.html) to analyze the images.
Open ITCN. When a dialog box appears that prompts the user with several parameters to estimate the size of the cell, set the minimum width of the cell of interest first. Click the "Detect Dark Peaks" option if the image is fluorescent and the cells are darkened.
Set the threshold value at 2 initially, and then hit the "Count" button. A newly counted version of the picture will appear with red dots delineating where cells have been counted.
Adjust the threshold and width parameter to get more accurate cellular data by defining the size of a "cell". Note: The number of cells counted will be in a dialog box on the right. Changing parameters will give different number of cells counted.
Continue to iterate through the threshold and width parameters until a good estimate is reached for the number of cells.
Representative Results
Using this protocol we show cell capture (Figure 3A) and cell release (Figure 3C) of MCF7GFP cells as well as live cell controls (Figure 4). We quantified the cell capture as 60% and 80% were released (Figure 3C). When we extended this approach to a mixture of RAW 264.7 macrophages and MCF7GFP cells, 50% of RAW macrophages were captured (Fig. 3D) and 80% of RAW macrophages were release with 20 mM biotin (Figure 3B). Since excess antibody can decrease cell capture, we optimized via titrating 0-10,000 nM of HLA antibody, and observe that the ideal antibody concentration is between 100-1,000 mM antibody (Fig. 1). Similarly, we determine that the ideal concentration of cells that can be captured is 1 x 105 and 1 x 106, since below that number of cells, values are lower than the background17 (Figure 2). The fluorescence data from each well is processed as described below.
![]()
Here, the average of 3 replicates is taken from a sample with no antibody (blank) and is subtracted from the fluorescence obtained from each sample. This value is then normalized to the average maximal fluorescence. This processing allows the researcher to clearly observe the low limit of cellular detection.
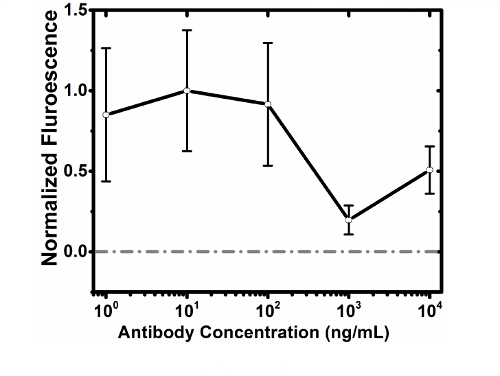 Figure 1.Normalized Blanked Antibody Titration. Human HLA-ABC is titrated across a constant quantity of MCF7GFP cells (125,000 cells per well) and the fluorescence of captured cells were quantified using a plate reader. Prior to exposure to the functionalized surface, the cells and antibodies were centrifuged to remove non-specific antibody attachment. Without this centrifugation, excess antibody will saturate surface and prevent cell pulldown, as shown with concentrations of 10,000 ng/ml where centrifugation was not sufficient to prevent antibody oversaturation of the surface, resulting in less cells binding to the surface. Gray line represents the control. Error bars represent standard error of the mean. Please click here to view a larger version of this figure.
Figure 1.Normalized Blanked Antibody Titration. Human HLA-ABC is titrated across a constant quantity of MCF7GFP cells (125,000 cells per well) and the fluorescence of captured cells were quantified using a plate reader. Prior to exposure to the functionalized surface, the cells and antibodies were centrifuged to remove non-specific antibody attachment. Without this centrifugation, excess antibody will saturate surface and prevent cell pulldown, as shown with concentrations of 10,000 ng/ml where centrifugation was not sufficient to prevent antibody oversaturation of the surface, resulting in less cells binding to the surface. Gray line represents the control. Error bars represent standard error of the mean. Please click here to view a larger version of this figure.
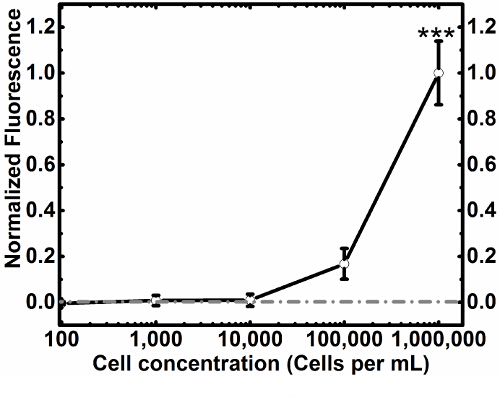 Figure 2.Normalized Blanked Cell Titration. Using optimized antibody concentration from the antibody titration, MCF7GFP cells were titrated to find optimized capture concentration while keeping functionalized surface and antibody concentrations constant. This can be used to calibrate cell capture for different applications. Columns in this figure are replicates, while rows change the concentration of cells to find the idea range for cellular capture. Gray line represents control, and error bars represent standard error of the mean. Please click here to view a larger version of this figure.
Figure 2.Normalized Blanked Cell Titration. Using optimized antibody concentration from the antibody titration, MCF7GFP cells were titrated to find optimized capture concentration while keeping functionalized surface and antibody concentrations constant. This can be used to calibrate cell capture for different applications. Columns in this figure are replicates, while rows change the concentration of cells to find the idea range for cellular capture. Gray line represents control, and error bars represent standard error of the mean. Please click here to view a larger version of this figure.
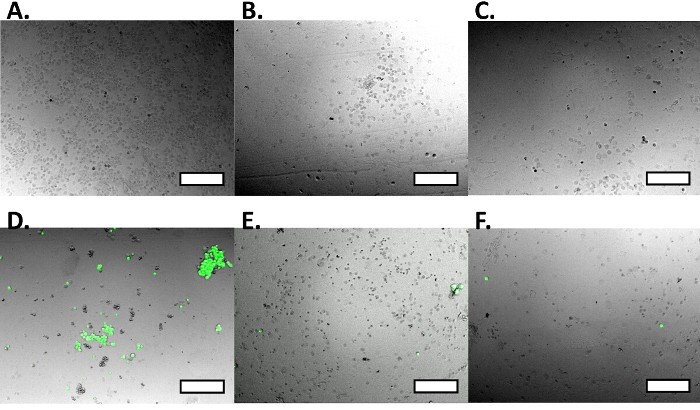 Figure 3.Capture and Release Experiments. A single cell mixture was exposed to the functionalized surface (A), capturing MCF7GFP cells using HLA-ABC antibody. When washed with HBSS (B), cells remained captured, but when exposed to a solution of 20 mM biotin (C), cells were released. A cellular mixture of MCF7GFP cells and RAW 264.7 macrophages were exposed to a functionalized surface and were captured using mCD11b antibody (D). Non-specific MCF7GFP cells are labeled in green. Fluorescence intensity of the MCF7GFP cells decreased when exposed to a neutral wash (E), which implies that the surface did not target them, and that they attached non-specifically. When exposed to a biotin wash (F), the targeted RAW macrophages were released from the surface. Scale bars are 250 µm. Please click here to view a larger version of this figure.
Figure 3.Capture and Release Experiments. A single cell mixture was exposed to the functionalized surface (A), capturing MCF7GFP cells using HLA-ABC antibody. When washed with HBSS (B), cells remained captured, but when exposed to a solution of 20 mM biotin (C), cells were released. A cellular mixture of MCF7GFP cells and RAW 264.7 macrophages were exposed to a functionalized surface and were captured using mCD11b antibody (D). Non-specific MCF7GFP cells are labeled in green. Fluorescence intensity of the MCF7GFP cells decreased when exposed to a neutral wash (E), which implies that the surface did not target them, and that they attached non-specifically. When exposed to a biotin wash (F), the targeted RAW macrophages were released from the surface. Scale bars are 250 µm. Please click here to view a larger version of this figure.
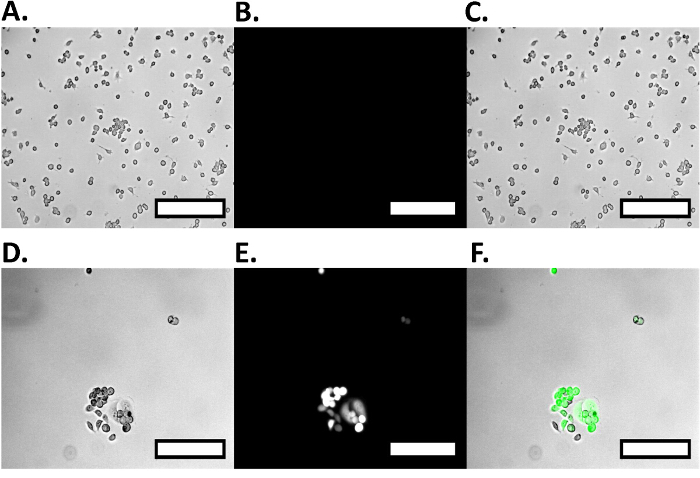 Figure 4.Positive and Negative Cell Control for Dual Cell Isolation. The positive control RAW Macrophages are imaged on a fluorescence microscope, showing no fluorescent activity as well as the relative sizing of cells. Brightfield RAW macrophages show cell sizing (A), while under GFP excitation, there is no fluorescence (B), which is shown in the merged image (C). The negative control MCF7GFP cells are imaged on a fluorescence microscope showing large fluorescent activity, as well as the MCF7GFP cells' relative size. Cells are imaged on brightfield (D), showing sizing, and while under GFP excitation (E) show large fluorescence, which can be clearly shown in the merged image (F). Scale bars are 250 µm. Please click here to view a larger version of this figure.
Figure 4.Positive and Negative Cell Control for Dual Cell Isolation. The positive control RAW Macrophages are imaged on a fluorescence microscope, showing no fluorescent activity as well as the relative sizing of cells. Brightfield RAW macrophages show cell sizing (A), while under GFP excitation, there is no fluorescence (B), which is shown in the merged image (C). The negative control MCF7GFP cells are imaged on a fluorescence microscope showing large fluorescent activity, as well as the MCF7GFP cells' relative size. Cells are imaged on brightfield (D), showing sizing, and while under GFP excitation (E) show large fluorescence, which can be clearly shown in the merged image (F). Scale bars are 250 µm. Please click here to view a larger version of this figure.
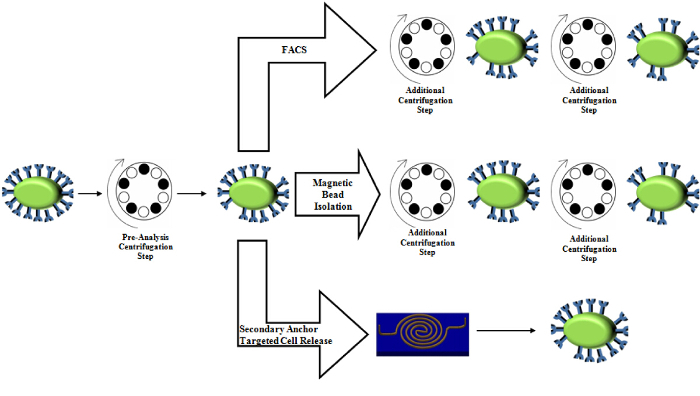 Figure 5.Comparison of centrifuge usage in standard purification techniques. The pre-analysis centrifugation step is conserved in sample preparation. Magnetic bead isolation as well as FACS involves several additional centrifugation steps, which introduces stress onto cells and may alter expression levels. Please click here to view a larger version of this figure.
Figure 5.Comparison of centrifuge usage in standard purification techniques. The pre-analysis centrifugation step is conserved in sample preparation. Magnetic bead isolation as well as FACS involves several additional centrifugation steps, which introduces stress onto cells and may alter expression levels. Please click here to view a larger version of this figure.
Supplemental Code File. Calculations. Please click here to download this file.
Discussion
Improvements in cell isolation techniques furthers scientific studies in structure-function relationships in neuroscience18, stem cell programming in regenerative biology, and angiogenic signaling in vascular biology19. Indeed, primary cell culture20 (e.g., HUVECs) in vascular biology is primarily done through the use of cell isolation techniques. Cell isolation was also recently used for quantitative flow (qFlow) cytometry analysis of plasma membrane receptors3,14,15,19,21. However, existing cell isolation methodologies affect cell-surface receptor levels and are costly in both personnel and reagents. We have advanced a new method in surface functionalization17, which allows for the creation of a gentle system of reproducible capture of a single cell type from a mixture of cell types to meet these shortcomings. This technique can be integrated into an iterative system, allowing the possibility of capturing different cell types in stages using specific antibodies. To further clarify the procedure, a number of troubleshooting questions and answers are offered below:
Functionalization Optimization: The functionalization processes has been optimized for improved uniformity and pull down of MCF7gfp cells. This protocol can alternatively be optimized and customized for any cell type and antibody configuration. This can be achieved through altering the concentrations of the base components in the capture surface, or by changing the type or concentrations of the antibodies. Most antibody types can be biotinylated using the above protocol. Once biotinylated, they can then be titrated (Figure 1) to find ideal concentration for pull down. The cell concentration can be titrated (Figure 2) to find the ideal concentration of cells to capture. Using this, the feasibility of capturing a certain cell type with the capture surface can be ascertained. For example, if a cell type of interest is on the scale of 100 cells per million cells, capturing concentrations of cells in that range using the functionalized surface would be ideal. If during the titration, the surface cannot capture cells of that small a concentration, then optimization of the surface or antibody is necessary in order to be able to filter out cells within that concentration range.
Which cell types are used in this protocol? How could someone modify this protocol to use a different cell type?
Currently, this protocol uses MCF7-GFP cells, and RAW 264.7 macrophages are often included to show an ability to pull out one cell type in a dual-cell mixture. These cells were selected as they were two of the most relevant cell types to mouse xenograft model (human tumor within murine environment). In order to calibrate the process for a variety of other cells, antibody specificity is paramount. There are several online resources available for selecting antibodies22.
What are some advantages of this method?
Gentle approach: The lack of force makes this approach gentle. Indeed, our calculated shear force is maximally, 24 x 10-6 pN17, which should not cause significant disruption to biomarkers, since hydrodynamic stresses of 2.09 Pa (656 pN assuming 314 pM2 cell surface area) induce necrosis while values of stress below 0.59 Pa (185 pN assuming 314 pM2 cell surface area) do not23. This gentle approach is thus advantageous over some commercially available options such as centrifugation-based approaches24, which exert up to 0.78 Pa25 of peak shear stress due to the sudden acceleration at values around 600 G, which may change protein expression patterns and morphologies25,26 and even induce cell necrosis23. Thus a process that could reduce the amount of centrifuge usages would reduce the amount of stresses that the cells would experience through purification and ultimately ensure more physiological data. Our current protocol uses centrifuges to purify and reconstitute the cell concentration prior to capture, however, this preparation process is standard in many purification techniques such as magnetic bead isolation27,28, flow cytometry29, and FACS30 (Figure 5). Our approach reduces all downstream centrifugation processes that the other techniques use in addition to the preparation step.
Biotin-avidin approach: The application of commonly used biomaterials (DSB-SAv and biotin-SAv) also renders this approach advantageous31,32. Avidin family proteins bind extremely selectively to the biotin family proteins and are used for a range of scientific and medical applications including: antibody-fluorophore attachment33, quantitative Qdot-polystyrene bead attachment34, and the creation of hydrogels that respond to stimuli in the environment to release encapsulated drugs32. Additionally, the relative ease of acquiring biotinylated antibodies makes this approach widely accessible and customizable.
Desthiobiotin approach without beads: The capture surface is able to implement the use of desthiobiotin without the use of harsh reagents and forces to release the cells. DSB has been used for reversible cell attachment and release by other systems in conjunction with DSB-antibodies and magnetic beads35. However, the harsh effects of the separation technique as well as the large cellular loss associated with preparing the samples27,28 result in differential receptor and chemokine expression28,36. This approach aims to mitigate and overcome these limitations by eliminating the use of both magnet and beads to create a much more gentle wash and release methodology.
What are the critical steps in this protocol? What can cause variability? How can that variability be controlled?
This process has four critical steps that can result in the decreased efficiency of the capture surface. The first critical step is the prevention of water to the APTES surface during the APTES functionalization steps, which would result in the destruction of the self-assembled surface. This is remedied by using ethanol as a solvent and baking the APTES in the oven to reduce hydrolysis from later aqueous solutions. The second critical step is the EDC reaction steps in which EDC catalyzes the reaction of the DSB with the APTES: allowing cross-linking of the two layers. If the EDC is excluded or not sufficiently added, the DSB will not be able to attach the floor, which will compromise the release mechanism. The third critical step is during the SAv functionalization, as maintaining the SAv layer is critical. Non-uniformity of the streptavidin layer results in the reduction in capture efficiency. The fourth critical step is in the biotinylation of the antibody, as the binding of the antibody to the capture surface is facilitated via the biotin-streptavidin interaction of the capture surface and antibody. If the antibody is non-biotinylated, then the streptavidin layer will be unable to capture it and pull it down, making the capture surface useless. Variability and inconsistency in the surface capture can be attributed to concentration non-regularity as well as stochasticity of layer attachment. This variation can be controlled by using precise concentrations of layer components calibrated to the surface and the intended targets of capture. In this design, concentrations are calibrated specifically to the capture of MCF7-GFP cells.
What are some drawbacks of this method?
Currently, the APTES functionalization is done by using a liquid phase immersion of the surface for 55 minutes followed by a baking step for up to 2 hours. Although this allows for a complete layer of APTES silane to bind to the surface, a more uniform process would be the vapor phase silanization37. This decreases APTES contact time, thus saving the researcher time, as well as increasing uniformity. Additionally, improved pull-down has been observed17 when overnight incubation of SAv is used, and currently, the surface functionalization requires two, overnight incubations, making the overall process take three days. So, optimizing the chemistry would offer significant time-savings for the researcher.
What are some warnings or precautions that may be helpful?
When functionalizing surfaces, it is imperative that proper care is taken to the risk of respiratory damage. Both APTES and mercaptoethanol can be extremely dangerous to the lungs and associated organs, thus it is necessary to do the functionalization process in a chemical hood to reduce interaction with the chemical fumes. Mercaptoethanol is a thiol which is especially pungent in odor. When using mercaptoethanol, allow for contaminated waste to sit in hood for a day or two before disposal.
What are the future aims and applications of this surface?
Our future aims involve customizing this surface to work with a variety of relevant cell types for angiogenic related diseases. Particularly, we intend to focus on Human Umbilical Vein Endothelial Cells in order to segue to using blood samples and separating out cells of interest from there. Additionally, we plan to integrate separation modalities such as aptamers into the design of the functionalized surface to further increase our capture and release percentages.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank the American Cancer Society, Illinois Division (282802) and the National Science Foundation CBET (1512598) for funding support. We also would like to thank Dr. Dianwen Zhang from the University of Illinois Beckman Institute for microscopy training. Finally, we would like to thank Jared Weddell, Stacie Chen, and Spencer Mamer for insightful discussions.
References
- Erdbruegger U, Haubitz M, Woywodt A. Circulating endothelial cells: a novel marker of endothelial damage. Clin. Chim. Acta. 2006;373(1-2):17–26. doi: 10.1016/j.cca.2006.05.016. [DOI] [PubMed] [Google Scholar]
- De Spiegelaere W, Cornillie P, Van Poucke M, Peelman L, Burvenich C, Van den Broeck W. Quantitative mRNA expression analysis in kidney glomeruli using microdissection techniques. Histol. Histopathol. 2011;26(2):267–275. doi: 10.14670/HH-26.267. [DOI] [PubMed] [Google Scholar]
- Chen S, Guo X, Imarenezor O, Imoukhuede PI. Quantification of VEGFRs, NRP1, and PDGFRs on Endothelial Cells and Fibroblasts Reveals Serum, Intra-Family Ligand, and Cross-Family Ligand Regulation. Cell. Mol. Bioeng. 2015;8(3):383–403. [Google Scholar]
- Cheung LSL, et al. Detachment of captured cancer cells under flow acceleration in a bio-functionalized microchannel. Lab Chip. 2009;9(12):1721–1731. doi: 10.1039/b822172c. [DOI] [PubMed] [Google Scholar]
- Privorotskaya N, et al. Rapid thermal lysis of cells using silicon-diamond microcantilever heaters. Lab Chip. 2010;10(9):1135–1141. doi: 10.1039/b923791g. [DOI] [PubMed] [Google Scholar]
- Park K, Akin D, Bashir R. Electrical capture and lysis of vaccinia virus particles using silicon nano-scale probe array. Biomed. Microdevices. 2007;9(6):877–883. doi: 10.1007/s10544-007-9101-3. [DOI] [PubMed] [Google Scholar]
- Galletti G, Sung M, Vahdat L. Isolation of breast cancer and gastric cancer circulating tumor cells by use of an anti HER2-based microfluidic device. Lab Chip. 2014;14(1):147–156. doi: 10.1039/c3lc51039e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schudel BR, Choi CJ, Cunningham BT, Kenis PJA. Microfluidic chip for combinatorial mixing and screening of assays. Lab Chip. 2009;9(12):1676–1680. doi: 10.1039/b901999e. [DOI] [PubMed] [Google Scholar]
- Lien KY, Chuang YH, et al. Rapid isolation and detection of cancer cells by utilizing integrated microfluidic systems. Lab Chip. 2010;10(21):2875–2886. doi: 10.1039/c005178k. [DOI] [PubMed] [Google Scholar]
- Stott SL, et al. Isolation of circulating tumor cells using a. PNAS. 2010;107(35):18392–18397. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Ting D, Stott S, Wittner B, Ozsolak F. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487(7408):510–513. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, Ogunwobi O, Chen T, Zhang J. >Capture, release and culture of circulating tumor cells from pancreatic cancer patients using an enhanced mixing chip. Lab Chip. 2014;14(1):89–98. doi: 10.1039/c3lc51017d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Cheung LSL, Schroeder JA, Jiang L, Zohar Y. A high-performance microsystem for isolating circulating tumor cells. Lab Chip. 2011;11(19):3269–3276. doi: 10.1039/c1lc20331b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoukhuede PI, Popel AS. Quantification and cell-to-cell variation of vascular endothelial growth factor receptors. Exp. Cell Res. 2011;317(7):955–965. doi: 10.1016/j.yexcr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoukhuede PI, Popel AS. Expression of VEGF receptors on endothelial cells in mouse skeletal muscle. PLoS One. 2012;7(9):e44791. doi: 10.1371/journal.pone.0044791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Kretzmer G, Schügerl K. Determination of a "critical shear stress level" applied to adherent mammalian cells. Enzyme Microb. Technol. 1992;14(3):209–213. doi: 10.1016/0141-0229(92)90068-y. [DOI] [PubMed] [Google Scholar]
- Ansari A, Lee-Montiel FT, Amos J, Imoukhuede PI. Secondary anchor targeted cell release. Biotechnol. Bioeng. 2015;112(11):2214–2227. doi: 10.1002/bit.25648. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Nashmi R, Imoukhuede P, Just H, McKinney S, Lester HA. Subcellular trafficking, pentameric assembly, and subunit stoichiometry of neuronal nicotinic acetylcholine receptors containing fluorescently labeled alpha6 and beta3 subunits. Mol. Pharmacol. 2008;73(1):27–41. doi: 10.1124/mol.107.039180. [DOI] [PubMed] [Google Scholar]
- Imoukhuede PI, Dokun AO, Annex BH, Popel AS. Endothelial cell-by-cell profiling reveals temporal dynamics of VEGFR1 and VEGFR2 membrane-localization following murine hindlimb ischemia. Am J Physiol Hear. Circ Physiol. 2013;4(8):H1085–H1093. doi: 10.1152/ajpheart.00514.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beijnum JR, Rousch M, Castermans K, van der Linden E, Griffioen AW. Isolation of endothelial cells from fresh tissues. Nat. Protoc. 2008;3(6):1085–1091. doi: 10.1038/nprot.2008.71. [DOI] [PubMed] [Google Scholar]
- Imoukhuede PI, Popel AS. Quantitative fluorescent profiling of VEGFRs reveals tumor cell and endothelial cell heterogeneity in breast cancer xenografts. Cancer Med. 2014;3(2):225–244. doi: 10.1002/cam4.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BD Biosciences. CD Marker Handbook: Human and Mouse. 2010. at: https://www.bdbiosciences.com/documents/cd_marker_handbook.pdf.
- Tanzeglock T, Soos M, Stephanopoulos G, Morbidelli M. Induction of mammalian cell death by simple shear and extensional flows. Biotechnol. Bioeng. 2009;104(2):360–370. doi: 10.1002/bit.22405. [DOI] [PubMed] [Google Scholar]
- Perritt D, Wong P, Macpherson JL, Henrichsen K, Symonds G, Pond S, inventors. Processing Blood. 2014. at: https://www.google.com/patents/US20140030238.
- Fukuda S, Schmid-Schönbein GW. Centrifugation attenuates the fluid shear response of circulating leukocytes. J. Leukoc. Biol. 2002;72(July):133–139. [PubMed] [Google Scholar]
- dela Paz NG, Walshe TE, Leach LL, Saint-Geniez M, D'Amore PA. Role of shear-stress-induced VEGF expression in endothelial cell survival. J. Cell Sci. 2012;125(Pt 4):831–843. doi: 10.1242/jcs.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard WJ, et al. Tumor Cells Circulate in the Peripheral Blood of All Major Carcinomas but not in Healthy Subjects or Patients With Nonmalignant Diseases Tumor Cells Circulate in the Peripheral Blood of All Major Carcinomas but not in Healthy Subjects or Patients With Nonmalignant diseases.". Clinical Cancer Research. 2005;10:6897–6904. doi: 10.1158/1078-0432.CCR-04-0378. [DOI] [PubMed] [Google Scholar]
- Nagrath S, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450(7173):1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Weddel J, Gupta P, Conard G, Parkin J, Imoukhuede PI. QFlow Cytometer-Based Receptoromic Screening: A High-throughput Quantification Approach Informing Biomarker Selection and Nanosensor. Submiss. 2016. [DOI] [PubMed]
- Vasa M, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 2001;89(1):E1–E7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- Hirsch JD, Eslamizar L, et al. Easily reversible desthiobiotin binding to streptavidin, avidin, and other biotin-binding proteins: uses for protein labeling, detection, and isolation. Anal. Biochem. 2002;308(2):343–357. doi: 10.1016/s0003-2697(02)00201-4. [DOI] [PubMed] [Google Scholar]
- Wilchek M, Bayer EA. Applications of Avidin-Biotin Technology: Literature Survey. Methods Enzymol. 1987;152(1):183–189. doi: 10.1016/0076-6879(90)84257-h. [DOI] [PubMed] [Google Scholar]
- Wu X, et al. Immunofluorescent labeling of cancer marker Her2 and other cellular targets with semiconductor quantum dots. Nat. Biotechnol. 2002;21(1):41–46. doi: 10.1038/nbt764. [DOI] [PubMed] [Google Scholar]
- Lee-Montiel FT, Imoukhuede PI. Engineering quantum dot calibration standards for quantitative fluorescent profiling. J. Mater. Chem. B. 2013;1:6434. doi: 10.1039/c3tb20904k. [DOI] [PubMed] [Google Scholar]
- Hornes E, Korsnes L, inventors. Oligonucleotide-linked magnetic particles and uses thereof. 1996. Available from: http://www.google.com/patents/US5512439.
- Naranbhai V, et al. Impact of blood processing variations on natural killer cell frequency, activation, chemokine receptor expression and function. J. Immunol. Methods. 2011;366(1-2):28–35. doi: 10.1016/j.jim.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav AR, Sriram R, Carter JA, Miller BL. Comparative study of solution-phase and vapor-phase deposition of aminosilanes on silicon dioxide surfaces. Mater. Sci. Eng. C. 2014;35(1):283–290. doi: 10.1016/j.msec.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]