

Abstract
Neural stem cells (NSCs) have the ability to self-renew and generate the three major neural lineages — astrocytes, neurons and oligodendrocytes. NSCs and neural progenitors (NPs) are commonly cultured in vitro as neurospheres. This protocol describes in detail how to determine the NSC frequency in a given cell population under clonal conditions. The protocol begins with the seeding of the cells at a density that allows for the generation of clonal neurospheres. The neurospheres are then transferred to chambered coverslips and differentiated under clonal conditions in conditioned medium, which maximizes the differentiation potential of the neurospheres. Finally, the NSC frequency is calculated based on neurosphere formation and multipotency capabilities. Utilities of this protocol include the evaluation of candidate NSC markers, purification of NSCs, and the ability to distinguish NSCs from NPs. This method takes 13 days to perform, which is much shorter than current methods to enumerate NSC frequency.
Keywords: Neuroscience, Issue 116, Neural stem cells, neural progenitors, clonal, neurospheres, multipotency, neural stem cell frequency
Introduction
Neural stem cells (NSCs) are cells of the central nervous system (CNS) that can self-renew and are multipotent. NSCs are first specified during development of the embryonic forebrain and continue to persist in the adult brain at specific regions such as the subventricular zone (SVZ) of the lateral ventricles and the dentate gyrus (DG) of the hippocampus. A number of NSC lines are being used in clinical trials for treatment of stroke and other neurological diseases such as Batten's disease1.
NSCs and neural progenitors (NPs) are propagated in culture as floating 3-dimensional spheroid structures called neurospheres. The neurosphere culture system was developed by Reynolds and Weiss in the early 1990s, when they found that embryonic and adult cortical cells could divide in the presence of epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF)2-4. Neurospheres consist of a heterogeneous mix of cells comprising of subclasses at different developmental stages5. It is challenging to specifically study NSCs using data obtained from neurospheres due to the presence of NPs. Hence, it is crucial to enrich NSCs from neurospheres. At present, four main methods have been used to enrich for NSCs in vitro. First is the use of cell surface markers. Lewis-X (LeX) and CD133 (also known as Prominin1) are the most prominent cell surface NSC markers6-9. Syndecan-1, Notch-1 and Integrin-beta1 are other surface proteins that enrich for NSCs10. Second is the dye exclusion. It has been shown that the side-population cells, which have the unique ability to pump out the fluorescent DNA-binding dye Hoechst 33342, are enriched for NSCs11. Third is the use of morphological selection. It has been demonstrated that cells with increased cell size and granularity harbor more NSCs than their counterparts5,12,13. Fourth is the addition of NSC survival factors to the culture medium. It has been shown that addition of Chondroitin sulphate proteoglycan and Apolipoprotein E enhances NSC survival and thus increases NSC frequency14,15. Although many markers including transcription factors have been associated with NSCs16,17, none of these markers are able to enrich NSCs to purity. Due to the lack of definitive NSC markers, it remains a challenge to quantify NSCs and distinguish them from NPs in vitro.
Initial studies used the neurosphere formation assay (NFA) to quantify NSCs7,11,13. In this assay, dissociated cells are cultured to form neurospheres and the number of neurospheres generated for every 100 cells plated is determined. This value is termed as the Neurosphere Formation Unit (NFU). The NFU equals the NSC frequency if all neurospheres arise from NSCs. However, it was shown that the NFU overestimates the NSC frequency as neurospheres are formed by both NSCs and early NPs5. Thus, it is inaccurate to enumerate NSCs solely based on neurosphere formation. It could be possible to quantify NSCs based on their ability to self-renew, proliferate extensively and generate multipotent neurospheres.
NSCs, which are EGF and bFGF responsive, usually survive for at least ten passages and thus display extensive self-renewal capacity in culture4,18-20. NPs, which are EGF and bFGF responsive as well, could also generate neurospheres for a few passages but not for an extended period of time. Hence, it has been widely accepted that bona fide NSCs can be enumerated with fair accuracy based on neurosphere formation for at least ten passages. In most studies, however, self-renewal is usually measured based on secondary or tertiary neurosphere formation due to the long experimental time required for ten passages. Hence, the secondary NFA can be used to broadly compare the self-renewal ability between populations, but cannot be used to accurately enumerate NSC frequency.
NSCs have a greater proliferative ability compared to NPs. This property of NSCs was used by Louis et al. to develop an assay for NSC enumeration — the neural colony-forming cell assay (NCFCA)21,22. In this assay, single cells are cultured for 3 weeks in a collagen-containing semisolid matrix. Under these culture conditions, it was shown that cells that form neurospheres above 2 mm in diameter are tripotent and can self-renew for long term. These cells are defined as NSCs. Therefore, the NSCs are effectively distinguished from NPs.
NSCs have the ability to differentiate into astrocytes, oligodendrocytes and neurons. For differentiation of neurospheres, growth factors are removed and serum is added to the culture medium. If all three neural lineages are observed in a neurosphere, then the cell that initiated that neurosphere is an NSC. However, the differentiation assay has some limitations. First, the culture conditions used for differentiation may not be optimal for generation of all three neural lineages. In fact, in a single neurosphere differentiation process, significant cell death occurs and mostly astrocyte generation occurs (Tham M and Ahmed S, unpublished). Second, there is the issue of clonality. For accurate enumeration of NSCs, formation and differentiation of neurospheres have to be performed under clonal conditions, where each neurosphere arises from a single cell. In bulk suspension cultures, aggregation occurs at both cellular and neurosphere levels23-25. Hence, it is possible for each neurosphere to arise from multiple cells or neurospheres, which complicates neurosphere counting and evaluation of multipotency. Recent evidence shows that aggregation of cells does not occur at low plating density of 0.5 cells/µL or below, and when culture plates are not moved during neurosphere formation15. Thus culturing cells at such low density would ensure clonality.
Currently, the NCFCA is the most commonly used method to distinguish NSCs from NPs and enumerate NSC frequency. The NCFCA, however, requires a relatively long time of three weeks. Here, we describe a protocol to enumerate NSC frequency based on the ability of NSCs to form multipotent neurospheres. This protocol takes only 13 days to perform. The NCFCA ensures clonality as the collagen matrix prevents movement of the neurospheres. The culture conditions used in this protocol also allows clonality to be maintained throughout. For instance, the use of 50-well chambered coverslips ensures that the neurospheres will differentiate without contacting each other. Furthermore, we use conditioned medium that supplies neurotrophic factors during differentiation to maximize the differentiation potential of the neurospheres (Figure 1).
Protocol
The treatment of animals was performed in accordance with the IACUC and NACLAR guidelines and approved by the animal department ((http://www.brc.astar.edu.sg/index.php?sectionID-11). NOTE: Neurosphere cultures were prepared from the forebrain of embryonic (E14) C57BL/6 mice as previously described5.
1. Culture of Clonal Sample Neurospheres (Day 1)
Prepare NSC growth medium by combining Dulbecco's Modified Eagle's Medium/Nutrient F-12 (DMEM/F12) medium with B27 supplement (final concentration: 1x), EGF (final concentration: 20 ng/mL), bFGF (final concentration:10 ng/mL) and penicillin/streptomycin (final concentration:1x).
- Dissociate E14.5 mouse neurospheres in 1 mL NSC growth medium and 1 mL 0.05 N Sodium Hydroxide for 7 min at RT. Pipette cells up and down occasionally to keep cells in suspension. Add 1 mL of 0.05 N Hydrochloric acid to neutralize the alkali. NOTE: Neurosphere cells are seeded at 20,000 cells/mL in a 10 cm culture dish and are cultured for 5 d at 37 °C, 5% CO2. The expected yield after is about 10 million cells per 10 cm dish. These cells are then dissociated and seeded at clonal density as described in steps 1.2 and 1.3, respectively.
- Perform cell count using Trypan Blue staining and a hemocytometer.
Seed single E14.5 mouse neurosphere cells at a density of 50 cells/mL or below in 100 µL NSC growth medium in a 96-well dish. Incubate the cells at 37 °C, 5% CO2 for 1 w to generate clonal neurospheres. NOTE: Clonality is ensured during neurosphere formation at this density as the cells do not contact each other.
2. Culture of Neurospheres for Conditioned Medium (Day 3)
Dissociate E14.5 mouse neurospheres as described in step 1.2.
Seed single cells from E14.5 mouse neurospheres at a density of 20,000 cells/mL in a 10 mL NSC growth medium in a 10 cm culture dish. Incubate the cells at 37 °C, 5% CO2 for 5 d to generate bulk cultured neurospheres.
3. Preparation of Neurosphere Conditioned Medium (Day 8)
Prepare coating solution by mixing 700 µL of 0.01% Poly-L-Lysine (PLL), 70 µL of 1 mg/mL Laminin and 6,230 µL of Phosphate Buffered Saline (PBS).
Coat a 10 cm culture dish with 7 mL of PLL/Laminin solution for 2 h at 37 °C.
Using a 10 mL serological pipette, transfer all the bulk cultured neurospheres to a 15 mL conical centrifuge tube. Centrifuge the neurospheres at 171 x g for 1 min at RT and remove the supernatant completely.
Prepare NSC differentiation medium by combining DMEM/F12 medium with B27 (final concentration: 1x) and Fetal Bovine Serum (FBS) (final concentration: 0.5%) Add 10 mL of NSC differentiation medium to neurospheres. Pipette up and down a few times to resuspend the neurospheres.
Aspirate PLL/Laminin from the 10 cm culture dish and wash once with 10 mL PBS. Transfer all the neurospheres to the PLL/Laminin-coated 10 cm culture dish. Incubate O/N at 37 °C, 5% CO2 for the neurospheres to adhere to the dish.
4. Determination of NFU of Sample Neurospheres (Day 8)
Count the number of neurospheres (seeded on day 1; section 1) formed in each well under a microscope. NOTE: Determine the number of neurospheres formed per 100 cells plated, which gives the NFU.
5. Transfer of Sample Neurospheres to 50-well Chambered Coverslip (Day 8)
Coat each well of a 50-well chambered coverslip with 10 µL of PLL/Laminin coating solution for 2 h at 37 °C. NOTE: Prepare coating solution as described in step 3.1.
Prepare 3 x 50 mL conical centrifuge tubes containing 30 mL of PBS each. Using sterile forceps, remove the coated 50-well chambered coverslip. Dip the chambered coverslip into the first tube of PBS, followed by the second and then the third tube of PBS, to wash off the coating solution.
Aspirate any remaining liquid from the wells of the chambered coverslip.
Gently pipette all medium and neurospheres from each well of the 96-well dish and transfer it to a single 10 cm culture dish.
Under a microscope, pick a sample neurosphere using a P10 micropipette fitted with a 10 µL pipette tip. Ensure that only a single neurosphere is picked each time by setting the pipette column to 2 µL. Use a new pipette tip for each neurosphere.
Transfer the neurosphere onto the center of a well of the coated 50-well chambered coverslip.
Repeat steps 5.5 and 5.6 until each well of the chambered coverslip contains a neurosphere. NOTE: The neurospheres may be picked under a microscope placed in a laminar flow hood or on a benchtop.
Add 10 µL of NSC differentiation medium to each well of the chambered coverslip. Place the chambered coverslip in a 10 cm culture dish and pipette PBS along the edges of the dish to prevent drying of the NSC differentiation medium. Place up to two chambered coverslips in one 10 cm culture dish. Incubate the chambered coverslip (contained in the 10 cm culture dish) overnight at 37 °C, 5% CO2.
6. Differentiation of Sample Neurospheres (Day 9)
Transfer the differentiation medium from the bulk cultured neurospheres (from section 3) to a 15 mL conical centrifuge tube. Ensure that 2-3 mL of differentiation medium remains in the culture dish to prevent adhered cells from detaching. Using a 20 mL sterile syringe, pass the differentiation medium through a 0.2 µm filter to remove any cells or debris.
Transfer the filtered differentiation medium back to the 10 cm culture dish containing the adhered cells.
Using sterile forceps, gently place the chambered coverslip onto the differentiation medium in an inverted manner such that the sample neurospheres are facing downwards. Incubate the chambered coverslip with the differentiation medium for 3 d at 37 °C, 5% CO2. NOTE: The sample neurospheres are in contact with the differentiation medium but not in contact with the differentiating cells beneath.
7. Fixing of Differentiated Neurospheres (Day 12)
Gently remove the chambered coverslip from the 10 cm culture dish using sterile forceps. Gently dip the coverslip into PBS to wash off the conditioned medium.
Peel off the silicone gasket from the coverslip. Perform this gently and slowly to avoid breaking the coverslip. Take note of the side where the differentiated neurospheres are adhered.
Pipette 1 mL of 4% paraformaldehyde (PFA) onto a piece of paraffin film cut to the size of the coverslip. Gently place the coverslip on the paraffin film with the neurospheres facing downwards in contact with the PFA. Fix the neurospheres for 20 min at RT. Caution: PFA is toxic both in liquid and vapor form and is combustible. Avoid inhalation of vapor and contact with skin and eyes. Always handle PFA in a ventilated fume hood.
Pipette 1 mL of PBS onto a piece of paraffin film. Wash the neurospheres by gently placing the coverslip with the neurospheres facing downwards in contact with PBS for 10 min at room temperature.
Repeat step 7.4 two more times.
8. O4 Staining of Differentiated Neurospheres (Day 12)
Prepare 3% bovine serum albumin (BSA) in PBS. Pipette 1 mL of the 3% BSA onto a piece of paraffin film. Block the neurospheres by gently placing the coverslip (with the neurospheres facing down) in contact with the BSA for 30 min at RT.
Pipette 1 mL of mouse anti-O4 IgM antibody (1:300 dilution in the 3% BSA) onto a piece of paraffin film and place the coverslip on the paraffin film with the neurospheres facing downwards in contact with the antibody. Allow O4 staining to occur for 2 h at RT.
Wash the coverslip twice with PBS as described in step 7.4.
Pipette 1 mL of green fluorescent dye conjugated-anti-mouse IgM secondary antibody (1:500 dilution in the 3% BSA) onto a piece of paraffin film and place the coverslip on the paraffin film with the neurospheres facing downwards in contact with the antibody. Leave for 1 h at RT in the dark. NOTE: Perform immunostaining in a dark environment from this step onwards.
Wash the coverslip twice with PBS as described in step 7.4.
9. Tuj1 and Glial Fibrillary Acidic Protein (GFAP) Staining of Differentiated Neurospheres (Day 12)
Prepare permeabilizing solution by adding Triton-X 100 (final concentration: 0.5%) to the 3% BSA. Pipette 1 mL of permeabilizing solution onto a piece of paraffin film. Place coverslip on the paraffin film with the neurospheres facing downwards in contact with the permeabilizing solution for 20 min at RT.
Wash the coverslip twice with PBS as described in step 7.4.
Pipette 1 mL of mouse anti-Tuj1 IgG2a antibody (1:500 dilution in the 3% BSA) and rabbit anti-GFAP IgG antibody (1:1,000 dilution in the 3% BSA) onto a piece of paraffin film and place coverslip on the paraffin film with the neurospheres facing downwards in contact with the antibodies. Allow Tuj1 and GFAP staining to occur for 2 h at RT. NOTE: Both antibodies can be prepared as a single solution.
Wash the coverslip twice with PBS as described in step 7.4.
Pipette 1 mL of red fluorescent dye conjugated-anti-mouse IgG2a (1:500 dilution in the 3% BSA) and far red fluorescent dye conjugated-anti-rabbit IgG (1:500 dilution in the 3% BSA) secondary antibodies onto a piece of paraffin film and place coverslip on the paraffin film with the neurospheres facing downwards in contact with the antibodies. Leave for 1 h at RT. NOTE: Both antibodies can be prepared as a single solution.
Wash the coverslip twice with PBS as described in step 7.4.
Dilute 4',6-diamidino-2-phenylindole (DAPI) stock solution to 300 nM in PBS. Pipette 1 mL of DAPI onto a piece of paraffin film and place coverslip on the paraffin film with the neurospheres facing downwards in contact with DAPI for 2 min at RT for nuclei staining.
Wash the coverslip once with PBS and twice with deionized water as described in step 7.4. Mount the coverslip on a glass slide using a suitable mounting medium and allow to dry O/N at RT.
10. Imaging of Differentiated Neurospheres (Day 13)
Image neurospheres under a confocal microscope using a 40X. Use the lasers at 488 nm, 594 nm and 647 nm to detect oligodendrocytes, neurons and astrocytes, respectively.
Determine the percentage of unipotent, bipotent and tripotent neurospheres. NOTE: Potency is determined by the ability of the neurosphere to differentiate into the three neural lineages - oligodendrocytes, neurons and astrocytes5,14,15,26. Oligodendrocytes are identified by positive staining for O4 (section 8). Neurons are identified by positive staining for Tuj1 (section 9). Astrocytes are identified by positive staining for GFAP (section 9). A unipotent neurosphere differentiates into only one of the lineage and should positively stain for either O4, GFAP or Tuj1. A bipotent neurosphere differentiates into any two lineages and should positively stain for O4 and GFAP or O4 and Tuj1 or GFAP and Tuj1. A tripotent neurosphere differentiates into all three lineages and should positively stain for O4, GFAP and Tuj1.
Determine NSC frequency in the population of interest by using the following formula: NSC frequency = (NFU x % of tripotent neurospheres) / 100%.
Representative Results
We have used this protocol to assess the ability of LeX, a known cell surface NSC marker7, to enrich for NSCs. Data from this study will be used to illustrate how NSCs are enumerated using the clonal assays in the protocol. E14.5 mouse neurosphere cells were incubated with anti-LeX antibody. Subsequently, cells that express LeX (LeX+) and cells that do not express LeX (LeX-) were seeded at 50 cells/mL by Fluorescence-Activated Cell Sorting (FACS) and left to generate clonal neurospheres for 1 wk. Time-lapse imaging shows that the neurospheres generated at 50 cells/mL are clonal (Figure 2). The NFU for LeX+ and LeX- cells was determined. It is shown that LeX+ cells have a significantly higher neurosphere formation capability compared to LeX- cells (Figure 3g). The neurospheres generated were then differentiated under clonal conditions, subsequently immunostained and imaged (Figure 3a-e). LeX+ cells generate significantly more tripotent neurospheres than LeX- cells (Figure 3f). The NSC frequency was then calculated based on the NFU and % of tripotent neurospheres (Figure 3g). Selection based on LeX, enriches NSC frequency by more than 14 fold, validating LeX as a NSC marker. We have also determined the NSC frequency of E14.5 mouse neurospheres using this protocol27 and NCFCA28. Both methods report comparable NSC frequency of about 2% in E14.5 mouse neurospheres.
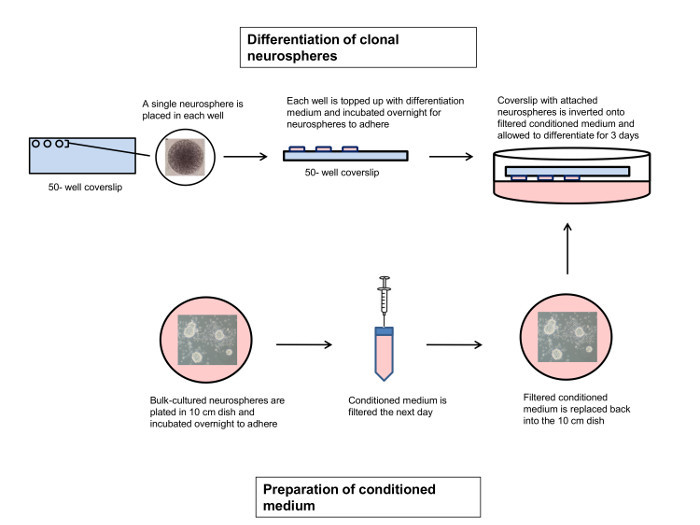 Figure 1.Protocol for Neurosphere Differentiation under Clonal Conditions. Sample neurospheres grown under clonal conditions are individually placed in each PLL/Laminin-coated well of a 50-well chambered coverslip, and allowed to adhere O/N. On the same day, bulk cultured neurospheres are plated in differentiation medium in a PLL/Laminin-coated 10 cm dish, and allowed to adhere O/N. The next day, conditioned medium from the bulk cultured neurospheres is filtered and placed back in the 10 cm dish. The sample neurospheres are then inverted onto the conditioned medium and allowed to differentiate for 3-4 days28. Please click here to view a larger version of this figure.
Figure 1.Protocol for Neurosphere Differentiation under Clonal Conditions. Sample neurospheres grown under clonal conditions are individually placed in each PLL/Laminin-coated well of a 50-well chambered coverslip, and allowed to adhere O/N. On the same day, bulk cultured neurospheres are plated in differentiation medium in a PLL/Laminin-coated 10 cm dish, and allowed to adhere O/N. The next day, conditioned medium from the bulk cultured neurospheres is filtered and placed back in the 10 cm dish. The sample neurospheres are then inverted onto the conditioned medium and allowed to differentiate for 3-4 days28. Please click here to view a larger version of this figure.
 Figure 2.Growth of a Clonal Neurosphere. Cells derived from E14.5 neurospheres were dissociated and seeded at 50 cells/mL in 96-well dish. Individual cells were tracked for 6 d by time-lapse imaging. Images of one such cell growing into a neurosphere is shown. Note that the neurosphere maintains clonality. (a) 0 days; (b) 1.19.45; (c) 2.95.37; (d) 3.32.76; (e) 4.46.53; (f) 5.61.44; where the first number refers to the day, second number refers to the fraction of the day, and third number refers to the fraction of the h. 10X magnification. Scale bar, 100 µm. Please click here to view a larger version of this figure.
Figure 2.Growth of a Clonal Neurosphere. Cells derived from E14.5 neurospheres were dissociated and seeded at 50 cells/mL in 96-well dish. Individual cells were tracked for 6 d by time-lapse imaging. Images of one such cell growing into a neurosphere is shown. Note that the neurosphere maintains clonality. (a) 0 days; (b) 1.19.45; (c) 2.95.37; (d) 3.32.76; (e) 4.46.53; (f) 5.61.44; where the first number refers to the day, second number refers to the fraction of the day, and third number refers to the fraction of the h. 10X magnification. Scale bar, 100 µm. Please click here to view a larger version of this figure.
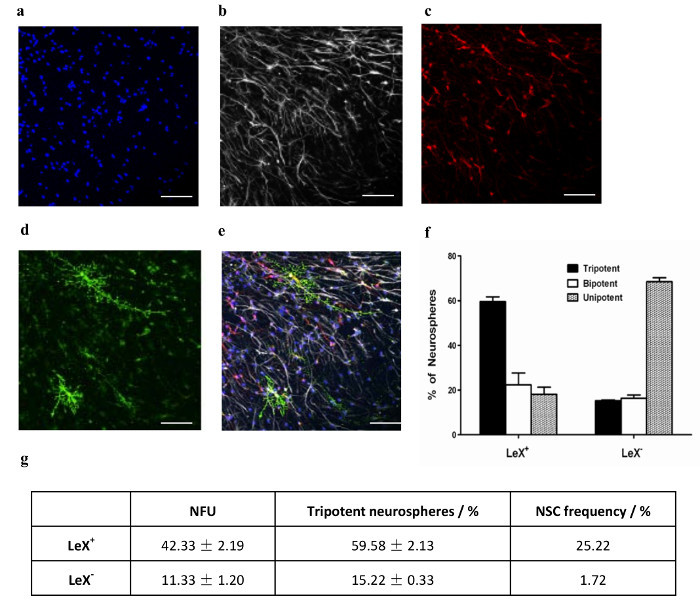 Figure 3. Differentiation of Clonal Neurospheres and Enumeration of NSC Frequency. Single neurospheres were picked and differentiated in 50-well chambered coverslip for 3 d. A representative image of a tripotent neurosphere stained with (a) DAPI and immunostained for (b) GFAP, (c) Tuj1 and (d) O4. (e) Shows the overlay of (a) - (d). Scale bar, 65 µm. (f) The % of unipotent, bipotent and tripotent neurospheres generated by LeX+ and LeX- cells (mean ±SEM; n = 3). (g) NSC frequency in LeX+ and LeX- cells calculated as the product of NFU and % of tripotent neurospheres. Please click here to view a larger version of this figure.
Figure 3. Differentiation of Clonal Neurospheres and Enumeration of NSC Frequency. Single neurospheres were picked and differentiated in 50-well chambered coverslip for 3 d. A representative image of a tripotent neurosphere stained with (a) DAPI and immunostained for (b) GFAP, (c) Tuj1 and (d) O4. (e) Shows the overlay of (a) - (d). Scale bar, 65 µm. (f) The % of unipotent, bipotent and tripotent neurospheres generated by LeX+ and LeX- cells (mean ±SEM; n = 3). (g) NSC frequency in LeX+ and LeX- cells calculated as the product of NFU and % of tripotent neurospheres. Please click here to view a larger version of this figure.
Discussion
We describe hee the enumeration of NSCs under clonal conditions. Critical steps include the use of fresh NSC growth and differentiation medium, and also the use of freshly prepared PLL/Laminin solution to coat the chambered coverslips, as well as the culture dishes. This ensures optimal growth and differentiation conditions of the neurospheres. The use of good quality antibodies to effectively detect the neurons and glia, as well as the selective picking of clonal neurospheres that are not in contact with other neurospheres, are critical. Furthermore, it is important to ensure the picked neurospheres do not dry up. It is recommended to add 10 µL of differentiation medium to each well after picking every 10 neurospheres. It is important to use one chambered coverslip in one 10 cm dish per sample to avoid cross talk between neurospheres from different samples. If two coverslips (each containing neurospheres from different samples) are placed in the same 10 cm dish, neurospheres from one coverslip might release factors that may alter the propensity of neurospheres in the other coverslip to differentiate into specific lineages. Two coverslips in the same 10 cm dish may also result in mix up between samples.
In the event the NFU or the NSC frequency is lower than expected, ensure that low passage number neurospheres are used. It is also important that healthy low passage neurospheres are used for generating conditioned medium. Next, if the neurospheres are cultured in wells with small diameter, such as in 96-well dish, then it will be difficult to maneuver and pick neurospheres using a micropipette. In this case, transfer the neurospheres to a larger culture dish, such as a 6-well dish, prior to picking. Some of the picked neurospheres may lift off from the chambered coverslip during culture and immunostaining. Minimizing the movement of the chambered coverslip during culture, and less intensive washing during immunostaining, would help to avoid detachment of neurospheres.
Initial studies quantified NSCs using only the NFA7,11,13. However, the NFA overestimates the NSC frequency as both NSCs and early NPs generate neurospheres5. Louis et al. developed another method to quantify NSCs known as the NCFCA21,22. The NCFCA involves culturing single cells in a collagen based matrix to ensure clonality, and it was shown that neurospheres above 2 mm in diameter are formed by NSCs only. Similar to the NCFCA, clonality is ensured throughout this protocol. This is achieved by generating neurospheres at clonal density and differentiating the neurospheres in chambered coverslips. The use of chambered coverslips confers a number of advantages. The chambered coverslip allows each sample neurosphere to differentiate without having physical contact with other sample neurospheres, thereby ensuring clonality during differentiation. The chambered coverslip can be placed suspended on the differentiation medium to allow the sample neurospheres to be continuously conditioned and to ensure maximum differentiation. In the absence of conditioned medium and serum during differentiation, survival of the neurospheres decreases and the neurospheres mostly generate astrocytes (Tham M and Ahmed S, unpublished). In addition, the immunostained neurospheres can be conveniently stored for long period of time and the chambered coverslips can be mounted directly on the microscope glass slides. Additionally, imaging of the immunostained neurospheres is made easy as one can quickly locate the neurosphere in the small chambered well, capture an image, and move on to the next well.
We have determined the NSC frequency in E14.5 mouse neurosphere culture using both the protocol described in this manuscript27 and NCFCA28. The NSC frequency in E14.5 mouse neurospheres determined by both methods are comparable. The NCFCA enumerates NSCs that are multipotent and have long-term self-renewal capacity. Since the NSC frequency from our method is comparable to that from NCFCA, the readout from our protocol does not seem to overestimate the NSC frequency. The NCFCA requires three weeks to be completed and mainly involves cell plating and medium replenishment. The protocol described here requires 13 days to perform and involves a different series of steps, e.g., neurosphere picking and immunostaining. This protocol could serve as an alternative method to enumerate NSCs. Researchers can use both NCFCA and this protocol to verify NSC frequency in their samples.
One limitation of this protocol is that a series of important steps have to be all performed on day 8. The preparation of the conditioned medium, determination of NFU of sample neurospheres, and transfer of sample neurospheres to the 50-well chambered coverslip have to be performed on day 8. One might take time to complete these steps. In addition, it requires practice to perform these steps in an efficient manner.
Neurospheres have been widely used to study NSC function and behavior. However, neurospheres consist of both NSCs and NPs. Hence, it is important to enrich NSCs from neurospheres to study NSC biology. Currently, cell surface markers, dye exclusion property as well as cell size and granularity have been used to enrich for NSCs6,7,9-13,29,30. This protocol could be used to quantify the enrichment of NSCs by potential NSC markers, and to facilitate the search for a definitive NSC marker. Four recent studies have used this protocol to enumerate NSC frequency5,14,15,26.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
We thank the Agency for Science, Technology and Research (A*STAR), Singapore for funding this research.
References
- Ahmed S. The culture of neural stem cells. J. Cell. Biochem. 2009;106(1):1–6. doi: 10.1002/jcb.21972. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 1992;12(11):4565–4574. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255(5052):1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Clonal and Population Analyses Demonstrate That an EGF-Responsive Mammalian Embryonic CNS Precursor Is a Stem Cell. Dev. Biol. 1996;175(1):1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- Narayanan G, et al. Single-Cell mRNA Profiling Identifies Progenitor Subclasses in Neurospheres. Stem Cells and Dev. 2012;21(18):3351–3362. doi: 10.1089/scd.2012.0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, et al. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. 2000;97(26):14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capela A, Temple S. LeX/ssea-1 Is Expressed by Adult Mouse CNS Stem Cells, Identifying Them as Nonependymal. Neuron. 2002;35(5):865–875. doi: 10.1016/s0896-6273(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Capela A, Temple S. LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Dev. Biol. 2006;291(2):300–313. doi: 10.1016/j.ydbio.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Corti S, et al. Isolation and characterization of murine neural stem/progenitor cells based on Prominin-1 expression. Exp. Neurol. 2007;205(2):547–562. doi: 10.1016/j.expneurol.2007.03.021. [DOI] [PubMed] [Google Scholar]
- Nagato M, et al. Prospective characterization of neural stem cells by flow cytometry analysis using a combination of surface markers. J. Neurosci. Res. 2005;80(4):456–466. doi: 10.1002/jnr.20442. [DOI] [PubMed] [Google Scholar]
- Kim M, Morshead CM. Distinct Populations of Forebrain Neural Stem and Progenitor Cells Can Be Isolated Using Side-Population Analysis. J. Neurosci. 2003;23(33):10703–10709. doi: 10.1523/JNEUROSCI.23-33-10703.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama A, Matsuzaki Y, Kawaguchi A, Shimazaki T, Okano H. Flow cytometric analysis of neural stem cells in the developing and adult mouse brain. J. Neurosci. Res. 2002;69(6):837–847. doi: 10.1002/jnr.10339. [DOI] [PubMed] [Google Scholar]
- Rietze RL, Valcanis H, Brooker GF, Thomas T, Voss AK, Bartlett PF. Purification of a pluripotent neural stem cell from the adult mouse brain. Nature. 2001;412(6848):736–739. doi: 10.1038/35089085. [DOI] [PubMed] [Google Scholar]
- Tham M, et al. CSPG Is a Secreted Factor that Stimulates Neural Stem Cell Survival Possibly by Enhanced EGFR Signaling. PLoS ONE. 2010;5(12):e15341. doi: 10.1371/journal.pone.0015341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan HT, Tham M, Hariharan S, Ramasamy S, Yu YH, Ahmed S. Identification of ApoE as an autocrine/paracrine factor that stimulates neural stem cell survival via MAPK/ERK signaling pathway. J. Neurochem. 2011;117(3):565–578. doi: 10.1111/j.1471-4159.2011.07227.x. [DOI] [PubMed] [Google Scholar]
- Ramasamy S, Narayanan G, Sankaran S, Yu YH, Ahmed S. Neural stem cell survival factors. Arch. Biochem. Biophys. 2013;534(1-2):71–87. doi: 10.1016/j.abb.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Ahmed S, et al. Transcription factors and neural stem cell self-renewal, growth and differentiation. Cell Adh. Migr. 2009;3(4):1–12. doi: 10.4161/cam.3.4.8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritti A, et al. Multipotent Neural Stem Cells Reside into the Rostral Extension and Olfactory Bulb of Adult Rodents. J. Neurosci. 2002;22(2):437–445. doi: 10.1523/JNEUROSCI.22-02-00437.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritti A, et al. Epidermal and Fibroblast Growth Factors Behave as Mitogenic Regulators for a Single Multipotent Stem Cell-Like Population from the Subventricular Region of the Adult Mouse Forebrain. J. Neurosci. 1999;19(9):3287–3297. doi: 10.1523/JNEUROSCI.19-09-03287.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vescovi AL, et al. Isolation and cloning of multipotential stem cells from the embryonic human CNS and establishment of transplantable human neural stem cell lines by epigenetic stimulation. Exp. Neurol. 1999;156(1):71–83. doi: 10.1006/exnr.1998.6998. [DOI] [PubMed] [Google Scholar]
- Azari H, Louis SA, Sharififar S, Vedam-Mai V, Reynolds BA. Neural-Colony Forming Cell Assay: An Assay To Discriminate Bona Fide Neural Stem Cells from Neural Progenitor Cells. J. Vis. Exp. 2011. p. e2639. [DOI] [PMC free article] [PubMed]
- Louis SA, et al. Enumeration of Neural Stem and Progenitor Cells in the Neural Colony-Forming Cell Assay. Stem Cells. 2008;26(4):988–996. doi: 10.1634/stemcells.2007-0867. [DOI] [PubMed] [Google Scholar]
- Coles-Takabe BL, et al. Don't look: growing clonal versus nonclonal neural stem cell colonies. Stem Cells. 2008;26(11):2938–2944. doi: 10.1634/stemcells.2008-0558. [DOI] [PubMed] [Google Scholar]
- Mori H, Fujitani T, Kanemura Y, Kino-Oka M, Taya M. Observational examination of aggregation and migration during early phase of neurosphere culture of mouse neural stem cells. J. Biosci. Bioeng. 2007;104(3):231–234. doi: 10.1263/jbb.104.231. [DOI] [PubMed] [Google Scholar]
- Singec I, et al. Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat. Methods. 2006;3(10):801–806. doi: 10.1038/nmeth926. [DOI] [PubMed] [Google Scholar]
- Yu YH, et al. Purification, Visualization, and Molecular Signature of Neural Stem Cells. Stem Cells and Dev. 2016;25(2):189–201. doi: 10.1089/scd.2015.0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YH. C1qR1 is comparable with LeX and Prom1 as a neural stem cell marker. Identification of mouse embryonic neural stem cell surface markers. 2012. Chapter 3.5, Figure 3.17.
- Tham M. Neural colony-forming cell assay. A role for chondroitin sulfate proteoglycan in regulating the survival and growth of neural stem cells. 2009. Chapter 3.6.3, Figure 3.22A.
- Yuan SH, et al. Cell-Surface Marker Signatures for the Isolation of Neural Stem Cells, Glia and Neurons Derived from Human Pluripotent Stem Cells. PLoS ONE. 2011;6(3):e17540. doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall PE, Lathia JD, Miller NGA, Caldwell MA, Ffrench-Constant C. Integrins Are Markers of Human Neural Stem Cells. Stem Cells. 2006;24(9):2078–2084. doi: 10.1634/stemcells.2005-0595. [DOI] [PubMed] [Google Scholar]