

Abstract
Fourier transform-infrared statistical models have the proven ability to identify subtle structural changes in DNA at various stages of tumor development. Using these models, we show evidence for a metastatic cancer DNA phenotype in histologically normal prostate tissues surrounding metastasizing tumors. Strikingly, the DNA base and backbone structures of the metastatic phenotype are indistinguishable from those of the metastasizing prostate tumors but distinctly different from the structure recently reported for the primary cancer DNA phenotype. These findings suggest that the DNA structure linked to the development of metastasis is preordained in progenitor cells relatively early in multistep tumorigenesis. The substantial structural differences found between the primary and metastatic cancer DNA phenotypes suggest that each evolves through a separate pathway. The metastatic phenotype is potentially an early predictor of metastatic disease. Interventions that inhibit its formation would be expected to also inhibit the development of metastatic tumors.
Keywords: metastasis, cancer prediction, cancer intervention, Fourier transform-infrared spectroscopy, DNA structure
Metastasis is commonly believed to result from the clonal selection of a few rare cells in a tumor population (1-3). An alternative mechanistic model for metastasis was suggested on the basis of DNA microarray studies implying that the proclivity for metastasis is hardwired in progenitor cells (4-6). One study (4) showed that gene expression profiles in primary breast tumors are strikingly similar to those in distant metastases of the same patients. Another study (7), which used laser capture microdissection in combination with DNA microarrays, found marked similarities at the transcriptional level among the distinct stages of breast tumor progression. Collectively, these studies (4-7) call into question classical theories of metastasis but support the concept that its characteristic features are preordained early in tumorigenesis (8).
Statistical models of Fourier transform-infrared (FTIR) spectra of DNA have the unique ability to discriminate between a variety of subtle changes in base functional group and backbone structures as well as in the conformational properties of DNA (9). These structural changes have been found in various stages of primary and metastatic tumor development (10-15). In a recent study (16), the FTIR technology effectively differentiated between the prostate DNA of histologically normal tissues, nonmetastasizing primary tumors, metastasizing primary tumors, and distant metastases of prostatic carcinomas. The first evidence for a primary cancer DNA phenotype in normal tissues was obtained in this study (16). FTIR technology also has been used to distinguish between the DNA of normal granulocytes and granulocytes from patients with myelodysplastic syndrome (17). It was also used recently to identify a primary cancer DNA phenotype in carcinogen-treated mice occurring ≈8 weeks before palpable tumors and to show that cyclophosphamide suppresses the formation of the phenotype and delays tumor formation (18).
Here we used the previously established statistical models (10, 16) to test the hypothesis that metastatic tumors of the human prostate are preceded by a metastatic cancer DNA phenotype in histologically normal tissues. We obtained DNA from primary tumor tissues with no evidence of metastasis (PT), primary tumor tissues with evidence of metastasis (MT), and histologically normal tissues surrounding the PT (NPT) and MT (NMT). The tumor and normal tissues were of high purity and were “matched” in that they were obtained from the same patient.
Materials and Methods
Tissue Acquisition. With institutional review board approval, we obtained frozen (-80°C) prostate tissues from the peripheral zone in which approximately two-thirds of tumors occur (19). Forty-two prostate samples were provided by the following donors: Baylor College of Medicine Specialized Program of Research Excellence tissue bank project, Houston (n = 18); Washington Pathology Consultants, Inc., Seattle (n = 20); Northwest Tissue Center, Seattle (n = 2); and the Cooperative Human Tissue Network, Pittsburgh (n = 2). The samples comprised the following groups: (i) tissues from patients, ages 50-75 years, with primary prostate cancer having no evidence of metastasis (PT), consisting of microscopically isolated tumor tissues (n = 9); (ii) matched histologically normal tissues microscopically isolated from the same patients with primary prostate tumors (NPT; n = 9); (iii) microscopically isolated metastasizing primary tumor tissues (MT; based on confirmed distant metastases), ages 55-71 years (n = 12); and (iv) matched microscopically isolated histologically normal tissues from the same patients with metastasizing primary prostate tumors (NMT; n = 12). The PTs and three of the MTs were used in a previous study (16).
Isolation of High-Purity Tissues. Samples obtained from the Baylor College of Medicine were isolated from fresh prostates by using a biopsy punch and evaluated for the presence or absence of tumors by using slides stained with hematoxylin/eosin. Tissue purity was determined by using two to three slides. The remaining samples were treated as follows. Frozen tumor slices were inked with different colors to denote posterior or anterior and right or left aspects. Two similarly inked, matching glass microscope slides of stained sections from the adjacent slices of the tumor also were obtained. Tumor foci were identified on the “sandwich” slides by dotting the tumor periphery and then extrapolated to the frozen slice such that essentially pure tumor tissue could be dissected. All isolated tumor tissues were estimated to be at least 90% pure. The histologically normal tissues surrounding the tumors showed no evidence of tumor cell contamination and were essentially pure.
DNA Extraction. As described in ref. 16, high-purity DNA (≈50 μg) was extracted from each prostate tissue (70-100 mg) with Qiagen (Valencia, CA) 100/G Genomic-tips by using a modification of the Qiagen extraction procedure: The DNA was passed through a 5.0-μm Cameo 30N filter (Osmonics, Minnetonka, MN) before precipitation. The Qiagen procedure is an ion-exchange system that does not constitute a source for artifactual oxidation of purines during extraction. In preparation for FTIR spectroscopy, the DNA was dissolved in 10-40 μl (depending on the size of the pellet) of optima-grade distilled water (Fisher Scientific). All samples were randomly selected for extraction and analysis to mitigate any batch effects.
FTIR Spectroscopy. FTIR analysis was performed with a microscope spectrometer (System 2000, PerkinElmer) as reported in refs. 9-11, 13, and 16-18. Briefly, a 0.2-μl aliquot of the DNA solution was spotted directly on a BaF2 plate, forming an outer ring that contained the DNA. Two separate spots (splits) were created for each DNA sample. To dry the DNA completely, the plate was placed in a lyophilizer for 1 h. By using the microscope spectrometer, 10 spectral determinations were made around each of the two rings per sample, and the percent transmittance values were converted (Fourier-transformed) into absorbance values. Each spectrum was baselined and then normalized to adjust for the optical characteristics of each sample (e.g., film thickness).
Statistical Analyses. The mean FTIR spectrum (1,750-700 cm-1) of each sample was obtained as described in ref. 16. A t test was performed to determine the statistical significance (P value) of differences at each wavenumber between the mean absorbance values for the DNA groups compared (e.g., PT vs. MT) (10, 13, 16).
Principal components (PCs) analysis, which involves ≈1 million correlations between spectral absorbances, integrates different properties of the spectra (e.g., varying peak heights, peak locations, and various combinations thereof). This analysis was performed on the mean spectrum of each sample, resulting in 10 PC scores per sample (10, 13, 16). A t test was used to determine significant differences between tissue groups for each PC score. PCs with significant differences (P ≤ 0.05) were used to construct two-dimensional scatter plots. Separation of sample clusters in the plots signifies that the groups are structurally dissimilar (10, 13, 16), whereas tightness of the points within the clusters is an indication of structural similarity.
Results and Discussion
The statistical models showed that the mean DNA spectrum of the PT was appreciably different from that of the MT, as reflected in significant differences (P ≤ 0.05) between the groups spanning 55.7% of the spectral range, 1,750-700 cm-1 (Fig. 1A), appreciably more than the 5% difference expected by chance (9). Marked differences between the groups were evident between 1,750 and 1,550 cm-1, a spectral area reflecting modifications in the base structure and perturbations in vertical base stacking properties (20). Substantial differences were found also in spectral areas assigned to backbone structures (≈1,350-750 cm-1) (Fig. 1A), notably the antisymmetric stretching vibration attributed to the 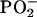 group (20). We found a substantive difference of four wavenumbers in the
group (20). We found a substantive difference of four wavenumbers in the 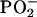 peak between the DNA from the PT (1,226 cm-1) and that of the MT (1,230 cm-1) (Fig. 1B). Differences between the groups were also found at ≈1,050 cm-1, an area of the spectrum assigned to ribose-phosphate main-chain vibrations (20). We showed previously, by using oligonucleotides and their derivatives (9), that changes in the functional group structure and conformational properties of the nucleotide bases can induce substantial modifications in the sugar-phosphate backbone.
peak between the DNA from the PT (1,226 cm-1) and that of the MT (1,230 cm-1) (Fig. 1B). Differences between the groups were also found at ≈1,050 cm-1, an area of the spectrum assigned to ribose-phosphate main-chain vibrations (20). We showed previously, by using oligonucleotides and their derivatives (9), that changes in the functional group structure and conformational properties of the nucleotide bases can induce substantial modifications in the sugar-phosphate backbone.
Fig. 1.

Significant differences in the base and backbone structures (notably the 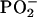 peak at ≈1,230 cm-1) exist between the DNA of the PT and MT and between the NPT and NMT. (A) P values from a t test between the mean DNA spectra of PT (n = 9) and MT (n = 12). (B) Comparison of the mean DNA spectra of the PT (solid line) and the MT (dashed line) between 1,260 and 1,200 cm-1.(C) P values from a t test between the mean DNA spectra of the NPT (n = 9) and NMT (n = 12). (D) Comparison of the mean DNA spectra of the NPT (solid line) and the NMT (dashed line) between 1,260 and 1,200 cm-1.
peak at ≈1,230 cm-1) exist between the DNA of the PT and MT and between the NPT and NMT. (A) P values from a t test between the mean DNA spectra of PT (n = 9) and MT (n = 12). (B) Comparison of the mean DNA spectra of the PT (solid line) and the MT (dashed line) between 1,260 and 1,200 cm-1.(C) P values from a t test between the mean DNA spectra of the NPT (n = 9) and NMT (n = 12). (D) Comparison of the mean DNA spectra of the NPT (solid line) and the NMT (dashed line) between 1,260 and 1,200 cm-1.
Most significantly, when the DNA of the NPT was compared with the DNA of the NMT, the spectral (structural) differences (P ≤ 0.05) between the groups (Fig. 1C) were virtually identical to those found between the PT and MT (Fig. 1A). The spectral differences between the DNA of these two histologically normal tissues spanned 71.5% of the spectral range (Fig. 1C). Importantly, the four-wavenumber difference in the 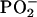 vibration (Fig. 1D) was identical to that found between the two tumor groups. These perturbations in the structure of the
vibration (Fig. 1D) was identical to that found between the two tumor groups. These perturbations in the structure of the 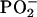 would be expected to profoundly alter the torsion angles of the phosphodiester-deoxyribose moiety and thus broadly affect the conformational properties of the molecule and its polymeric architecture to include the integrity of the nucleotide base structures (9).
would be expected to profoundly alter the torsion angles of the phosphodiester-deoxyribose moiety and thus broadly affect the conformational properties of the molecule and its polymeric architecture to include the integrity of the nucleotide base structures (9).
To obtain additional insight into the structural differences between the DNA groups, we analyzed the individual spectra by using the sensitive and highly discriminating statistical technique of PCs analysis. This approach allowed each DNA spectrum to be visualized as a point in two-dimensional space. Significant PC scores PC2 (P = 0.035) and PC3 (P < 0.001) were plotted to compare the DNA structure of the two tumor groups (Fig. 2A). Within each tumor group, the relatively tight clustering of points signifies a commonality of structure, whereas the high degree of discrimination between the groups reflects structural differences. An analogous comparison between the PC scores for the DNA from the NPT and NMT produced a very similar plot (PC2 and PC3, P < 0.01; Fig. 2B).
Fig. 2.

PC plots show a high degree of structural discrimination between the DNA of the PT and MT and the NPT and NMT. (A) Plot of PC2 vs. PC3 from the PT (▵) and the MT (▴). (B) Plot of PC2 vs. PC3 from the NPT (⋄) and the NMT (♦) (see text for details).
Overall, we found pronounced structural differences in the DNA between the PT and the MT, consistent with our previous prostate study (16). More than 100 genes have been identified in several solid tumors that are considered to be predictive of a poor prognosis (6, 21, 22). Thus, different combinations of these genes (including oncogenes and tumor suppressor genes), together with various epigenetic changes (e.g., resulting from free radicals) (16), probably account for most of the DNA structural differences between the PT and the MT (Fig. 1A). Strikingly, we found that the histologically normal tissues surrounding the PT and the MT also showed comparable differences in DNA structure (Fig. 1C), indicating the presence of two distinct and identifiable phenotypes: a primary cancer DNA phenotype and a metastatic cancer DNA phenotype.
We show that the DNA structure from the NPT is indistinguishable from that of the PT (<5% of the spectral range had P ≤ 0.05; Fig. 3A). This finding with the matched samples is consistent with our earlier study in which a primary cancer DNA phenotype was identified in the prostates of ≈42% of cancer-free men ages 50-80 years (16). Moreover, in a related study using the FTIR models in which the hind legs of mice were injected with the carcinogen 3-methylcholanthrene, we identified a comparable, early developing primary cancer DNA phenotype in histologically normal tissues (18). Administration of cyclophosphamide substantially suppressed the development of this phenotype concomitantly with a significant delay (30%) in tumor formation, thus demonstrating an association between the appearance of the primary phenotype and the subsequent development of a primary tumor (18).
Fig. 3.

Significant differences do not exist (<5% of the spectral range had P ≤ 0.05) between the DNA structures of the NPT and PT or between the NMT and MT. (A) NPT (n = 9) vs. PT (n = 9). (B) NMT (n = 12) vs. MT (n = 12).
The spectral models used here provide initial evidence for the evolution of a metastatic phenotype by showing that the structure of the NMT is virtually identical to that of the MT (<5% of the spectral range had P ≤ 0.05; Fig. 3B). In contrast, we found that this metastatic phenotype in NMT is distinctly different from the prostate DNA of healthy men (≈70% of the spectral range had P ≤ 0.05) (data not shown). These findings indicate that the metastatic phenotype is preordained in progenitor cells and potentially gives rise to metastatic prostate tumors. That is, a metastatic cancer DNA phenotype evolves in which the essential features of metastasis are hardwired in the DNA. As with the primary phenotype identified in the carcinogen-injected mice (18), this metastatic phenotype may similarly occur early in multistep tumorigenesis (4-8).
The question arises as to how it is possible that the DNA structures of both the metastatic cancer DNA phenotype and the MT could have identical base and backbone properties (Fig. 3B). Our study with oligonucleotides (9) suggested that modifications in the structure of the nucleotide bases induce readily identifiable conformational changes in the sugar-phosphate backbone (e.g., the 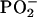 ), consistent with the polymeric nature of DNA. Thus, in DNA replication, the backbone structures would be faithfully reproduced according to the “blueprint” established by the nucleotide bases. Hence, the DNA structure of the metastatic phenotype would be expected to match that of the MT. These circumstances do not suggest that the metastatic phenotype is derived from the primary phenotype because of the substantial differences existing between their base and backbone structures (Fig. 1C). Rather, it seems that both phenotypes evolve through independent pathways.
), consistent with the polymeric nature of DNA. Thus, in DNA replication, the backbone structures would be faithfully reproduced according to the “blueprint” established by the nucleotide bases. Hence, the DNA structure of the metastatic phenotype would be expected to match that of the MT. These circumstances do not suggest that the metastatic phenotype is derived from the primary phenotype because of the substantial differences existing between their base and backbone structures (Fig. 1C). Rather, it seems that both phenotypes evolve through independent pathways.
Conclusions
Regardless of the etiology of the metastatic phenotype, the pronounced structural differences between the primary phenotype and the metastatic phenotype would be expected to translate into distinct differences in their respective biological properties. It is notable that the unique functional group and conformational properties of the base and backbone structures of the metastatic phenotype may allow (e.g., because of changes in torsion angles) for the desuppression of genes endowed with the proclivity for metastasis.
Identification of a metastatic cancer DNA phenotype is an attractive basis for the early detection of preneoplastic and neoplastic tissues at high risk for metastasis. Detection of the metastatic phenotype (e.g., by means of biopsy) could signal the need for intervention at an early stage of tumorigenesis when there is the greatest possibility for a successful outcome. Alternative intervention strategies may include suppressing or disrupting the formation of the metastatic phenotype, thereby thwarting or delaying metastatic tumor development. Finally, we believe our findings would benefit from the complementary perspective provided by gene expression profiles.
Acknowledgments
We thank Drs. Jordan U. Gutterman, Shuk-mei Ho, and Thomas R. Sutter for helpful comments and Brenda Duer and Nhan Vo for technical assistance. We thank the Cooperative Human Tissue Network, the Northwest Tissue Center, Washington Pathology Consultants, and the Specialized Program of Research Excellence at Baylor College of Medicine (funded by National Cancer Institute Grant CA58204) for providing tissues and pathology data. This study was supported by National Cancer Institute Grant CA79690.
Abbreviations: FTIR, Fourier transform-infrared; PT, primary tumor tissues with no evidence of metastasis; MT, primary tumor tissues with evidence of metastasis; NPT, histologically normal tissues surrounding the PT; NMT, histologically normal tissues surrounding the MT; PC, principal component.
References
- 1.Yokota, J. (2000) Carcinogenesis 21, 497-503. [DOI] [PubMed] [Google Scholar]
- 2.Fidler, I. J. & Kripke, M. L. (1977) Science 197, 893-895. [DOI] [PubMed] [Google Scholar]
- 3.Poste, G. & Fidler, I. J. (1980) Nature 283, 139-146. [DOI] [PubMed] [Google Scholar]
- 4.Weigelt, B., Glas, A. M., Wessels, L. F. A., Witteveen, A. T., Peterse, J. L. & van't Veer, L. J. (2003) Proc. Natl. Acad. Sci. USA 100, 15901-15905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weigelt, B. & van't Veer, L. J. (2004) Cell Cycle 3, E87-E88. [DOI] [PubMed] [Google Scholar]
- 6.Ramaswamy, S., Ross, K. N., Lander, E. S. & Golub, T. R. (2003) Nat. Genet. 33, 49-54. [DOI] [PubMed] [Google Scholar]
- 7.Ma, X. J., Salunga, R., Tuggle, J. T., Gaudet, J., Enright, E., McQuary, P., Payette, T., Pistone, M., Stecker, K., Zhang, B. M., et al. (2003) Proc. Natl. Acad. Sci. USA 24, 5974-5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernards, R. & Weinberg, R. A. (2002) Nature 418, 823. [DOI] [PubMed] [Google Scholar]
- 9.Malins, D. C., Polissar, N. L., Ostrander, G. K. & Vinson, M. A. (2000) Proc. Natl. Acad. Sci. USA 97, 12442-12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Closas, M., Hankinson, S. E., Ho, S., Malins, D. C., Polissar, N. L., Schaefer, S. N., Su, Y. & Vinson, M. A. (2000) J. Natl. Cancer Inst. Monogr. 27, 147-156. [DOI] [PubMed] [Google Scholar]
- 11.Malins, D. C., Polissar, N. L., Schaefer, S., Su, Y. & Vinson, M. (1998) Proc. Natl. Acad. Sci. USA 95, 7637-7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malins, D. C., Polissar, N. L. & Gunselman, S. J. (1997) Proc. Natl. Acad. Sci. USA 94, 259-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malins, D. C., Polissar, N. L., Su, Y., Gardner, H. S. & Gunselman, S. J. (1997) Nat. Med. 3, 927-930. [DOI] [PubMed] [Google Scholar]
- 14.Malins, D. C., Polissar, N. L. & Gunselman, S. J. (1996) Proc. Natl. Acad. Sci. USA 93, 14047-14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malins, D. C., Polissar, N. L., Nishikida, K., Holmes, E. H., Gardner, H. S. & Gunselman, S. J. (1995) Cancer 75, 503-517. [DOI] [PubMed] [Google Scholar]
- 16.Malins, D. C., Johnson, P. M., Barker, E. A., Polissar, N. L., Wheeler, T. M. & Anderson, K. M. (2003) Proc. Natl. Acad. Sci. USA 100, 5401-5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malins, D. C., Anderson, K. M., Polissar, N. L., Ostrander, G. K., Knobbe, E. T., Green, V. M., Gilman, N. K. & Spivak, J. L. (2004) Proc. Natl. Acad. Sci. USA 101, 5008-5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malins, D. C., Anderson, K. M., Gilman, N. K., Green, V. M., Barker, E. A. & Hellström, K. E. (2004) Proc. Natl. Acad. Sci. USA 101, 10721-10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirby, R. S., Christmas, T. J. & Brawer, M. K. (1996) Prostate Cancer (Mosby, London).
- 20.Tsuboi, M. (1969) Appl. Spectrosc. Rev. 3, 45-90. [Google Scholar]
- 21.van de Vijver, M. J., He, Y. D., van't Veer, L. J., Dai, H., Hart, A. A. M., Voskuil, D. W., Schreiber, G. J., Peterse, J. L., Roberts, C., Marton, M. J., et al. (2002) N. Engl. J. Med. 347, 1999-2009. [DOI] [PubMed] [Google Scholar]
- 22.van't Veer, L. J., Dai, H., van de Vijver, M. J., He, Y. D., Hart, A. A. M., Mao, M., Peterse, H. L., van der Kooy, K., Marton, M. J., Witteveen, A. T., et al. (2002) Nature 415, 530-536. [DOI] [PubMed] [Google Scholar]