

Abstract
Patient derived tumor xenograft (PDTX) models provide a necessary platform in facilitating anti-cancer drug development prior to human trials. Human tumor pieces are injected subcutaneously into athymic nude mice (immunocompromised, T cell deficient) to create a bank of tumors and subsequently are passaged into different generations of mice in order to maintain these tumors from patients. Importantly, cellular heterogeneity of the original tumor is closely emulated in this model, which provides a more clinically relevant model for evaluation of drug efficacy studies (single agent and combination), biomarker analysis, resistant pathways and cancer stem cell biology. Some limitations of the PDTX model include the replacement of the human stroma with mouse stroma after the first generation in mice, inability to investigate treatment effects on metastasis due to the subcutaneous injections of the tumors, and the lack of evaluation of immunotherapies due to the use of immunocompromised mice. However, even with these limitations, the PDTX model provides a powerful preclinical platform in the drug discovery process.
Keywords: Cancer Research, Issue 115, PDTX, CRC, stroma, heterogeneity, tumor bank, preclinical
Introduction
Colorectal cancer (CRC) is a significant contributor to cancer deaths in the United States. In 2015, there were an estimated 132,700 new cases of CRC with 49,700 deaths 1. Although the prognosis in patients with localized disease is excellent, patients with advanced disease have poor outcomes, making this a major priority in the development of novel therapies. Despite standard of care chemotherapeutic regimens and newer biologics that are deployed against this disease, there has been only an incremental increase in overall survival. Accordingly, there is a significant effort in understanding the driver pathways involved in facilitating tumor growth in this disease. The Cancer Genome Atlas Network has recently identified numerous main pathways that are implicated in CRC dysregulation and include: WNT, phosphoinositide 3-kinase (PI3K), RAS, transforming growth factor-β (TGF- β) and TP53 2. Together, with investigations describing other pathways that potentiate growth in CRC have ignited the development of newer therapies aimed at significantly improving the survival in this patient population 3-5. Utilizing preclinical models in oncology drug development have been essential in this process in predicting the clinical activity of these novel compounds.
Various preclinical models have been utilized in the drug development process. Considering that preclinical transgenic animal models and immortalized cell lines have been unsuccessful in determining the clinical activity of novel oncology therapies, largely due to their inability to reflect the complexity of human tumors, patient-derived tumor xenograft (PDTX) models have been established. The greatest advantage of this model is that tumor heterogeneity remains intact and closely reflects the molecular characteristics and clonality of the originating patient tumor 6-9. PDTX models provide an excellent in vivo preclinical platform to study novel agents, drug resistance pathways, combinational strategies, and cancer stem cell biology 10.
A general overview of the PDTX process is illustrated in Figure 1. It begins in the clinic, consenting patients to allow some of their excess tumor tissue to be used for this research. Next, at surgery, a piece of the tumor is grossed by a pathologist and put into media to be transported to research personnel. Immediately after this, a section of the tumor is cut into small pieces and transplanted into immunodeficient mice subcutaneously. Once the tumor grows, it is passaged into different generations of mice in order to maintain the tumor10. Typically, after the F3 generation the tumor can be expanded into a treatment study where novel compounds and/or combinational therapies are evaluated. Utilizing Next Gen Seq (Exome Seq, RNA Seq and SNP array) potential predictive biomarkers are discovered that assist in the selection of patients that may derive benefit from a particular treatment.
The overarching goals of using PDTX models are to: 1) evaluate the efficacy of novel therapies as single agent or in combination and 2) identify predictive biomarkers of sensitivity or resistance prior to clinical investigation. In this manuscript, we provide the methodology in the initiation and maintenance of a CRC PDTX bank and provide the advantages and limitations of this model in drug development discovery.
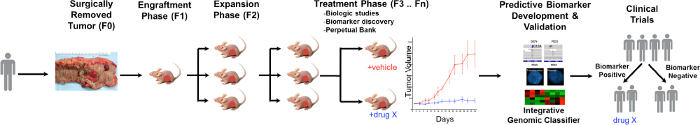 Figure 1. Overview of the CRC PDTX Model Protocol. A patient derived tumor is received from surgery and immediately injected into athymic nude mice subcutaneously. Once the tumor grows it is expanded into subsequent generations and eventually expanded for treatment studies. Treatment responses are assessed and predictive biomarkers are identified that may aid in patient selection. Please click here to view a larger version of this figure.
Figure 1. Overview of the CRC PDTX Model Protocol. A patient derived tumor is received from surgery and immediately injected into athymic nude mice subcutaneously. Once the tumor grows it is expanded into subsequent generations and eventually expanded for treatment studies. Treatment responses are assessed and predictive biomarkers are identified that may aid in patient selection. Please click here to view a larger version of this figure.
Protocol
Ethics Statement: Patient-derived colorectal adenocarcinoma tumor specimens were obtained from consenting patients at the University of Colorado Hospital in accordance with a protocol approved by the Colorado Multiple Institutional Review Board (08-0439). All animal work was performed under animal protocols approved by the University of Colorado Denver Institutional Animal Care and Use Committee (IACUC, Protocol # 51412(06)1E and 96813(04)1E).
1. Receiving and Preparing Patient Blood
Collect 1 - 2 ml of blood in a blood/cell separation tube containing sodium citrate (tube phases included are plasma, lymphocyte and monocyte band, density gradient fluid, gel barrier, and erythrocytes and neutrophils). Caution, follow Blood Borne Pathogen guidelines with blood or tissue. Note: PBMCs and plasma could be used for future studies that may include: comparing germline genetic variation with tumor mutations, isolating circulating tumor cells, evaluating cell free DNA (cfDNA), examining proteins and microRNAs, etc.
Centrifuge the blood/cell separation tube at 2,500 x g for 15 min with no brake at RT.
- Collect peripheral blood mononuclear cells (PBMCs in lymphocyte and monocyte band of tube) and put into a 1.5 ml microcentrifuge tube (clear, autoclavable, DNase, RNase, and pyrogen free), and fill to the top of the tube with sterile Phosphate-Buffered Saline (PBS).
- Centrifuge at 2,300 x g for 3 min at RT to form a pellet of PBMCs at the bottom of the tube. Next, remove the supernatant carefully. Add 1 ml of sterile PBS to wash the pellet and centrifuge at 3,000 x g for 30 sec and then remove supernatant.
Use a pipette to collect plasma from the blood/cell separation tube (in plasma phase of tube) in a 1.5 ml sterile cryogenic vial (DNase and RNase free, no human DNA, endotoxin free) and put the plasma and pellet of PMBC's into a -80 °C freezer for storage.
2. Receiving and Preparing Patient Tumor Sample
Prepare either RPMI or DMEM media with 1:100 (10 %) of Penicillin-Streptomycin and Non-essential Amino Acids, 1:1000 (1%) Plasmocin, and 10 % inactivated Fetal Bovine Serum (complete media) and add 20 - 25 ml to a sterile collection cup for the tumor specimen.
Retrieve tumor sample in a sterile cup containing complete media on ice or keep at 4 °C. Note: The tumor sample is a piece of excess patient tumor tissue that is removed by a surgeon, processed by a pathologist, and placed into a sterile collection cup with complete media. Caution, follow Blood Borne Pathogen guidelines with blood or tissue. Also, ideally to inject five mice, the tumor should be approximately 1 cm³.
- Bring the tumor sample to the animal facility for injecting into immunocompromised mice. Note: It is best to process the tumor sample as soon as possible.
- In a laminar flow hood, place the tumor sample into a sterile plastic cutting dish from the tumor cup. Use autoclaved-small scissors and forceps to cut into 10 - 12 approximately 3 x 3 x 3 mm pieces and place into an autoclaved 1.5 ml microcentrifuge tube filled with 300 µl of gelatinous protein mixture solution. Keep tube on ice.
- As a priority inject tumor into mice, but if there is any tumor left over, collect flash frozen vials (FF), formalin fixed paraffin embedded (FFPE) cups, and a viable tumor vial.
- For FF, collect small pieces of tumor tissues from a sterile cryogenic vial for further analysis such as protein, RNA, or DNA isolation. Place FF immediately into a liquid nitrogen dewar and store long-term in a -80 °C freezer.
- For FFPE, if there is enough tumor, place 3 small pieces into a 10 ml 10 % formalin cup and process into paraffin embedded blocks once tumor has been in formalin for at least 24 hr. Note: 24 hr is based off our Histology Core suggestions 11.
- Lastly, place the remaining tumor into a cryogenic tube. Make viable media by adding 10 % Dimethyl Sulfoxide (DMSO) to complete media. Next, add 1 ml to a cryogenic tube and store on ice. Note: Do not keep on ice for long periods of time.
- Cut any leftover tumor into small pieces that ideally will be enough for 10 tumors to be injected into 5 mice in the future. Then, place the viable tumor tube on ice until it can be placed into an Isopropyl Alcohol freezing container (slowly freezes the tumor 1°C at a time) and put into -80 °C freezer to ensure that the tumors do not die during the freezing process. Note: For long-term storage the tumors are removed (after 2 - 3 days) and placed into a large liquid nitrogen dewar. Please note that viable tubes should not be allowed to thaw unless it is being taken out to inject into mice.
3. Injection of Patient Derived Tumor Xenografts
Use five 6 - 8 week old male and/or female athymic nude mice (T-cell deficient) for each separate patient derived tumor. Inject a total of 10 tumors (2 injections per mouse) subcutaneously (SQ). Note: The original human tumor is designated as F0 and then once injected into mice the next generation is F1. Also,use autoclaved forceps, scissors, and trocars for each unique explant.
- Load pieces of tumor from the gelatinous protein mixture solution into autoclaved 12 G trocars and ensure that the tumor is completely pushed into the trocar.
- In a sterile laminar flow hood, place 5 mice into an anesthesia box that is connected to an Isoflurane anesthesia machine (started at the initial rate of 5% Isoflurane, 3 - 4% oxygen, and 2 - 3% Isoflurane for maintenance of anesthesia) and charcoal filter (absorbs excess gas). Note: An experienced technician should be able to perform the entire procedure on a cage of 5 mice within approximately 5 min total (roughly 30 sec per mouse). Therefore additional thermal support and eye lubrication is not generally used. For less experienced individuals or during training, it is highly recommended that individuals either perform the procedure on less animals at one time and/or use a warming pad/eye lubrication during procedure if animals will be anesthetized longer than 5 min. Procedure is based upon IACUC standards at our university.
- Pinch a mouse toe to ensure the mouse is no longer responsive, then take the mouse out of the Isoflurane box. Place the mouse onto a clean field to inject tumors on the middle dorsal neck region and sliding the trocar down subcutaneously until the flank region is reached with autoclaved 12 G trocars.
- Deliver one tumor subcutaneously to each side of the mouse's flank. Pinch the tumor when the trocar is pulled out, ensuring that the tumor stays in the desired flank region. The gelatinous protein mixture hardens with the mouse’s body temperature and encapsulates the tumor for approximately 1 week to help the tumor grow and secure it to the SQ connective tissue. The lesion made by the trocar is no larger than 4 mm and there is no need to close the skin with staples. Note: Our veterinarians determined no closure is needed based on very small size of the lesion, quick healing time (middle dorsal neck region reduces any skin tension or grooming of lesion), no infections noted, and close monitoring of the animals during this time period.
- Before the mouse is fully awake inject Meloxicam 2 mg/kg SQ (pain medication) away from the site of tumor injection. Next, place the mouse into the cage and monitor until the mouse is awake and moving. Note: Do not leave recovering animals unattended until fully awake. Meloxicam dosage is based upon IACUC standards at our university.
- Next, repeat the same procedure with the 4 remaining mice and monitor their breathing. Note: Please note this is a very quick procedure and mice wake up very soon after Meloxicam injection, but adjust Isoflurane as needed. Each time a mouse is taken out, turn down the Isoflurane. The Meloxicam will last 24 hr and the lesions from the trocars will fully heal in 1 week.
4. Maintenance of Patient Derived Tumor Xenograft Bank
Monitor growth and health of mice at least once per week. Use a spreadsheet (or other tracking system) to track tumor sizes, tumor generation, date of tumor injection, and health of mice. Note: If mice have lost 15% of their original body weight, low body condition scores (≥ 2), have tumor ulcerations, tumors reaching 2,000 mm3 or total tumors 3,000 mm3, or are sickly (hunched, cold, lethargy, etc.) in any way, mice are euthanized via CO₂ or anesthetized followed by cervical dislocation as a secondary method. Euthanize via anesthesia and cervical dislocation if collecting tumor, otherwise mice are euthanized via CO₂ and cervical dislocation.
- When a tumor is approximately 1,500-2,000 mm3 anesthetize the mice as described above and then perform cervical dislocation to euthanize.
- Check that the mouse has no heartbeat. Excise the SQ tumor with autoclaved scissors and forceps. NOTE: Allow tumors to grow for up to 1 year and if no tumor is seen then euthanize the mice via CO2, followed by cervical dislocation as a secondary method.
- Passage the best growing tumor into the next generation (aka a new set of 5 mice). Use instructions above to collect 10 - 12 tumors for passing and then collect the leftover tumor as also described above. Note: Collecting numerous viable tubes is very important in the early generations (F1 - F8); therefore collect several viable tubes, FF tubes, and 1 FFPE per generation.
- Keep the remaining mice until a new generation of mice has tumor growth of approximately 300 mm3. When remaining mice have tumors that are large, continue to collect as described above.
Continue to passage tumors and collect at each stage until F15. At this point, take a viable tube of the earliest generation possible out of liquid nitrogen and let thaw on ice and follow tumor-injecting procedures described above. Note: Tumors that grow from viable tubes take longer to grow in mice compared to passing from generation to generation, so keep that in mind when planning. If a particular PDTX model is no longer needed at any passage, euthanize and collect tumor, to ensure numerous viable tubes are collected for future use.
5. Developmental Therapeutics with Patient Derived Tumor Xenografts
Note: Most tumors at F3 generation have good growth kinetics (grow faster and more consistent), therefore, proceed to PDTX drug efficacy studies.
- When the desired tumor is very large (1,500 - 2,000 mm3), follow procedures above to collect tumor to expand into desired amount of mice.
- Depending on the hypothesis being tested determine the number of tumors needed per treatment group. Note: 1 ml of gelatinous protein mixture solution can approximately fit 30 small tumor pieces (depending on tumor morphology) for injecting into mice.
Check mice with tumors weekly and when most tumors are visibly small (approximately between 50 - 300 mm³) measure the tumors with calipers to determine the tumor volume. Tumor volume = [width² (smallest measurement) x length (largest measurement)] x 0.52. Note: The gelatinous protein mixture solution surrounds the injected tumor pieces for one week, then after one-week tumor growth will be accurate.
Use tumor volumes between 50 - 300 mm³ and then average the left and right tumors. Then randomize into treatment groups with 10 tumors per group and a group average within a few numbers of each other. Next, sort the mice into groups and begin the desired treatment study.
Dose the mice with drug (schedule dependent on drug), weigh and measure (tumor) twice per week. At the end of study, euthanize mice via anesthesia and cervical dislocation and collect tumor for future pharmacodynamic analysis in the lab. Note: The study lasts for 30 days depending on vehicle tumor size and health of mice.
6. Organization of a PDTX bank
- Keep a well-documented form to prevent repeating of research and appropriate use of animals. NOTE: This is key to a successful PDTX bank.
- Use spreadsheets to keep track of a tumor from the human patient in the clinic, to mice in the PDTX bank, treatments, data, and what is collected. Note: this includes freezer, liquid nitrogen dewars, and FFPE storage as well.
Representative Results
Similarities of Common Mutations in the CRC PDTX Models and the TCGA
We investigated whether the percentage of common mutations (KRAS, NRAS, BRAF, PIK3CA, APC, CTNNB1 and TP53) in the CRC PDTX bank were representative to the mutation frequency seen in the CRC patient population. As shown in Figure 2A (TCGA) and B (CRC PDTX bank), the frequency of mutations in these genes were very similar between the TCGA (n= 276 patients) and the CRC PDTX bank (n= 59 CRC patients). The biggest difference observed was in the APC gene whereby a 23 % difference was seen. These results suggest that common mutations observed in the CRC patient population is well represented in the CRC PDTX model.
Evaluation of the Stability of Treatment Responses Between Different Generations
In this CRC PDTX model, we set out to determine whether treatment effects were similar between different generations. Tumors were expanded in athymic nude mice (10 tumors/group) and the efficacy of the standard of care agents such as cetuximab (0.4 mg/mouse IP twice per week) and irinotecan (15 mg/kg IP once per week) were examined in 4 unique CRC PDTX models in two separate generations. As illustrated in Figure 3A and B, CRC026 (F3) was more resistant to cetuximab treatment, while CRC010 (F6) exhibited treatment sensitivity. Similar findings were observed when these CRC explants were treated in different generations; CRC026 (F9) was resistant and CRC010 (F7) was sensitive to cetuximab. To determine the efficacy of irinotecan in the PDTX model, we investigated treatment effects on tumor growth in 2 CRC PDTX models. While CRC098 (F8) was resistant to irinotecan treatment, CRC036 (F5) exhibited sensitivity (Figure 3C and D). As was observed with cetuximab, treatment with irinotecan in different generations did not change the treatment response in these tumors; CRC098 (F12) was resistant and CRC036 (F10) was sensitive to irinotecan (Figure 3C and D). Although the growth kinetics of untreated tumors was sometimes different between generations, the treatment responses to irinotecan and cetuximab remained the same, indicating the stability of this model in evaluating anti-cancer therapies.
Investigation of the Stroma Component in the CRC PDTX Model
Next, we were interested in determining if the stroma component within this CRC explant model was comprised of human and/or mouse derived cells. We used a dual-color Cot-1 FISH assay that consisted of mouse Cot-1 DNA (green fluorophore) and human Cot-1 DNA (red fluorophore) to determine mouse and human cells in 10 separate CRC explants between F0 and F1 generations 25. As shown in Figure 4A, in the F1 generation the human stroma is replaced by the mouse stroma, since the human tumor is now surrounded by all mouse stroma. These findings were evident in all 10 CRC explants that were subjected to Cot-1 FISH between the F0 and F1 generations. In addition to Cot-1 FISH, we investigated protein levels of mouse and human HGF and VEGF ligands in the F0 and F1 generations using mouse and human ELISAs for these ligands. As displayed in Figure 4B and C, whereas all human derived HGF in the F0 generation is replaced with mouse HGF in the F1 generation, VEGF ligand consisted of both human and mouse in the F1 generation. Together these experiments suggest that in the CRC PDTX mouse model that the mouse stroma overtakes the human stroma in the F1 generation and the ligands secreted may vary with respect to being human and/or mouse derived.
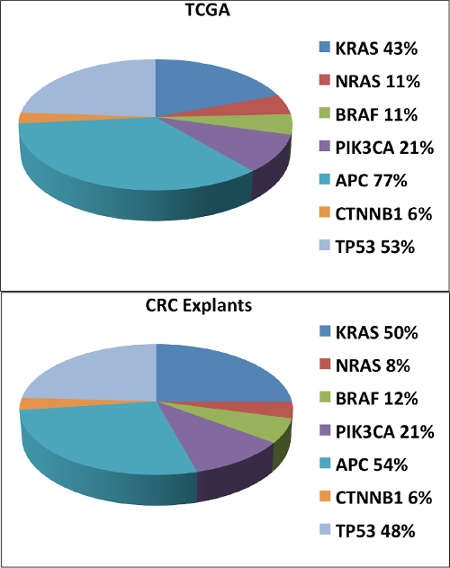 Figure 2. Comparison of Common Mutations in the CRC PDTX Bank and the TCGA. We observed very similar mutation frequencies between the TCGA (A) and the CRC PDTX model (B) with respect to KRAS, NRAS, BRAF, PIK3CA, CTNNB1 and TP53. Please click here to view a larger version of this figure.
Figure 2. Comparison of Common Mutations in the CRC PDTX Bank and the TCGA. We observed very similar mutation frequencies between the TCGA (A) and the CRC PDTX model (B) with respect to KRAS, NRAS, BRAF, PIK3CA, CTNNB1 and TP53. Please click here to view a larger version of this figure.
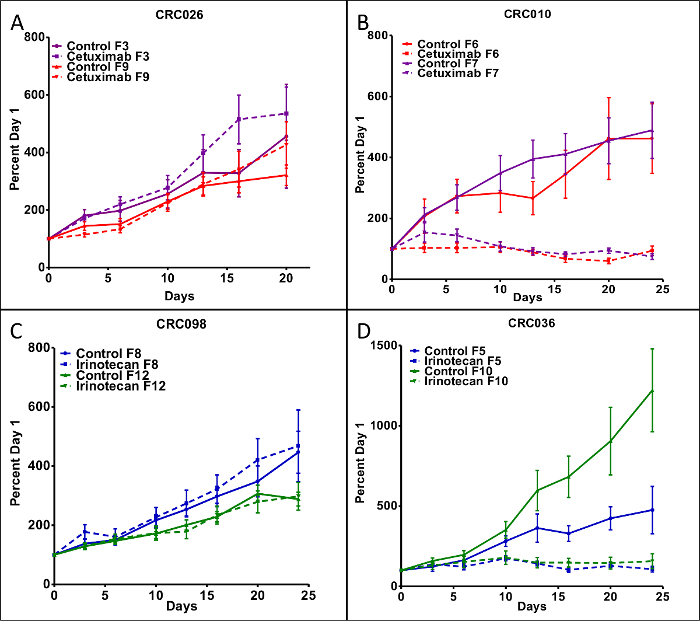 Figure 3. The Effects of Cetuximab and Irinotecan Treatment on Tumor Growth.(A) CRC026 displayed resistance to cetuximab when evaluated in the F3 and F9 generations and (B) CRC010 exhibited sensitivity to cetuximab in the F6 and F7 generations. (C) CRC098 showed resistance to irinotecan in the F8 and F12 generations, while (D) CRC036 was sensitive to irinotecan in the F5 and F10 generations. Each data point represents an average of 10 tumors per treatment group. Mice were treated with cetuximab (100 μl IP 400 μg/mouse) twice a week and irinotecan (100 μl IP 20 mg/kg) was dosed once a week. Data presented as mean ± SEM. Please click here to view a larger version of this figure.
Figure 3. The Effects of Cetuximab and Irinotecan Treatment on Tumor Growth.(A) CRC026 displayed resistance to cetuximab when evaluated in the F3 and F9 generations and (B) CRC010 exhibited sensitivity to cetuximab in the F6 and F7 generations. (C) CRC098 showed resistance to irinotecan in the F8 and F12 generations, while (D) CRC036 was sensitive to irinotecan in the F5 and F10 generations. Each data point represents an average of 10 tumors per treatment group. Mice were treated with cetuximab (100 μl IP 400 μg/mouse) twice a week and irinotecan (100 μl IP 20 mg/kg) was dosed once a week. Data presented as mean ± SEM. Please click here to view a larger version of this figure.
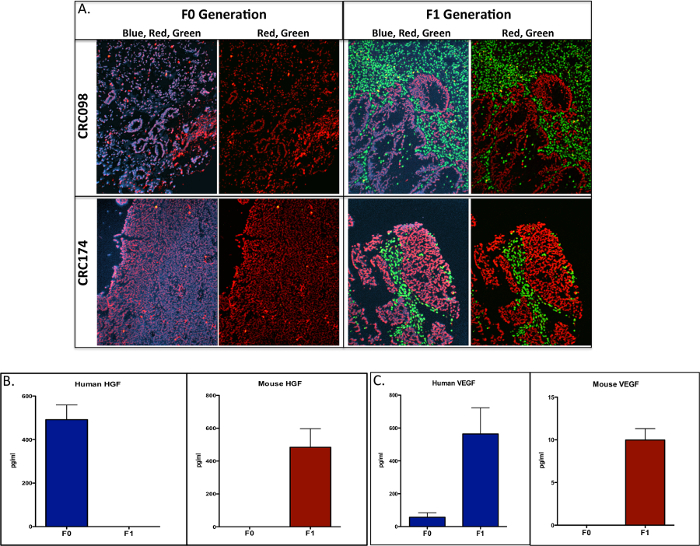 Figure 4. Evaluation of Mouse and Tumor Cell Components in the F0 and F1 Generations.(A) Dual-color FISH for the human cot-1 DNA (red) and mouse cot-1 DNA (green) was used to investigate differences between tumors in the F0 and F1 generation. Human stroma (F0) is replaced by mouse stroma (F1) in CRC098 and CR174 (scale = 20x). Necrotic cells were identified by reduced or lack of DAPI intercalation and red fluorescence. Investigation of mouse and human HGF and VEGF ligand expression by ELISA (mouse and human ELISAs) between F0 and F1. (B) Human HGF was replaced by mouse in the F1 generation and (C) human and mouse VEGF were evident in the F1 generation (picograms/milliliter [pg/ml]). Data presented as mean ± SEM. Please click here to view a larger version of this figure.
Figure 4. Evaluation of Mouse and Tumor Cell Components in the F0 and F1 Generations.(A) Dual-color FISH for the human cot-1 DNA (red) and mouse cot-1 DNA (green) was used to investigate differences between tumors in the F0 and F1 generation. Human stroma (F0) is replaced by mouse stroma (F1) in CRC098 and CR174 (scale = 20x). Necrotic cells were identified by reduced or lack of DAPI intercalation and red fluorescence. Investigation of mouse and human HGF and VEGF ligand expression by ELISA (mouse and human ELISAs) between F0 and F1. (B) Human HGF was replaced by mouse in the F1 generation and (C) human and mouse VEGF were evident in the F1 generation (picograms/milliliter [pg/ml]). Data presented as mean ± SEM. Please click here to view a larger version of this figure.
Discussion
The PDTX drug discovery platform offers an improved model to the shortcomings of other preclinical models that are unreliable in predicting clinical activity of novel compounds. Importantly, tumors in this model are biologically stable, retain metastatic potential, and exhibit similar drug responsiveness from generation to generation. In this model, patient derived tumors are injected into athymic nude mice, passaged, and subsequently used in therapeutic evaluation. There are several critical steps for a successful PDTX bank that include: 1) a cohesive clinical team to identify/consent patients and for the removal and grossing of tumor tissue for the PDTX model and 2) a strong research group with excellent laboratory and animal technical skills for injecting tumors, organization and maintenance of the PDTX bank and monitoring the health of mice. A significant advantage in our PDTX models is injecting tumors with the trocar procedure. The alternative method of cutting a tumor pocket and then suturing or using skin clips is more time consuming for personnel, requires more pain medication for the mice, and monitoring of the sutured or wound clipped area. The trocar procedure requires less training, is very quick, the mice are under anesthesia for less time, and less pain medications are needed. Therefore, in our experience injecting tumors with trocars is the best method versus alternative methods. These factors will significantly influence the overall success of the PDTX bank and in the drug discovery process. Although this in vivo model is considerably more costly than testing agents in cancer cell line cultures, PDTX models offer a more clinically relevant approach in testing oncology compounds.
In the CRC PDTX bank, we have received 99 tumor samples that were injected into athymic nude mice. There were 68 out of 99 (68.7% take rate) tumors that grew in mice and were passed into multiple generations. There are several reasons why some tumors that we received did not grow in mice. For instance, we sometimes received only very small pieces of tissue that only allowed us to inject into only one mouse decreasing the chances of establishing a tumor. Another issue was that at times we received patient tissue that was normal and did not contain tumor cells evident on the H&E slide. In addition, some poor tissue quality can be due to receiving already necrosed tumor where there was a patient response to treatment. Therefore, it is important to have a reliable surgical and pathology team when grossing the tumor. Considering some of these issues, we have been able to establish one of the largest CRC PDTX banks of fully annotated tumors with respect to mutations, gene expression, and clinical characteristics, making this a valuable model for the evaluation of novel therapies with the ultimate goal of improving patient outcomes.
Numerous biological and combinational therapies have been investigated in this model with the objective of determining efficacy, drug resistance mechanisms, as well as treatment effects on the cancer stem cell population. Our group has examined the efficacy of numerous novel biological pathway inhibitors using this preclinical model 12-18, which has provided valuable insight into further clinical development of these compounds. Many of these studies have identified predictive biomarkers 12-14,17,18 that may aid in patient selection in future clinical trials. In addition, other studies have further determined mechanisms of treatment resistance using this model, which has led to the development of rational combinations 19-24. For instance, Bardelli and colleagues 19 demonstrated that cetuximab treatment induced MET amplification and that Met may be an underlying mechanism of treatment resistance to cetuximab in CRC. 19In a separate study, Bertotti et al. 20 identified Her2 as a target in CRC tumors that were resistant to cetuximab. Finally, we and another group have shown that treatment with a Notch pathway inhibitor in combination with irinotecan reduced the CRC cancer stem cell population and tumor recurrence after treatment was discontinued 25,26. Together, these studies demonstrate the potential power of utilizing PDTX models in the drug development process that may significantly impact the further development of novel compounds.
Despite the major advantages using patient derived tumors in determining the efficacy of new compounds, there are several limitations in this model. As shown experimentally in this paper, the human stroma from the originating tumor (F0) is replaced with the mouse stroma at the F1 generation in the CRC PDTX model. Depending on the particular drug target, this may be an issue when mouse ligands are unable to activate the receptor(s) on human tumor cells. For instance, we show that in the F1 generation, HGF is derived from the mouse stroma and studies have determined that mouse HGF is incapable of functionally activating the human c-Met receptor 27,28. As a result, c-Met inhibitors may not exhibit anti-tumor growth effects in these PDTX models. In fact, humanized HGF SCID mice have been developed to address this potential problem 28. Another disadvantage of using subcutaneous tumors is the inability to study treatment effects on the metastatic potential of tumors. Using orthotopic models, although more technically difficult to establish and image tumor growth, will likely provide better insight into the treatment effects on metastasis. A final limitation of this model is the inability to investigate the role of the immune system in potentiating tumor growth and its function in facilitating resistance to treatment. Moreover, with the excellent activity of immunotherapies recently demonstrated in the clinic, using immunodeficient mice prevents the investigation of immune targeted agents. Accordingly, humanized mouse models have been developed to address these limitations, which may provide value in understanding the fundamental role of immune-tumor interactions as well as allow for the investigation of immunotherapies in combination with other novel and approved compounds.
In conclusion, we provide a method on developing and maintaining a CRC PDTX model that is invaluable in determining therapeutic efficacy, predictive biomarkers and drug resistance pathways of novel anti-cancer agents. Although this model has inherent challenges, the utility of this model is that it closely recapitulates the tumor heterogeneity of the original tumor, which offers a more precise and clinically relevant investigation of novel therapies prior to clinical evaluation. Future evaluation of the clinical impact in drug development of this preclinical PDTX model will ultimately determine the power of this model in predicting the clinical activity of therapies in cancer.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by grant 1R01CA152303-01.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics. 2015. CA Cancer J Clin. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcaroli JJ, et al. Tumours with elevated levels of the Notch and Wnt pathways exhibit efficacy to PF-03084014, a gamma-secretase inhibitor, in a preclinical colorectal explant model. Br J Cancer. 2013;109(3):667–675. doi: 10.1038/bjc.2013.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard J, Grothey A. Antiangiogenesis agents in colorectal cancer. Curr Opin Oncol. 2010;22(4):374–380. doi: 10.1097/CCO.0b013e328339524e. [DOI] [PubMed] [Google Scholar]
- van Es JH, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435(7044):959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- Cassidy JW, Caldas C, Bruna A. Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts. Cancer Res. 2015;75(15):2963–2968. doi: 10.1158/0008-5472.CAN-15-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, et al. Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin Transl Oncol. 2010;12(7):473–480. doi: 10.1007/s12094-010-0540-6. [DOI] [PubMed] [Google Scholar]
- Julien S, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18(19):5314–5328. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73(17):5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentler JJ, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9(6):338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson FL. Histotechnology: A Self-Assessment Workbook. Chicago, IL: American Society of Clinical Pathologists Press; 1996. [Google Scholar]
- Arcaroli JJ, et al. Common PIK3CA mutants and a novel 3' UTR mutation are associated with increased sensitivity to saracatinib. Clin Cancer Res. 2012;18(9):2704–2714. doi: 10.1158/1078-0432.CCR-11-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcaroli JJ, et al. A NOTCH1 gene copy number gain is a prognostic indicator of worse survival and a predictive biomarker to a Notch1 targeting antibody in colorectal cancer. Int J Cancer. 2016;138(1):195–205. doi: 10.1002/ijc.29676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcaroli JJ, et al. Gene array and fluorescence in situ hybridization biomarkers of activity of saracatinib (AZD0530), a Src inhibitor, in a preclinical model of colorectal cancer. Clin Cancer Res. 2010;16(16):4165–4177. doi: 10.1158/1078-0432.CCR-10-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu CH, et al. Antitumor activity of a potent MEK inhibitor, TAK-733, against colorectal cancer cell lines and patient derived xenografts. Oncotarget. 2015;6(33):34561–34572. doi: 10.18632/oncotarget.5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts TM, et al. Association of the epithelial-to-mesenchymal transition phenotype with responsiveness to the p21-activated kinase inhibitor, PF-3758309, in colon cancer models. Front Pharmacol. 2013;4:35. doi: 10.3389/fphar.2013.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song EK, et al. Potent antitumor activity of cabozantinib, a c-MET and VEGFR2 inhibitor, in a colorectal cancer patient-derived tumor explant model. Int J Cancer. 2015;136(8):1967–1975. doi: 10.1002/ijc.29225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentler JJ, et al. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Mol Cancer Ther. 2010;9(12):3351–3362. doi: 10.1158/1535-7163.MCT-10-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardelli A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–673. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotti A, et al. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1(6):508–523. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- Davis SL, et al. Combined inhibition of MEK and Aurora A kinase in KRAS/PIK3CA double-mutant colorectal cancer models. Front Pharmacol. 2015;6:120. doi: 10.3389/fphar.2015.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli MP, et al. Preclinical activity of the rational combination of selumetinib (AZD6244) in combination with vorinostat in KRAS-mutant colorectal cancer models. Clin Cancer Res. 2012;18(4):1051–1062. doi: 10.1158/1078-0432.CCR-11-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts TM, et al. Dual pharmacological targeting of the MAP kinase and PI3K/mTOR pathway in preclinical models of colorectal cancer. PLoS One. 2014;9(11):e113037. doi: 10.1371/journal.pone.0113037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreafico A, et al. Rational combination of a MEK inhibitor, selumetinib, and the Wnt/calcium pathway modulator, cyclosporin A, in preclinical models of colorectal cancer. Clin Cancer Res. 2013;19(15):4149–4162. doi: 10.1158/1078-0432.CCR-12-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcaroli JJ, et al. ALDH+ tumor-initiating cells exhibiting gain in NOTCH1 gene copy number have enhanced regrowth sensitivity to a gamma-secretase inhibitor and irinotecan in colorectal cancer. Mol Oncol. 2012;6(3):370–381. doi: 10.1016/j.molonc.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoey T, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009;5(2):168–177. doi: 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- Ikebuchi F, et al. Dissociation of c-Met phosphotyrosine sites in human cells in response to mouse hepatocyte growth factor but not human hepatocyte growth factor: the possible roles of different amino acids in different species. Cell Biochem Funct. 2013;31(4):298–304. doi: 10.1002/cbf.2898. [DOI] [PubMed] [Google Scholar]
- Zhang YW, et al. Enhanced growth of human met-expressing xenografts in a new strain of immunocompromised mice transgenic for human hepatocyte growth factor/scatter factor. Oncogene. 2005;24(1):101–106. doi: 10.1038/sj.onc.1208181. [DOI] [PubMed] [Google Scholar]