

Abstract
Transmission electron microscopy (TEM) provides details of the cellular organization and ultrastructure. However, TEM analysis of rare cell populations, especially cells in suspension such as hematopoietic stem cells (HSCs), remains limited due to the requirement of a high cell number during sample preparation. There are a few cytospin or monolayer approaches for TEM analysis from scarce samples, but these approaches fail to get significant quantitative data from the limited number of cells. Here, an alternative and novel approach for sample preparation in TEM studies is described for rare cell populations that enables quantitative analysis.
A relatively low cell number, i.e., 10,000 HSCs, was successfully used for TEM analysis compared to the millions of cells typically used for TEM studies. In particular, Evans blue staining was performed after paraformaldehyde-glutaraldehyde (PFA-GA) fixation to visualize the tiny cell pellet, which facilitated embedding in agarose. Clusters of numerous cells were observed in ultra-thin sections. The cells had a well preserved morphology, and the ultra-structural details of the Golgi complex and several mitochondria were visible. This efficient, easy and reproducible protocol allows sample preparation from a low cell number and can be used for qualitative and quantitative TEM analysis on rare cell populations from limited biological samples.
Keywords: Cellular Biology, Issue 116, Transmission electron microscopy, TEM, stem cells, HSC, rare populations, Evans blue, osmium tetraoxide
Introduction
Ultra-structural and sub-organelle details of cells have been mainly revealed by transmission electron microscopy (TEM) studies from tissues or cell pellets1,2. Solid pieces of tissues can be easily utilized for electron microscopy studies. However, TEM analyses1,3 on cells in suspension remain challenging and necessitate high cell numbers, i.e., millions of cells. Because of this, ultra-structural analyses of rare cell populations in suspension, e.g., hematopoietic stem cells (HSCs), are not easily assessed. Multiple attempts for TEM analysis from scarce samples using cytospin or monolayer approaches fail to get significant quantitative data from the limited number of cells. Thus, the requirement of a high cell number limits the use of this powerful tool to understand the subcellular ultrastructural details of rare cell populations.
A key limitation for TEM studies with a limited number of cells in suspension is the localization of the cells for processing and thus, TEM studies from limited cells with particularly small sizes remain challenging. Several alternative approaches have been adopted to counter this limitation: the BSA/bisacrylamide (BSA-BA) mediated polymerization of cell suspension, the staining of cells with a dye to make them visible on a thin film support including coverslips,and TEM analyses from cytospin preparations4-6. However, very limited success was achieved, as very few cells were found in the sections after ultra-sectioning. The key challenge of identifying sparse cells persists because the cell monolayer remains mostly invisible in the solidified gel for sectioning. Furthermore, cytospin preparation of cells may alter their cellular organization due to cell spreading and the fragility of the cell structure. Hence, these inherent drawbacks warrant a novel approach to perform TEM studies from rare cell populations with more consistency. To overcome this problem, we have described a novel and alternative sample preparation method for TEM studies from rare cell populations7.
Here, we report an efficient sample preparation protocol to perform TEM from scarce biological samples with consistent qualitative and quantitative results. Evans blue staining was performed after fixation to localize a tiny cell pellet from low number cells, i.e., 10,000 bone marrow hematopoietic stem and progenitor cells that would otherwise have remained invisible, and the pellet was embedded into agarose before dehydration and resin embedding processes. This method clusters the cells together and enables the efficient analysis of the ultrastructure and subcellular organization of hematopoietic stem cells (identified as Lin- Sca-1+ c-Kit+ Flt3- CD34-; HSC), a rare 0.2 - 0.5% cell population in the bone marrow. This experimental protocol can be useful to perform ultra-structural studies and obtain quantitative results on many rare but highly important populations.
Protocol
All experimental procedures were approved by the institutional animal committee at the Cincinnati Children's Research Foundation. For this study, hematopoietic stem cells were isolated from the bone marrow of C57Bl/6 inbred mice aged 2 - 4 months. Cell sorting using FACS after staining of BM with different surface makers including Lineage, c-Kit, Sca-1, Flt3 and CD34 was used for purification of HSC based on Lin- Sca-1+ c-Kit+ Flt3- CD34- gating strategy as standard protocol described before8.
CAUTION: Several highly toxic chemicals are used during this procedure. These are toxic by inhalation and skin contact. Please wear gloves and protective clothing. Work in fume hood while working with these chemicals.
1. Cells, Fixation, Staining and Pre-embedding
Sort 10,000 hematopoietic stem cells (using Lin- Sca-1+ c-Kit+ Flt3- CD34- gating strategy; LT-HSC population) in a 1.5 ml microcentrifuge tube containing 600 µl Iscove's Modified Dulbecco's Medium (IMDM) with 10% fetal bovine serum using FACS.
Spin down sorted HSCs at 1,000 x g for 10 min in a 1.5 ml microcentrifuge tube using a centrifuge with swing-out buckets. Remove medium by gentle aspiration and leave 200 µl medium in the tubes at each step. At this stage, the cell pellet remains invisible in the tube.
- Fix the cells by adding 0.2 ml of 2x fixative solution at room temperature (RT) for 1 hr1,9.
- Make the fixative solution fresh before use. At 1x, the solution is comprised of 3% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer.
Centrifuge the cells at 1,000 x g for 10 min at 30 °C using a centrifuge with swing-out buckets.
Wash cells with 600 µl of 0.1 M cacodylate buffer using centrifugation and leave 200 µl buffer in the tube after centrifugation.
Stain the fixed cells by adding 200 µl of 2 mg/ml solution of Evans Blue in cacodylate buffer and incubate for 20 min at RT. The cells will be colored blue.
Wash cells 3 times with 600 µl of 0.1 M cacodylate buffer using centrifugation and leave 200 µl buffer in the tube each time.
Re-suspend the cells in 200 µl buffer and transfer the cell suspension to a 0.5 ml tube. Centrifuge at 1,000 x g for 10 min using a table top centrifuge. Remove buffer gently without disturbing the now visible tiny cell pellet, and leave 50 µl buffer in the tube.
Add 200 mg low melting agarose to 5 ml of 0.1 M cacodylate buffer in the 15 ml tube for a 4% agarose solution. Melt the agarose by transferring the tube to boiling water in a 100 ml glass bottle and keep lukewarm until used.
Add 200 µl of 4% low melting agarose dissolved in 0.1 M cacodylate buffer to the cell suspension in 0.5 ml tube and immediately centrifuge at 1,000 x g for 10 min at 30 °C using a table top centrifuge.
After confirming that cells pelleted at bottom of the 0.5 ml tube into the semi-solid agarose after centrifugation, immediately transfer the tube to 4 °C or ice for 20 min to solidify the agarose. This step is a very critical, as a small visible cell pellet at this step results in a good cluster of cells under the electron microscope.
Carefully transfer the solidified agarose containing the cell pellet from the tube to a 35 mm plastic Petri dish containing buffer using a 27 G needle3,10.
Trim the solidified agarose containing the cell pellet into one piece of about 1 - 2 mm with a scalpel. Transfer the agarose piece containing the cell pellet to a new 1.5 ml tube.
Wash 2 - 3 times using 600 µl of 0.1 M cacodylate buffer by adding and removing buffer using pipette without centrifugation. Use this agarose piece containing the cell pellet for the next step. At this step, the sample can be stored at 4 °C overnight before moving to next step.
2. Osmification, Dehydration, Embedding
Remove the cacodylate buffer from the 1.5 ml tube with the cell pellet containing agarose using a pipette. Add 1.0 ml of 1% osmium tetraoxide (OsO4) solution in a fume hood, and incubate for 1 hr at 4 °C.
Wash three times with 600 µl of 0.1 M cacodylate buffer and transfer the sample to a 20 ml glass scintillation vial with a cap.
- Process the sample for dehydration, infiltration and embedding into resin (e.g., LX-112) with serial changes of the following solutions in a 6 well plate. Move the cell-pellet agarose piece using forceps.
- Immerse the sample in 25% ethanol at RT for 15 min.
- Immerse the sample in 50% ethanol at RT for 15 min.
- Immerse the sample in 75% ethanol at RT for 15 min.
- Immerse the sample in 95% ethanol at RT for 15 min.
- Immerse the sample in 100% ethanol at RT for 15 min, twice.
- Immerse the sample in ethanol: resin (3:1) at RT for 30 min.
- Immerse the sample in ethanol: resin (1:1) at RT for 30 min.
- Immerse the sample in ethanol: resin (1:3) at RT for 30 min.
- Immerse the sample in pure resin at RT for 60 min, twice.
Transfer the sample to the bottom of pyramid tip shaped rubber mold using forceps carefully and add more pure resin on top of the sample to fill the mold. Incubate the sample at 60 °C for 72 hr for resin polymerization.
Remove the polymerized resin pyramid from the molds by twisting the rubber mold. A small black cluster of cells should be visible in the polymerized resin pyramid, as described earlier7. Attach the polymerized resin pyramid on the mounting cylinder using cyanoacrylate.
3. Sectioning and Transmission Electron Microscopy
Trim the pyramid block with a razor blade and a diamond knife after mounting the block on an ultra-microtome. Make a short wide trapezoidal shape with the top and the bottom of block parallel to each other, and with evenly angled sides. This shape of the pyramid helps for serial sectioning.
Further, trim the block around the cell pellet with the diamond knife and remove the plastic sections around the cell pellet agarose piece.
Cut 1 µm sections using an ultra-microtome. Move the 1 µm sections from the water boat to a glass slide using an eyelash tool and a metal loop tool as previously reported7. Transfer sections on slides and transfer slides to the hot plate (37 °C) for drying.
Add a drop of toluidine blue solution to the sections using a syringe equipped with 0.22 µm filter and incubate for 3 min. Rinse the slides with distilled water and let the sections dry. Check with a light microscope to identify the position of the cells.
Cut ultra-thin sections (70 - 100 nm) using a diamond knife. Move 2 - 3 sections on 200-mesh grids from the water boat to a glass Petri dish using an eyelash tool.
Transfer the glass Petri dish containing grids with sections to a hot plate at 30 °C to dry sections for 30 min.
Stain the grids with drops of 1% uranyl acetate at RT for 10 min and then rinse in distilled water 6 - 8 times. Stain with Reynolds lead citrate, at RT for 5 min and rinse in distilled water 6 - 8 times. Transfer the glass Petri dish containing the grids to a hot plate at 37 °C to dry sections.
Analyze the sections with an electron microscope, at 80 kV7.
Representative Results
An efficient and consistent protocol for sample preparation to perform TEM analysis from low number of cells is described. Post-fixation staining with Evans blue and the transfer of cells to a 0.5 ml microcentrifuge tube helped visualize the tiny cell pellet7. Osmification with osmium tetraoxide in agarose led to an easy detection of the dark cell pellet during the dehydration and embedding.
Semi-thin sections from the blocks containing a tiny cell pellet confirmed a cluster of numerous cells together (Figure 1A and 1B). Ultra-thin sections were cut from the cell-clustered area and analyzed under electron microscopy (Figure 1C). The morphology and ultra-structure of these cells were well preserved (Figure 1D). The electron micrograph of individual cell showed high details on the intracellular ultra-structures (Figure 1E). Furthermore, the high magnification (50,000X) images showed a well-preserved structure and the integrity of sub-cellular organelles, such as mitochondria (Figure 1F).
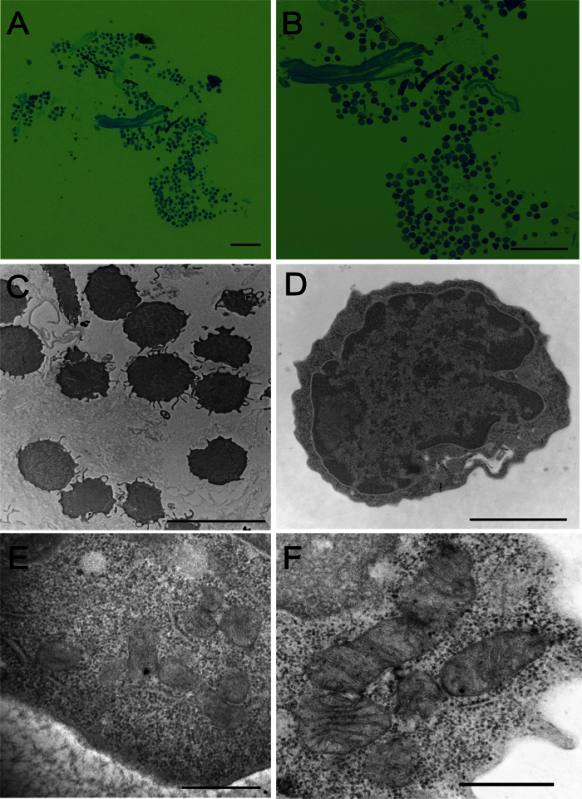 Figure 1:Image Analysis Under Light and Electron Microscope. (A) Low magnification image of a representative field of a toluidine blue stained section of 1 µm thickness under light microscope. (B) Light microscope image showing the intact morphology of cells in cluster after toluidine blue staining of 1 µm thickness section. (C) An overview field of the cell cluster observed under electron microscope after staining with uranyl acetate and lead citrate. (D) A single LT-HSC under electron microscope. (E) Higher magnification image of HSC showing subcellular structures. (F) Higher magnification image of mitochondria from HSPC. Scale bar: A/B, 100 µm; C, 10 µm; D, 2 µm; E/F, 500 nm. Please click here to view a larger version of this figure.
Figure 1:Image Analysis Under Light and Electron Microscope. (A) Low magnification image of a representative field of a toluidine blue stained section of 1 µm thickness under light microscope. (B) Light microscope image showing the intact morphology of cells in cluster after toluidine blue staining of 1 µm thickness section. (C) An overview field of the cell cluster observed under electron microscope after staining with uranyl acetate and lead citrate. (D) A single LT-HSC under electron microscope. (E) Higher magnification image of HSC showing subcellular structures. (F) Higher magnification image of mitochondria from HSPC. Scale bar: A/B, 100 µm; C, 10 µm; D, 2 µm; E/F, 500 nm. Please click here to view a larger version of this figure.
Discussion
This method enables TEM analysis on low cell numbers, by using Evans blue staining and agarose embedding to localize a tiny cell pellet during the dehydration, resin embedding and sectioning processes. Importantly, it maintains cell clusters together and preserves the cell ultra-structure, which is desirable to examine multiple cells for quantification of data.
In TEM studies, a cell pellet containing millions of cells is often required for efficient embedment into an agarose/gelatin matrix and for washes, dehydration and infiltration to obtain data from numerous cells. Researchers have adopted immobilization of the cell suspension in a BSA-bisacrylamide polymerized resin, staining of monolayers on a thin support, or cytospins for TEM analysis on low cell numbers/scarce samples4-6. However, it remains a tedious job to localize and cut cells under the TEM because of the sparse distribution of the cells within the matrix and possible alterations of the cell morphology, due to spreading and a cleavage between the cells during cytospin preparation that often required re-embedding before sectioning. Furthermore, the monolayer remains challenging to localize for sectioning11. Thus, a cell pellet even from scarce biological samples of low cell number is much easier for TEM analysis1.
This method represents a significant advantage to previous published methods4,5 in terms of qualitative and quantitative ultra-structural analysis from limited samples. Together, this protocol describes an efficient way to localize the cell pellets for TEM using Evans blue, which makes the sample processing from low cell numbers easy for TEM to enable qualitative and quantitative data analysis. Typically, the sample preparation for TEM requires a larger number of cells than what is usually required for light microscopy, because of the multiple steps in sample processing for TEM and proper orientation of the cell block for cutting at the right position1,10.
It is known that post-fixation osmification provides a better contrast in images, particularly those of membranes and glycogen particles under an electron microscope9, and further support staining with uranyl acetate, a relatively non-specific stain for proteins, and lead citrate that stains membranes, nucleic acids and glycogen. However, osmification is almost impossible to perform with scarce biological samples in suspension after pre-embedding in BSA-BA due to the high likelihood of sample loss during sectioning and matrix darkening. Thus the quality of the electron micrographs was compromised in previous studies4,9. Osmification here led to darkening of the cell pellet with minimal change in the color of the agarose. Thus, the present protocol allows osmification after pre-embedding in agarose in order to provide good contrast in TEM images.
Cell clusters were quite easy to analyze with electron microscopy using this method. The pre-staining with Evans blue helped to localize tiny cell pellets during sample preparation without affecting sub-organelle cell details. This method offers an alternative approach for sample preparation to successfully perform TEM analysis from low cell numbers. In this method, TEM analysis was effectively performed using cells of a small size (5 - 8 µm). Thus, this approach can easily be adopted with even lower cell number (~ 1,000 cells) of larger cells such as fibroblasts and endothelial cells. Although Evans blue did not affect sub-organelle cell details in the present study, the use of Evans blue for immuno-gold electron microscopy with low cell numbers has not been tested.
Together, an efficient method for sample preparation to perform TEM studies from a low cell number is described. TEM studies are required to reveal critical information on the morphology and the ultra-structures of cells. Hence, ultra-structural information is often missing on various scarce cell populations. This alternative and novel approach for sample preparation will be helpful to perform TEM studies from any rare cell population. Furthermore, this method allows qualitative and quantitative data from the analysis of cell clusters. As electron microscopy offers unsurpassed details of the cell ultra-structure and organization, this method will be quite useful to understand many biological processes on key, but rare, cell populations.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank the Pathology Research Core for assistance with electron microscope analysis studies at Cincinnati Children's Hospital Medical Center. The work was supported by NIH (American Society of Hematology Bridge award to-MDF; R01 DK102890 to MDF).
References
- Bozzola JJ. Conventional specimen preparation techniques for transmission electron microscopy of cultured cells. Methods Mol Biol. 2007;369:1–18. doi: 10.1007/978-1-59745-294-6_1. [DOI] [PubMed] [Google Scholar]
- Leapman RD. Novel techniques in electron microscopy. Curr Opin Neurobiol. 2004;14:591–598. doi: 10.1016/j.conb.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Anderson DR. A method of preparing peripheral leucocytes for electron microscopy. J Ulstract Res. 1965;13:263–268. doi: 10.1016/s0022-5320(65)80075-2. [DOI] [PubMed] [Google Scholar]
- Taupin P. Processing scarce biological samples for light and transmission electron microscopy. Eur J Histochem. 2008;52:135–139. doi: 10.4081/1203. [DOI] [PubMed] [Google Scholar]
- Mather J, Stanbridge CM, Butler EB. Method for the removal of selected cells from cytological smear preparations for transmission electron microscopy. J Clin Pathol. 1981;34:1355–1357. doi: 10.1136/jcp.34.12.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oorschot V, de Wit H, Annaert WG, Klumperman J. A novel flat-embedding method to prepare ultrathin cryosections from cultured cells in their in situ orientation. J Histochem Cytochem. 2002;50:1067–1080. doi: 10.1177/002215540205000809. [DOI] [PubMed] [Google Scholar]
- Kumar S, Ciraolo G, Hinge A, Filippi MD. An efficient and reproducible process for transmission electron microscopy (TEM) of rare cell populations. J Immunol Methods. 2014;404:87–90. doi: 10.1016/j.jim.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, et al. Enrichment of hematopoietic stem cells with SLAM and LSK markers for the detection of hematopoietic stem cell function in normal and Trp53 null mice. Exp Hematol. 2008;36:1236–1243. doi: 10.1016/j.exphem.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopwood D. The reactions between formaldehyde, glutaraldehyde and osmium tetroxide, and their fixation effects o bovine serum albumin and on tissue blocks. Histochemistry. 1970;24:50–64. doi: 10.1007/BF00310003. [DOI] [PubMed] [Google Scholar]
- Saini R, et al. Nitric oxide synthase localization in the rat neutrophils: immunocytochemical, molecular, and biochemical studies. J Leukoc Biol. 2006;79:519–528. doi: 10.1189/jlb.0605320. [DOI] [PubMed] [Google Scholar]
- Kumar S, et al. The small GTPase Rap1b negatively regulates neutrophil chemotaxis and transcellular diapedesis by inhibiting Akt activation. J Exp Med. 2014;211:1741–1758. doi: 10.1084/jem.20131706. [DOI] [PMC free article] [PubMed] [Google Scholar]