

Abstract
The intravenous injection of C1498 cells into syngeneic or congenic mice has been performed since 1941. These injections result in the development of acute leukemia. However, the nature of this disease has not been well documented in the literature. Here, we provide a technical protocol for characterizing C1498 cells in vitro and for determining the nature of the induced leukemia in vivo. The first part of this procedure is focused on determining the hematopoietic lineage and the stage of differentiation of cultured C1498 cells. To achieve this, multi-parametric flow cytometric staining is used to detect hematopoietic cell markers. Immunofluorescence microscopy, cytochemistry and a May-Grünwald Giemsa staining are then performed to assess the expression of myeloperoxidase, the activity of esterases and cellular morphology, respectively. The second part of this protocol is dedicated to describing the leukemia disease that is induced in vivo. The latter can be achieved by determining the frequencies of leukemic and inherent cells in the blood, hematopoietic organs (e.g., bone marrow and spleen) and non-lymphoid tissues (e.g., the liver and lungs) using specific staining and flow cytometry analyses. The nature of the leukemia is then confirmed using May-Grünwald Giemsa staining and staining for specific esterases in the bone marrow. Here, we present the results that were obtained using this protocol in age-matched C1498- and PBS-injected mice.
Keywords: Cancer Research, Issue 116, Acute myeloid leukemia, cell line, mice, bone marrow, spleen, thymus, flow cytometry, May-Grünwald Giemsa staining, immunofluorescence microscopy, esterases cytochemistry
Introduction
Acute myeloid leukemia (AML) is characterized by the uncontrolled proliferation of hematopoietic myeloid cells that are blocked at different stages of maturation. This dysregulation can affect the granulocytic, monocytic, erythrocytic or megaryocytic differentiation pathways1. AML cells accumulate in the bone marrow, leading to impaired hematopoiesis, which results in thrombopenia, lymphopenia and anemia. The leukemic cells also invade the blood and non-lymphoid organs.
The C1498 mouse model has been used for decades as a model for acute leukemia since cancer cells were isolated from a leukemic 10-month-old C57BL/6 (H-2b) female mouse in 1941. The literature describes the invasion into the blood, hematopoietic organs (e.g., the spleen and lymph nodes) and non-hematopoietic organs (e.g., the liver, lungs, ovaries, and kidneys) by highly proliferative C1498 cells after they were injected via an intravenous, subcutaneous or intra-peritoneal route into susceptible mice2-4. However, this mouse model was reported to induce either granulocytic2,5 or myelomonocytic6 leukemia. More recently, a study published in 2002 described this type of cancer as murine NKT cell leukemia7. Thus, the literature differs concerning the nature of this C1498 cell line and the associated leukemia it induces in mice. These discrepancies are mainly due to a lack of detailed and updated published information about the cells and the leukemic disease in general because many studies were performed in the 1950 - 70's.
Here we provide a detailed protocol to describe how to characterize C1498 cells and analyze the nature of the leukemic disease that is induced by their intravenous injection into mice. The first section of this protocol is dedicated to a description of C1498 cells that have been cultured in vitro. Fluorescent antibodies directed against surface and intracellular hematopoietic markers were used to determine their phenotype using flow cytometry. The presence of myeloperoxidase was assessed using immunofluorescence microscopy, their hematopoietic lineage and differentiation stage were evaluated using cytochemistry to assess the activity of esterases, and May-Grünwald Giemsa staining was performed. The C1498 cells were then injected into mice, and the acute leukemia disease that was induced is described in the second section of this manuscript. The same techniques were used to determine the frequencies and the phenotypes of leukemic and inherent cells in the bone marrow, peripheral blood, spleen and non-hematopoietic organs (the liver and lungs).
This protocol is highly reproducible, and the data presented here will help researchers to assess the effects of new therapeutic strategies. This leukemia mouse model has already been used to test immunotherapy approaches and different cancer chemotherapeutic drugs8,9. Their efficacy was evaluated by determining the evolution of tumor burden and survival rates. This protocol can be used to provide additional information about the distribution and subsistence of leukemic and other hematopoietic cell populations during treatment.
Protocol
Animal housing and all experimental procedures were approved by the local Animal Care Ethical Committee, CEEA.NPDC (agreement no.512012), and all experiments were performed in accordance with the French and European guidelines for the Care and Use of Laboratory Animals.
1. In vitro Characterization of the C1498 Cell Line
- In vitro culture of C1498 cells
- Prepare complete RPMI (Roswell Park Memorial Institute) 1640 medium by adding 50 ml of fetal bovine serum (FBS), 5 ml of penicillin (100 U/ml)-streptomycin (100 µg/ml), 500 µl of 50 mM β-mercaptoethanol, 5 ml of N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES), 5 ml of non-essential amino acids and 5 mL of sodium pyruvate to 500 ml of RPMI medium.
- Grow the C1498 cell line in complete RPMI. Harvest the cells in suspension by pipetting, and transfer the cells to a 50 ml tube. Centrifuge at 350 x g for 10 min, and remove the supernatant.
- Add 20 ml of phosphate-buffered saline (PBS) (1x) solution, centrifuge at 350 x g for 10 min, and remove the supernatant.
- Resuspend the cells in 10 ml of Fluorescence-Activated Cell Sorter (FACs) buffer (2.5 g of bovine serum albumin (BSA) powder and 2 ml of 0.5 M ethylenediaminetetraacetic acid (EDTA) solution in 500 ml of PBS solution). Count the cells using a Thoma cell counting chamber after staining the cells with trypan blue.
- Phenotypic characterization of the C1498 cell line using immunostaining and flow cytometry analysis
- Cell surface staining
- Prepare FACs buffer.
- Adjust the harvested cells in FACs buffer to 107 cells/ml and dispense 106 cells (in 100 µl) for each staining experiment into flow cytometry tubes.
- Label the cells with 100 µl of the following antibodies or their associated isotype controls diluted in FACs buffer:
- For markers of precursor and differentiated cells, label the cells with anti-CD11b/anti-CD18 (1), anti-Ly-6G (1), anti-CD19, anti-B220 (2), anti-NK1.1, anti-CD49b, anti-CD4 (1), anti-CD8 (2), anti-CD3 (3), anti-CD21/35, anti-CD115 and anti-TCRVβ antibodies.
- For hematopoietic stem/progenitor cells markers, use a combination of anti-CD34/anti-CD117/anti-Sca-1, anti-CD150/anti-CD117/anti-Sca-1, anti-CD117/anti-CD127 or anti-CD16/32-biotin antibodies alone.
- For markers of cell functions (e.g., adhesion, antigen presentation, co-stimulatory molecules and receptors), stain the cells with anti-CD18 (2)/anti-CD11a, anti-MHC class I, anti-MHC class II, anti-CD31, anti-CD44, anti-CD80-biotin, anti-CD86, and anti-CD274 antibodies.
- Incubate all of the flow cytometry tubes at 4 °C for 30 min.
- Wash the cells twice by adding 2 ml of FACs buffer to each tube, centrifuge at 350 x g for 5 min, and remove the supernatant.
- Add 100 µl of FACs buffer to each tube and proceed to secondary staining by adding 100 µl of fluorescent streptavidin (1/100 in FACs buffer for a final dilution of 1/200) to the biotinylated-conjugated antibodies. Incubate the tubes at 4 °C for 30 min.
- Wash the cells twice as follows: add 2 ml of FACs buffer to each tube, centrifuge the tubes at 350 x g for 5 min, and remove the supernatant by pipetting.
- Resuspend the cells in 500 µl of cold PBS and place the cells on ice, keeping them in the dark by using aluminum foil to cover the tubes. Analyze results using a cytometer10.
- Intracellular staining
- Prepare fixation buffer by adding 125 ml of a 4% paraformaldehyde (PFA) solution to 375 ml of PBS solution. NOTE: To prepare 500 ml of 4% PFA, heat 400 ml of PBS solution to approximately 60 °C on a stir plate in a ventilated hood. Add 20 g of PFA powder, and raise the pH until the PFA is dissolved. Allow the solution to cool, adjust the pH to 6.9, and make up the volume to 500 ml with PBS.
- Prepare the permeabilizing buffer by adding 0.5 g of saponin and 0.5 g of BSA to 500 ml of PBS solution.
- Adjust the harvested cells in FACs buffer to 107 cells/ml and distribute 106 of the cells (100 µl) for each staining experiment into flow cytometry tubes. Centrifuge the cells at 350 x g for 5 min, and remove the supernatant.
- Fix the cells in 200 µl of 1% PFA solution and incubate for 10 min at 4 °C.
- Add 2 ml of permeabilizing buffer to each tube, centrifuge the tubes at 350 x g for 5 min, and remove the supernatant by pipetting. Add 100 µl of permeabilizing buffer to each tube.
- Label the cells with 100 µl of the following antibodies or their corresponding isotype controls after diluting them in permeabilizing buffer: anti-CD3 (2)/anti-CD8 (1), anti-CD3 (3)/anti-CD4 (2), anti-CD107b and anti-CD3 (3)/anti-TCRVβ.
- Incubate the cells at 4 °C for 30 min.
- Wash the cells twice by adding 2 ml of permeabilizing buffer to each tube. Centrifuge the tubes at 350 x g for 5 min, and remove the supernatant.
- Add 100 µl of permeabilizing buffer to the cells. Proceed to secondary staining by adding 100 µl of fluorescent streptavidin diluted in permeabilizing buffer to biotinylated-conjugated antibodies. Incubate the tubes at 4 °C for 30 min.
- Add 2 ml of permeabilizing buffer to each tube, centrifuge the tubes at 350 x g for 5 min, and remove the supernatant. Repeat this step one more time.
- Resuspend the cells in 500 µl of cold PBS, and then place the cells on ice in the dark. Analyze the results using a flow cytometer10.
- Preparation of a cell suspension on slides for microscopy
- Wash 106 of the harvested C1498 cells (obtained in step 1.1.4) with 5 ml of cold FACs buffer twice, and dilute the cells in 1 ml of cold FACs buffer. Place the tubes on ice.
- Place slides into disposable chambers with pre-attached filter cards and place these into a cytocentrifuge.
- Add 100 µl of FACs buffer to each chamber and filter card, and spin them for 2 min at 4.52 x g.
- Add 100 µl of cells to each chamber and filter card, and spin the cells at 4.52 x g for 2 min.
- Carefully remove the slides from the chambers and air-dry the slides before staining them with myeloperoxidase (step 1.4), esterases (step 1.5) or May-Grünwald Giemsa (step 1.6).
- Myeloperoxidase staining for immunofluorescence
- Fix the cells on the slides by immersing the slides in a cold methanol:acetone (1:1) solution for 2 min, and then air-dry the slides.
- To confine the liquid to the area of the slide containing the cells, draw a circle around the cells using a water-repellent pen.
- Rinse the cells in 200 µl of cold PBS solution for 10 min.
- Block the cells in 200 µl of 3% BSA/PBS buffer containing 10 µl of normal donkey serum and 10 µg/ml of purified anti-CD16/32 antibodies.
- Apply 200 µl of the anti-mouse myeloperoxidase (diluted to 20 µg/ml in 3% BSA/PBS buffer). Incubate the cells O/N at 4 °C in a humidity chamber.
- Wash the cells with 200 µl of cold 0.1% BSA/PBS.
- Apply 200 µl of anti-goat IgG antibodies diluted at 1/250 in 3% BSA/PBS buffer. Incubate the cells for 2 hr at RT in a humidity chamber.
- Wash the cells 3 times with 200 µl of 0.1% BSA/PBS buffer and twice in cold PBS.
- Stain the cell nuclei with 200 µl of Hoechst diluted at 1/1,000 in PBS (to a final concentration at 1 µg/mL) for 2 min at RT.
- Wash the slides with water and allow them to air-dry before mounting. Apply one drop of mounting medium 1 to the cells, place one edge of a cover-glass onto the slide, and carefully lower it onto the cells using forceps. Press gently on the cover-glass to remove any air bubbles.
- Esterase cytochemistry Note: Pre-warm all reagents to RT.
- Fixative preparation
- To prepare the Citrate-Acetone-Formaldehyde (CAF) solution, add 2.5 ml of citrate solution, 6.5 ml of acetone and 0.8 ml of 37% formaldehyde to a glass bottle. Mix gently and store at 4 °C.
- Naphthol AS-D Chloroacetate esterase (CAE) activity assay
- Warm deionized water to 37 °C.
- In a 50 ml tube, add 1 ml of sodium nitrite solution to 1 ml of dye solution. Mix gently and let stand for 2 min. Add 40 ml of pre-warmed deionized water, 5 ml of pH 6.3 buffer concentrate and 1 ml of Naphthol AS-D Chloroacetate solution. Mix and transfer into a Coplin jar.
- Fix the cells onto the slides (see section 1.3) for 30 sec with CAF solution (see step 1.5.1.1), and wash the slides for 45 sec with deionized water.
- Transfer the slides into the solution that was prepared in step 1.5.2.2, and incubate the slides at 37 °C for 30 min in a humidity chamber protected from light.
- Dry the slides and then rinse them via immersion in deionized water for 2 min.
- Counterstain the cells by adding few drops of Hematoxylin solution and incubating them for 1 min.
- Wash the slides with neutral water (pH 7) and allow them to air-dry. Apply one drop of mounting medium 2 to the cells, place one edge of a cover-glass onto the slide and carefully lower it onto the cells using forceps. Press gently on the cover-glass to remove any air bubbles.
- Alpha-Naphthyl butyrate esterase (NBE) activity assay
- Warm α-naphthyl butyrate solution to 37 °C before use.
- Dilute one tablet of sodium nitrite in 6.25 ml of deionized water.
- In a 50 ml tube, add 1.5 ml of a sodium nitrite tablet solution and 1.5 ml of a Pararosaniline solution. Mix gently and allow the solution to stand for 5 min. Supplement the solution with 40 ml of phosphate-buffered solution. Bring to pH 6 by carefully adding 10 N NaOH dropwise. Add 5 ml of the α-naphthyl butyrate solution, mix the entire solution, and transfer it into a Coplin jar.
- Fix the cells onto the slides for 10 sec using the CAF solution at RT and rinse for 45 sec with deionized water.
- Transfer slides into the Coplin jar containing the solution that was prepared in step 1.5.3.2 and incubate together for 1 hr at 37 °C in a humidity chamber while protected from light.
- Rinse the slides for 2 min in neutral water (pH 7) and air-dry.
- Counterstain the cells with Methylene blue solution by adding few drops on the slide and incubate for 4 min.
- Immerse the slides in deionized water for 2 min and allow them to air-dry. To mount the slides, apply one drop of mounting medium 2 to the cells, place one edge of the cover-glass onto the slide, and carefully lower it onto the cells using forceps. Press gently on the cover-glass to remove any air bubbles.
- May-Grünwald Giemsa (MGG) staining
- Stain the cells (prepared in section 1.3) by immersing the slides into a Coplin jar containing May-Grünwald solution for 3 min.
- Transfer the slides into a Coplin jar containing the pH 6.8 buffer solution for 1 min.
- Stain the slides by placing them into a Coplin jar containing the Giemsa R solution (diluted to 1/20 in pH 6.8 buffer solution) for 10 min. Wash the slides with neutral water (pH 7) for 10 sec.
- Drain and air-dry the slides. Mount the slides by applying one drop of mounting medium 2 onto the cells. Place one edge of the cover-glass onto the slide and carefully lower it onto the cells using forceps. Press gently on the cover-glass to remove any air bubbles.
2. In Vivo Development and Characterization of Acute Leukemia
NOTE: Four-week-old female congenic C57BL/6J-Ly5.1 mice were maintained under specific pathogen-free conditions (i.e., in a sterile environment). The mice were injected when they were between 5 and 6 weeks old.
- Intravenous injection with C1498 cells
- Harvest the cultured C1498 cells in suspensions by pipetting. Transfer the cells to a 50 mL tube and centrifuge at 350 x g for 10 min. Wash the cells in 10 ml of cold PBS twice, and prepare a cell suspension of 107 cells/ml in PBS. Place the cellular suspension on ice before performing the injection.
- Place the mouse in a restrainer and perform the injection under sterile conditions in a laminar flow hood.
- Use a 29G needle with a syringe to inject the cells into the tail vein. Grasp the tail at the distal end, and disinfect it with a gauze sponge soaked in 70% ethanol. Check to be sure that there are no air bubbles in the syringe, and then slowly inject 100 µl of the C1498 cell suspension (106 cells) into the tail vein.
- After the injection, remove the needle from the tail, and control any bleeding by applying pressure with a sterile gauze sponge at the injection site. Return the animal to its cage, and carefully check its health over the next hours and days.
- Retro orbital blood collection
- Monitor the behavior of the PBS- and C1498-injected mice for signs of leukemic disease (e.g., piloerection, isolation from the group, and reduced or no movements in the cage). NOTE: This usually occurs between 17 to 19 days after the cells are injected.
- Perform retro orbital blood collection just before euthanasia (see step 2.2.7) under sterile conditions in a laminar flow hood and under a heating lamp to prevent hypothermia.
- For anesthesia, use ketamine at 150 mg/kg and xylazine at 10 mg/kg. Prepare the anesthetic solution by diluting 1.5 ml of ketamine and 0.5 ml of xylazine in 18 ml of PBS solution.
- Anesthetize the control and leukemic mice. Proceed with an intraperitoneal injection of 200 µl of the anesthetic solution per 10 g of mouse using a 26G needle and a 1 ml syringe. Check for the loss of the pedal reflex to confirm anesthesia.
- Insert a capillary tube into the medial canthus of the eye. Blood will rise from the orbital sinus into the capillary tube. Control the bleeding by gently applying pressure onto the eye with a sterile gauze sponge. NOTE: A volume of 100 to 200 µl of blood can be collected using this technique.
- Collect the blood into an EDTA tube, and store the sample on ice before isolating the mononuclear cells.
- Euthanize the mouse using cervical dislocation, and proceed to isolate the organs (section 2.3).
- Organs and cells isolation
- Organs isolation
- Place the euthanized mouse onto its back on a plastic board and use needles to pin the animal's feet to facilitate organ isolation. Disinfect the mouse using 70% ethanol before performing an incision.
- Using sterile scissors, perform a ventral incision from the abdominal skin to the neck. Cut through the abdominal wall to access the liver. Cut through the rib cage and the diaphragm to access the lungs. Move the intestine to the side and remove the spleen using sterile scissors and forceps.
- To isolate the bone marrow, cut the legs at the top of the femurs above the joint using sterile scissors. Disconnect the tibia from the femur by pulling gently, and remove the skin and muscles from the bones using forceps and scissors.
- Place each organ and bone in a 50 ml tube containing cold PBS, and place them on ice.
- Isolation of cells from the organs
- Weigh the spleen before disrupting the cells. Mechanically disrupt the spleen, lungs and liver by pressing them through a 70 µm strainer using a syringe plunger in a 50 ml tube, and collect the cells into 30 ml of cold PBS.
- To collect bone marrow cells, put the femurs and tibias in a Petri dish on ice, cut the extremities using sterile scissors, and flush out the bone marrow by inserting a 26G needle attached to a 10 ml syringe containing 5 ml of cold PBS.
- Disrupt the bone marrow cells by passing the cell suspension through the needle/syringe, and filter the cell suspension through a 70 µm strainer in a 50 ml tube.
- Centrifuge all of the tubes containing each of the organs and the bone marrow cells at 350 x g for 10 min. Discard the supernatant, and resuspend the cells collected from the lungs and bone marrow in 2 ml of lysis buffer (1x) and the cells isolated from the liver and spleen in 5 ml of lysis buffer (1x) by gently pipetting the mixture up and down. Fill the tubes to 50 mL with cold PBS.
- Centrifuge the cells at 350 x g for 10 min. Resuspend the cells in FACs buffer for flow cytometry analysis or to prepare the cells for microscopy. Count the cells using a Thoma cell counting chamber after staining them with trypan blue.
- Cell surface staining of cells isolated from organs for flow cytometry analysis
- In a flow cytometry tube, label 106 cells that were isolated from organs with 10 µg/ml of purified anti-CD16/32 antibodies in 100 µl of FACs buffer.
- To 106 bone marrow cells, add 100 µl of the following antibodies or combinations of antibodies and their corresponding isotype controls diluted in FACs buffer: anti-CD11b/anti-CD3 (1)/anti-Ly6C/anti-Ly6G (2), anti-B220 (1)/anti-CD45.2/anti-CD19, anti-CD115/anti-CD3 (1)/anti-Ly6C/anti-Ly6G (2), anti-CD45.2, anti-Ly6G (2), anti-CD11b, anti-CD115 or anti-CD19 alone for compensation settings.
- To splenocytes, add 100 µl of the following antibodies and their corresponding isotype controls diluted in FACs buffer: a combination of anti-CD11b/anti-CD3 (1)/anti-Ly6C/anti-Ly6G (2), anti-B220 (1)/anti-CD45.2/anti-CD19, anti-CD45.2, anti-Ly6G (2), anti-CD11b or anti-CD19 for compensation settings.
- To lung and liver cells, add 100 µl of anti-CD45.2 antibodies and its corresponding isotype control diluted at 1/100 in FACs buffer.
- Incubate all of the cell solutions for 30 min at 4 °C.
- Wash the cells by adding 2 ml of FACs buffer to each tube. Centrifuge the tubes for 5 min at 350 x g and discarding the supernatant by pipetting. Repeat this step one more time.
- Resuspend the labeled cells in 500 µl of cold PBS. Keep the cells on ice and protected from light before performing flow cytometry for acquisition and analysis10.
- Isolation of blood mononuclear cells and immunofluorescent staining for flow cytometry analysis
- Before starting the protocol, pre-warm the separating solution to RT.
- Transfer the blood sample (100 to 200 µl; obtained from step 2.2) into a microcentrifuge tube, and add PBS/1 mM EDTA solution until the solution volume is 500 µl. Carefully layer 500 µl of separating solution under the solution containing the blood using a 30G needle and a 1 ml syringe. Do not mix the blood and the separating solution.
- Centrifuge the tubes at 800 x g (without brake) for 20 min at RT. After centrifugation, collect the cellular ring (the opaque white layer) using a pipette. Transfer the cells to a microcentrifuge tube. NOTE: The opaque white layer contains lymphocytes as well as monocytes and appears between the lower layer - the separating solution - and the upper layer.
- Add 1 ml of PBS solution, and centrifuge the tube at 350 x g for 10 min. Resuspend the cells in 600 µl of FACs buffer.
- Add 10 µg/ml of purified anti-CD16/32 antibodies and distribute 100 µl of the cell suspension into six separate tubes (100 µl each).
- Label the cells with 100 µl of the following antibodies or their associated isotype controls diluted in FACs buffer: a combination of anti-CD3 (1)/anti-B220 (1)/anti-CD45.2 and anti-Ly6C/anti-CD115/anti-CD45.2 or anti-CD45.2 and anti-CD115 alone for compensation settings.
- Incubate all of the tubes at 4 °C for 30 min.
- Wash the cells by adding 2 ml of FACs buffer to each tube, and then centrifuge the tubes for 10 min at 350 x g, and discard the supernatant using a pipette.
- Resuspend the labeled cells in 500 µl cold PBS. Keep the cells on ice and protected from light before performing flow cytometry acquisition and analysis10.
- Preparation of bone marrow cell suspensions on slides for microscopy
- Follow the steps described in section 1.3, but in step 1.3.1, use 105 bone marrow cells, and in step 1.3.4, spin the cells into each chamber at 72.26 x g for 10 min.
- Esterase activity assays using bone marrow cells
- To perform the bone marrow esterase cytochemistry assays, proceed from steps 1.5 to 1.5.3.7.
- May-Grünwald Giemsa staining of bone marrow cells
- To stain bone marrow cells, follow the protocol described in section 1.6, but in step 1.6.1, incubate the slides in May-Grünwald solution for 5 min.
Representative Results
To characterize the C1498 mouse model, we proceeded with two major steps. First, the C1498 cells were characterized to determine their hematopoietic lineage and maturation stage in vitro (Figure 1). These cells were then injected into congenic mice, and the nature of the induced leukemic disease was assessed to determine different features: leukemic cell infiltration, their phenotype, a quantification of the hematopoietic cells (mature and progenitors/precursors) in bone marrow, the frequencies of C1498 cells and mature hematopoietic cells in the blood and an evaluation of organ swelling (in the spleen, liver, and lungs) and cellular composition.
To characterize the C1498 cell phenotypes in vitro, the cells were labeled with antibodies directed against molecules that are expressed by hematopoietic precursors and mature cells (Table 1), and the results were analyzed using flow cytometry. The C1498 cells were positive for cell surface expression of Mac-1 (CD11b/CD18) (∼ 7%), B220 (> 25%), and they displayed intracellular expression of CD3ε, T-Cell Receptor (TCR) Vβ chains and Mac-3 (Figures 2A and B). The cells were negative for the cell surface markers Ly6G, Ly6C, CD115, CD21/CD35, CD19, CD3, CD4, CD8, NK1.1, and pan-NK molecules and for the intracellular expression of CD4 and CD8 (data not shown). They were then examined for markers of hematopoietic stem cells and progenitors (Table 1). They were also negative for cell surface expression of CD117, CD34, Sca-1, CD150 and CD16/32 (data not shown). These leukemic cells were then tested to determine the expression of adhesion, antigen presentation and co-stimulatory molecules. The cells expressed the surface markers LFA-1 (CD11a/CD18), CD44, CD31 (PECAM-1), and H-2Db and were negative for MHC class II, CD80, CD86 and CD274 (data not shown). C1498 cells therefore expressed both myeloid (Mac-1, Mac-3) and lymphoid markers (B220, CD3, TCR).
To better characterize their hematopoietic lineage, myeloperoxidase expression was assessed using immunofluorescence microscopy. All of the cells were positive for the myeloperoxidase, which verified their myeloid origin (Figure 3A). A majority of the cells also stained positive for α-naphthyl butyrate esterases (Figure 3B, left panel), and some of them stained for the naphthol AS-D chloroacetate esterases (black arrows) (Figure 3B, right panel). The results indicate that the cells contained mixtures of monocytic and granulocytic cells. After May-Grünwald Giemsa staining was performed, the C1498 cells were observed to display a blast-like morphology with a high nucleo-cytoplasmic ratio, 3 to 5 nucleoli in the nucleus, a perinuclear halo, numerous vacuoles and a basophilic cytoplasm (Figure 3C). Thus, the C1498 cell line is composed of monoblasts and myeloblasts.
The C1498 cells (CD45.2+) were then intravenously injected into CD45.1+ mice. The mice succumbed 17 to 19 days after the cells were injected. These mice were sacrificed so that their leukemia type could be analyzed before they died from the disease. The control mice, which were injected with PBS, were analyzed at the same time points for comparison. The C1498 cell-injected mice displayed massive infiltration of C1498 cells into their bone marrow, as demonstrated by the blast-like appearance of the cells after May-Grünwald Giemsa staining was performed (Figure 4A). They also preserved their monocytic and granulocytic phenotypes (Figure 4B and C), demonstrating an accumulation of monoblastic and myeloblastic cells that is characteristic of acute myelomonocytic leukemia.
To determine whether medullary hematopoietic cells numbers were lower following leukemic cells invasion, CD45.2+ C1498 cells, B lymphocytic, monocytic and granulocytic populations (including progenitors, precursors and mature cells), were quantified using immunofluorescent staining and multi-parametric flow cytometry analysis. Leukemic cells represented 16 to 36% of the hematopoietic cells (data not shown). The other cell types were all present in significantly lower numbers in the C1498-injected mice than in the PBS-injected mice (by 5-fold on average for B cell subsets, 4-fold on average for granulocytic cells and 3-fold on average for monocytic subsets) (Figure 5A to C).
An investigation of the frequencies of mononuclear cells in leukemic and control mouse blood samples showed that they contained a comparable percentage of lymphocytes (Figure 6A) but a higher frequency of monocytic and leukemic cells. These characteristics are representative of acute myelomonocytic leukemia11 (Figure 6B).
Among the other features of acute myelomonocytic leukemia12, the C1498-injected mice presented with swollen livers (hepatomegaly), lungs and spleens (splenomegaly) (Figure 7A). Various frequencies of CD45.2+ C1498 cells were detected in these organs using immunofluorescent staining and flow cytometry analysis (Figure 7B). As splenomegaly can result from high numbers of infiltrated monocytes, we also estimated the proportions of splenic populations. The numbers of cells in the B lymphocytic, monocytic and granulocytic cell fractions were significantly larger, by an average of 2-fold, 2.5-fold and 3-fold, respectively, in leukemic spleens than in control spleens (Figure 7C).
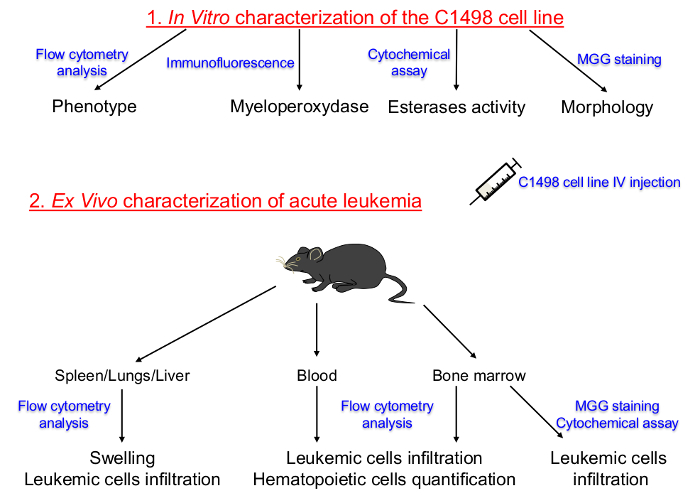 Figure 1. Schematic Representation of the Protocol Set Up for Characterizing in vitro Cultured C1498 Cell Lines and in vivo Descriptions of Acute Leukemia. The hematopoietic lineage and the differentiation stage of tissue-cultured C1498 cells were first determined. C1498 cells were then injected into congenic mice to induce the development of acute leukemia. The isolation of bone marrow, peripheral blood, spleen, liver and lung tissues was performed to determine the frequencies, phenotypes and morphological changes after the C1498 cells infiltration. IV: Intravenous MGG: May-Grünwald Giemsa.
Please click here to view a larger version of this figure.
Figure 1. Schematic Representation of the Protocol Set Up for Characterizing in vitro Cultured C1498 Cell Lines and in vivo Descriptions of Acute Leukemia. The hematopoietic lineage and the differentiation stage of tissue-cultured C1498 cells were first determined. C1498 cells were then injected into congenic mice to induce the development of acute leukemia. The isolation of bone marrow, peripheral blood, spleen, liver and lung tissues was performed to determine the frequencies, phenotypes and morphological changes after the C1498 cells infiltration. IV: Intravenous MGG: May-Grünwald Giemsa.
Please click here to view a larger version of this figure.
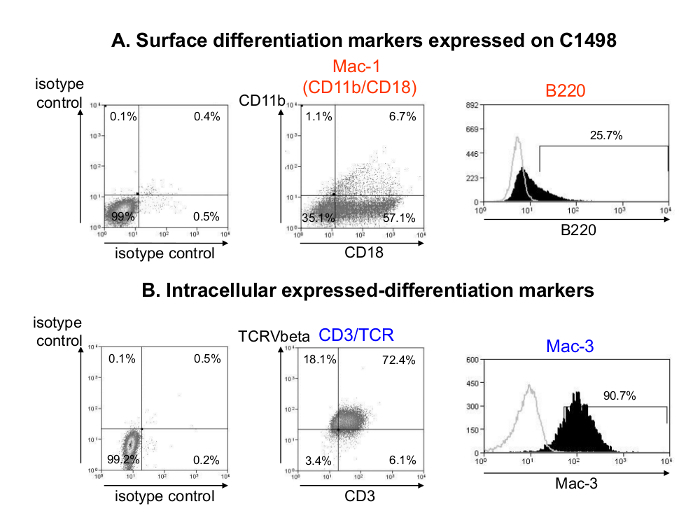 Figure 2. Phenotypic Analysis of C1498 Cells after in vitro Culture. Representative flow cytometry dot plots and histograms of cell surface (A) and intracellular (B) C1498-expressed molecules that were associated with hematopoietic mature cell differentiation are shown. C1498 cells were harvested from cultures, washed and labeled using fluorescent antibodies that were specific for the cell surface CD11b, CD18 and B220 markers or their isotype controls. For intracellular staining, the cells were fixed, permeabilized and labeled using antibodies directed against Mac-3, CD3ε, and a common epitope of the TCR (T-Cell Receptor) Vβ chain or their isotype controls. Analyses were performed using gating with live cells. Please click here to view a larger version of this figure.
Figure 2. Phenotypic Analysis of C1498 Cells after in vitro Culture. Representative flow cytometry dot plots and histograms of cell surface (A) and intracellular (B) C1498-expressed molecules that were associated with hematopoietic mature cell differentiation are shown. C1498 cells were harvested from cultures, washed and labeled using fluorescent antibodies that were specific for the cell surface CD11b, CD18 and B220 markers or their isotype controls. For intracellular staining, the cells were fixed, permeabilized and labeled using antibodies directed against Mac-3, CD3ε, and a common epitope of the TCR (T-Cell Receptor) Vβ chain or their isotype controls. Analyses were performed using gating with live cells. Please click here to view a larger version of this figure.
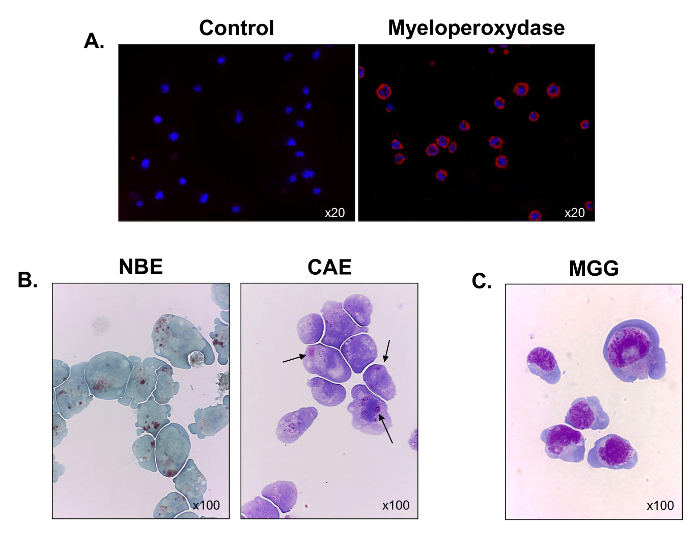 Figure 3. Functional and Morphological Characterization of Cultured C1498 Cells. C1498 cells were harvested from cultures and centrifuged on slides for microscopy. (A) Staining for myeloperoxidase expression was performed using immunofluorescence. (B) Cytochemical reactions were used to analyze the α-naphthyl butyrate esterase (NBE) and naphthol AS-D chloroacetate esterase (CAE) activities in C1498 cells. Cells were considered to be positive for each label when brown and red-purple, large cytoplasmic granules, respectively, were observed. (C) May-Grünwald Giemsa (MGG) staining of C1498 cells. For each staining experiment, the microscopy objective magnification is indicated. Each image is representative of three separate experiments. Please click here to view a larger version of this figure.
Figure 3. Functional and Morphological Characterization of Cultured C1498 Cells. C1498 cells were harvested from cultures and centrifuged on slides for microscopy. (A) Staining for myeloperoxidase expression was performed using immunofluorescence. (B) Cytochemical reactions were used to analyze the α-naphthyl butyrate esterase (NBE) and naphthol AS-D chloroacetate esterase (CAE) activities in C1498 cells. Cells were considered to be positive for each label when brown and red-purple, large cytoplasmic granules, respectively, were observed. (C) May-Grünwald Giemsa (MGG) staining of C1498 cells. For each staining experiment, the microscopy objective magnification is indicated. Each image is representative of three separate experiments. Please click here to view a larger version of this figure.
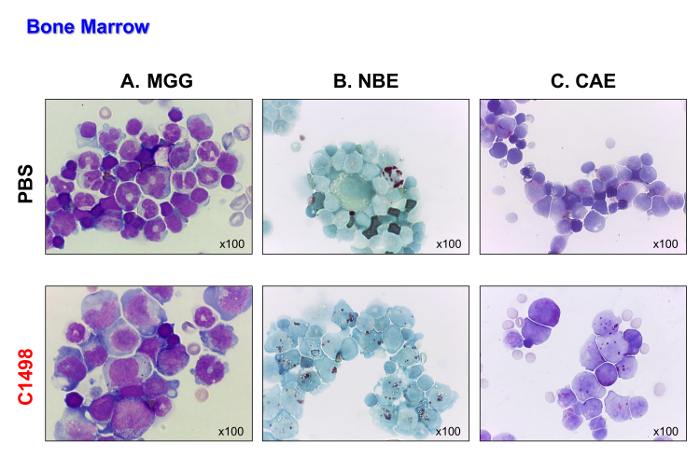 Figure 4. Bone Marrow Morphologies in PBS- and C1498-injected Mice. Bone marrow cells were isolated from PBS- and C1498 cell-injected mice and centrifuged onto slides for microscopy. (A) May-Grünwald Giemsa (MGG) staining. (B) α-naphthyl butyrate esterase (NBE) and (C) naphthol AS-D chloroacetate esterase (CAE) functions were evaluated using cytochemistry. In panel A, the band (immature) or segmented (mature) neutrophils are less visible in the bone marrow of the C1498-injected mice than the PBS-injected mice. Panel B and C indicate that there was an accumulation of monocytic and granulocytic cells in the leukemic bone marrow compared to the numbers observed in the control bone marrow. All microscopic analyses were performed using a 100X magnification objective. Please click here to view a larger version of this figure.
Figure 4. Bone Marrow Morphologies in PBS- and C1498-injected Mice. Bone marrow cells were isolated from PBS- and C1498 cell-injected mice and centrifuged onto slides for microscopy. (A) May-Grünwald Giemsa (MGG) staining. (B) α-naphthyl butyrate esterase (NBE) and (C) naphthol AS-D chloroacetate esterase (CAE) functions were evaluated using cytochemistry. In panel A, the band (immature) or segmented (mature) neutrophils are less visible in the bone marrow of the C1498-injected mice than the PBS-injected mice. Panel B and C indicate that there was an accumulation of monocytic and granulocytic cells in the leukemic bone marrow compared to the numbers observed in the control bone marrow. All microscopic analyses were performed using a 100X magnification objective. Please click here to view a larger version of this figure.
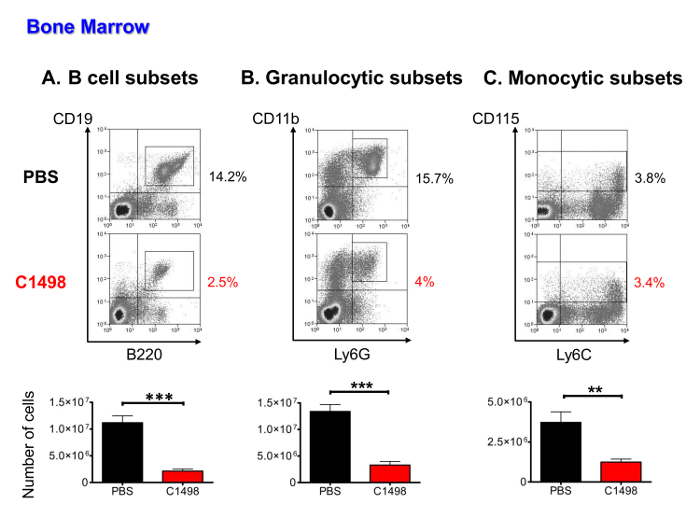 Figure 5. Quantitative Analysis of Medullary Populations in PBS- and C1498-injected Mice. Bone marrow cells were isolated from PBS- and C1498 cell-injected mice and estimated after cell counting was performed. The frequencies of the different cell populations were determined after immunostaining and live cell gated flow cytometry analysis. (A) The B cell subsets included CD19+B220+cells in stages from pro-B cells to mature B lymphocytes (B) granulocytic cells in the CD3- and CD11b+Ly6G+ lineages, which included precursors and immature and mature granulocytes. (C) The monocytic subsets were defined as CD3-CD115+ and included cells in the progenitor to mature monocyte stages. n = 7 mice/group, and the data are presented as histograms showing the means ± SEM. ***, p< 0.0001 and **, p< 0.01, unpaired Student's t-test comparing PBS- and C1498-injected mice. Please click here to view a larger version of this figure.
Figure 5. Quantitative Analysis of Medullary Populations in PBS- and C1498-injected Mice. Bone marrow cells were isolated from PBS- and C1498 cell-injected mice and estimated after cell counting was performed. The frequencies of the different cell populations were determined after immunostaining and live cell gated flow cytometry analysis. (A) The B cell subsets included CD19+B220+cells in stages from pro-B cells to mature B lymphocytes (B) granulocytic cells in the CD3- and CD11b+Ly6G+ lineages, which included precursors and immature and mature granulocytes. (C) The monocytic subsets were defined as CD3-CD115+ and included cells in the progenitor to mature monocyte stages. n = 7 mice/group, and the data are presented as histograms showing the means ± SEM. ***, p< 0.0001 and **, p< 0.01, unpaired Student's t-test comparing PBS- and C1498-injected mice. Please click here to view a larger version of this figure.
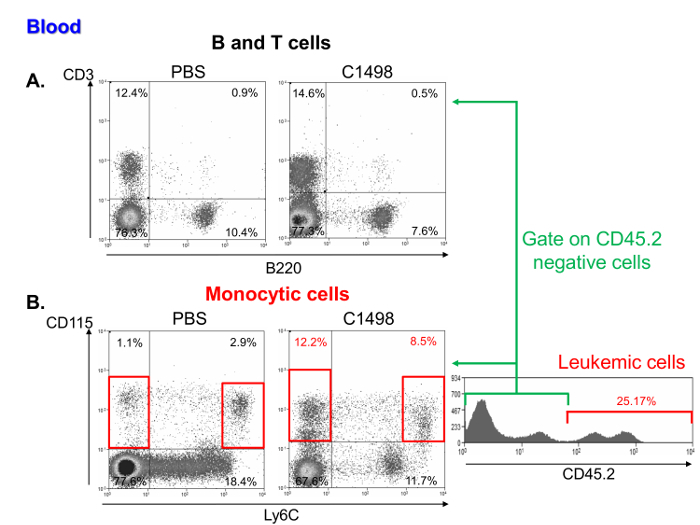 Figure 6. Blood Analysis of Mononuclear Cell Subsets in PBS- and C1498-injected mice. Representative flow cytometry dot plots of (A) T and B lymphocyte percentages, which were respectively defined as CD3+ and B220+ cells in PBS- and C1498 cell-injected mice. (B) Monocytic cell frequencies in C1498 leukemic and control (PBS) mice were determined by analyzing CD115+Ly6C- and CD115+Ly6Chigh cells. The analysis was performed by gating live cells. To compare leukemic and control mice, CD45.2+ C1498 cells were excluded. Please click here to view a larger version of this figure.
Figure 6. Blood Analysis of Mononuclear Cell Subsets in PBS- and C1498-injected mice. Representative flow cytometry dot plots of (A) T and B lymphocyte percentages, which were respectively defined as CD3+ and B220+ cells in PBS- and C1498 cell-injected mice. (B) Monocytic cell frequencies in C1498 leukemic and control (PBS) mice were determined by analyzing CD115+Ly6C- and CD115+Ly6Chigh cells. The analysis was performed by gating live cells. To compare leukemic and control mice, CD45.2+ C1498 cells were excluded. Please click here to view a larger version of this figure.
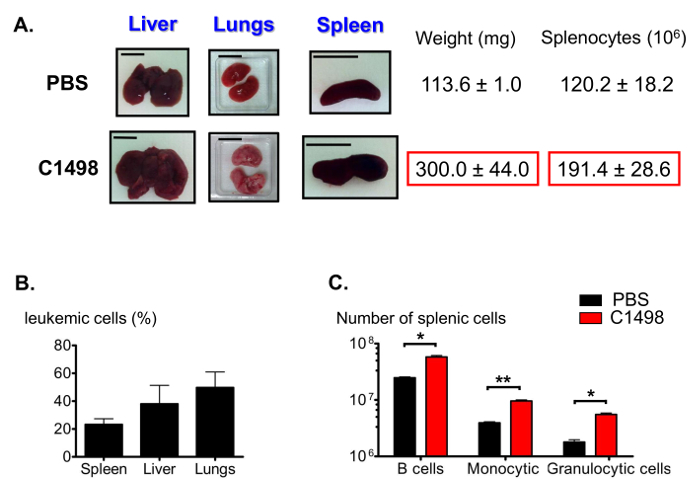 Figure 7. Estimation of Splenic Populations in Leukemic and Control Mice. (A) Representative photographs of liver, lung and spleen swelling in leukemic mice compared to control mice. Spleens were collected and weighed, and splenocytes were counted following tissue disruption. (B) Histogram representing leukemic cell frequencies in different organs after immunostaining was performed for CD45.2+ cells and the results were analyzed using flow cytometry. (C) Estimations of splenic B, granulocytic and monocytic cell numbers after immunostaining and flow cytometry analysis gating were performed to identify live CD19+B220+, CD3-CD11b+Ly6G+, CD3-CD11b+Ly6C- and CD3-CD11b+Ly6Chigh cells. The scale bars shown for the lungs, spleens and livers indicate 1 cm. n = 5 - 8 mice/group, and the data are represented in histograms as the means ± SEM. *, p< 0.05; **, p= 0.0033, unpaired Student's t-test comparing PBS- and C1498-injected mice. Please click here to view a larger version of this figure.
Figure 7. Estimation of Splenic Populations in Leukemic and Control Mice. (A) Representative photographs of liver, lung and spleen swelling in leukemic mice compared to control mice. Spleens were collected and weighed, and splenocytes were counted following tissue disruption. (B) Histogram representing leukemic cell frequencies in different organs after immunostaining was performed for CD45.2+ cells and the results were analyzed using flow cytometry. (C) Estimations of splenic B, granulocytic and monocytic cell numbers after immunostaining and flow cytometry analysis gating were performed to identify live CD19+B220+, CD3-CD11b+Ly6G+, CD3-CD11b+Ly6C- and CD3-CD11b+Ly6Chigh cells. The scale bars shown for the lungs, spleens and livers indicate 1 cm. n = 5 - 8 mice/group, and the data are represented in histograms as the means ± SEM. *, p< 0.05; **, p= 0.0033, unpaired Student's t-test comparing PBS- and C1498-injected mice. Please click here to view a larger version of this figure.
| Cell Type | Membrane or Intracellular Molecules |
| Precursors and Mature cells | |
| NK cells | NK1.1+, pan-NK+ |
| NKT cells | NK1.1+, pan-NK+, TCR Vbeta+(8.2), CD3+ |
| T lymphocytes | TCR Vbeta+, CD3+, CD4+, CD8+ |
| B cells precursors and B lymphocytes | B220+, CD19+, CD21/35+ |
| granulocytic precursors and granulocytes | Ly6G+, Mac-1+, CD11b+ |
| monocytic precursors and monocytes/macrophages | CD11b+, Mac-1+, Mac-3+, CD21/35+, CD115+, Ly6Chi |
| Progenitors | |
| multipotent progenitors | CD117+ Sca-1+ CD34+ (Lin- CD150-) |
| lymphoid-primed multipotent progenitors | CD117hi Sca-1hi CD127+ (Lin- ) |
| common lymphoid progenitors | CD117lo Sca-1lo CD127+ (Lin- ) |
| common myeloid progenitors | CD16/32lo CD117+ CD34int (Lin- Sca-1-) |
| granulocyte-macrophage progenitors | CD16/32hi CD117+ CD34hi (Lin- Sca-1-) |
| megakaryocyte-erythroid progenitors | CD16/32lo CD117+ CD34lo (Lin- Sca-1-) |
| Hematopoietic stem cells | CD117+ Sca-1+ CD150+ (Lin- CD34-) |
Table 1. Markers of Hematopoietic Cell Lineages and Differentiation. CD: cluster of differentiation; Lin: markers of mature cells; lo: low expression; hi: high expression; int: intermediate expression; NK: natural killer cells; TCR: T-cell receptor.
Discussion
In previous studies, the C1498 cell line was described as an inducer of acute granulocytic5, myelomonocytic6 or NKT7 cell leukemia. However, demonstrative data in the literature were either absent or incomplete. The protocol presented here uses different techniques, such as flow cytometry, immunofluorescence, MGG staining and cytochemical assays, to characterize cultured C1498 cells and to determine the nature of the leukemia that is induced in mice after they are injected.
When we phenotyped in vitro cultured C1498 cells after immunostaining and flow cytometry analyses were performed, we observed some limitations because these cells expressed few cell surface hematopoietic markers that have been previously described in the literature6,7. In agreement with our results, Labelle et al. did not observe the cell surface expression of mature TCR on C1498 cells using flow cytometry staining. However, they considered them to be a NKT cell line after they detected CD3ε and TCRVβ8.2 mRNAs7. We also observed the intracellular expression of TCRVβ chains and CD3ε molecules in most of the cells (> 70%), but their hematopoietic lineages could not be determined because there was also concomitant intracellular expression of the Mac-3 molecule.
Myeloperoxidase, MGG staining and assessments to analyze functional esterases using cytochemistry demonstrated that the C1498 cell line had a myeloid origin and was composed of monoblasts and myeloblasts. These results were concordant with the percentage of Mac-3+ cells that were obtained in flow cytometry staining analysis. Although not quantitative, these steps represent key experiments to be performed. Indeed, they remain, so far, the best existing methods for characterizing the lineage and differentiation stage of hematopoietic cells that express no or few specific phenotypic markers.
Flow cytometry staining was helpful for demonstrating the development of acute leukemia in congenic mice after C1498 cells were intravenously injected. The CD45.2+ C1498 cells that infiltrated into the peripheral blood and various organs were isolated, and their frequencies were determined. Quantification was also performed to analyze inherent medullary and splenic cells after immunophenotyping. Limitations were encountered when the C1498 cell phenotype was examined in organs as they expressed few hematopoietic markers (only a few of them were B220+). To define the nature of the observed acute leukemia, May-Grünwald Giemsa staining and an analysis of the activities of monocytic and granulocytic esterases were performed using bone marrow. The results showed that C1498 cells preserved their myeloblastic and monoblastic morphology and function, revealing the onset of myelomonocytic leukemia.
In consideration for the critical steps described in this protocol, particular attention should be given to pH when performing cytochemical reactions and MGG staining because errors in pH can lead to incorrect interpretations of results. For instance, α-naphthyl butyrate esterase activity is specific to monocytic cells only at a pH of 6.0 because granulocytes and lymphocytes can also stain positive for this test at higher pH values. Fixating the cells is not recommended before performing MGG staining, and we showed that only CAF fixation provided satisfactory results when performing esterases cytochemical reactions using C1498 cells. To preserve the expression of the CD115 molecule and its detection by flow cytometry, all of the samples (e.g., blood, bone marrow, and spleen cells) should be kept on ice during the procedure. If no staining is observed in flow cytometry or/and immunofluorescence experiments, the reference of the antibodies, their storage recommendations and their dilutions should be checked. The references specified in the materials/equipment table have been selected for flow cytometry or immunofluorescence applications. The primary/secondary antibodies or their conjugated fluorophores might have lost their activity due to inappropriate storage (e.g., exposure to light or heat), improper dilution, extensive freezing/thawing or the use of contaminated buffers. Run positive controls to ensure that they are working properly. Use mouse bone marrow or spleen-derived cells that are known to express the proteins of interest. To avoid high background and non-specific staining, make sure that the cells are washed properly and kept at high humidity (for immunofluorescence) and that the antibodies are diluted as instructed. Use the same concentration and dilution for the isotype control antibody and the primary antibody to accurately determine the background level in the sample. For esterase cytochemistry experiments, the reagents can be tested by using positive and negative control slides containing purified mouse splenic granulocytic (Ly6G+) and monocytic (CD115+) cells.
The procedure described in this study showed that many of the leukemic features observed in mice after the injection of C1498 cells shared common hallmarks with human acute myelomonocytic leukemia11,12. The invaded leukemic cells resulted in a reduction of mature and immature (progenitors and precursors) medullary hematopoietic cells. C1498 cells are present at high frequency (> 20%) in the peripheral blood, as are monocytic cells. Hepatomegaly and splenomegaly were observed to result from the infiltration of leukemic cells, and significant increases in B lymphocytes and myeloid cells were also observed to accompany splenomegaly. Thrombopenia was also observed when blood platelets numbers were estimated using a hematology analyzer.
It was shown, using in vitro experiments, that C1498 cells inhibit normal murine hematopoiesis by secreting soluble factors13. In several tumor mouse models, immature myeloid cells (including monocytic and granulocytic cells) have also been shown to migrate from the bone marrow to the spleen, where they inhibit anti-tumor specific T cell activation and proliferation14. Thus, the reduction in hematopoietic cells that was observed in the bone marrow could have resulted from either a deficiency in hematopoiesis and/or from their emigration. This latter mechanism could explain the presence of monocytosis in the peripheral blood or the observation of enlarged myeloid fractions in the spleen. It is also conceivable that these cells could have been derived from improved splenic hematopoiesis. Indeed, under steady-state conditions, some subsets of splenic B cells were identified as precursors of mature B lymphocytes15. Moreover, under inflammatory conditions, medullary stem and progenitors cells have been shown to relocate to the spleen to induce the production of mature monocytes16. This protocol does not allow us to draw conclusions regarding the mechanisms that are involved in the development of leukemia, and additional functional as well as molecular assays should be employed to do so. However, these data include detailed information about the clinical features of acute myelomonocytic leukemia and will help researchers to evaluate and understand the effects of new therapeutic agents.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors would like to acknowledge the "Ligue Nationale contre le Cancer" (Comité du Septentrion), the SIRIC ONCOLille (Grant INCa-DGOS-INSERM 6041) and the Institut pour la Recherche sur le Cancer de Lille (IRCL) for supporting this work. They would like to thank Delphine Taillieu and the animal facility staff for housing the mice and maintaining their welfare. We also thank Raphaëlle Caillerez and Nathalie Jouy for their respective help in microscopy and flow cytometry.
References
- Forthun RB, Hinrichs C, Dowling TH, Bruserud Ø, Selheim F. The past, present and future subclassification of patients with acute myeloid leukemia. Curr. Pharm. Biotechnol. 2016;17(1):6–19. doi: 10.2174/1389201016666150907113653. [DOI] [PubMed] [Google Scholar]
- Goldie H, Butler CH, Anderson MM, Maxwell MC, Hahn PF. Growth characteristics of free C1498 (granulocytic leukemia) tumor cells in the peritoneal fluid and the blood of C57 mice. Cancer Res. 1953;13(2):125–129. [PubMed] [Google Scholar]
- Tanaka KK, Roberts E. Biological studies of E.L.4 lymphoma and C Leukemia in susceptible (C57BL) and resistant (B10.D2) mice. Cancer Res. 1964;24(1498):1785–1797. [PubMed] [Google Scholar]
- Law LW. Characterization of an influence affecting growth of transplantable leukemias in mice. Cancer Res. 1944;4:257–260. [Google Scholar]
- Graham JD, McMahon Welch C, Patchen ML. Studies of an implanted murine myelogenous leukemia C1498. Ohio J. Sci. 1975;75(4):202–208. [Google Scholar]
- Boyer MW, Orchard PJ, Gorden KB, Anderson PM, Mclvor RS, Blazar BR. Dependency on intercellular adhesion molecule recognition and local interleukin-2 provision in generation of an in vivo CD8+ T-cell immune response to murine myeloid leukemia. Blood. 1995;85(9):2498–2506. [PubMed] [Google Scholar]
- Labelle JL, Truitt RL. Characterization of a murine NKT cell tumor previously described as an acute myelogenous leukemia. Leuk. Lymphoma. 2002;43(8):1637–1644. doi: 10.1080/1042819021000002974. [DOI] [PubMed] [Google Scholar]
- Bradner WT, Pindell MH. Myeloid leukemia as a screen for cancer chemotherapeutic agents. Cancer Res. 1966;26(4):375–390. Pt 2. [PubMed] [Google Scholar]
- Lin JM, Li B, Rimmer E, VanRoey M, Jooss K. Enhancement of the anti-tumor efficacy of a GM-CSF-secreting tumor cell immunotherapy in preclinical models by cytosine arabinoside. Exp. Hematol. 2008;36(3):319–329. doi: 10.1016/j.exphem.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Robinson JP. Handbook of Flow Cytometry Methods. New York, NY: Wiley-Liss, Inc; 1993. [Google Scholar]
- Xu Y, McKenna RW, Wilson KS, Karandikar NJ, Schultz RA, Kroft SH. Immunophenotypic identification of acute myeloid leukemia with monocytic differentiation. Leukemia. 2006;20(7):1321–1324. doi: 10.1038/sj.leu.2404242. [DOI] [PubMed] [Google Scholar]
- Hassan K, Qureshi M, Shafi S, Ikram N, Akhtar MJ. Acute myeloid leukemia-FAB classification and its correlation with clinico-haematological features. J. Pak. Med. Assoc. 1993;43(10):200–203. [PubMed] [Google Scholar]
- Quesenberry PJ, Rappeport JM, Fountebouni A, Sullivan R, Zuckerman K, Ryan M. Inhibition of normal murine hematopoiesis by leukemic cells. N.Engl.J. Med. 1978;299(2):71–75. doi: 10.1056/NEJM197807132990204. [DOI] [PubMed] [Google Scholar]
- Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008;181(8):5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loder F, et al. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J. Exp. Med. 1999;190(1):75–89. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins CS, et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125(2):364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]