

Abstract
Protein-protein interactions play an essential role in the function of a living organism. Once an interaction has been identified and validated it is necessary to characterize it at the structural and mechanistic level. Several biochemical and biophysical methods exist for such purpose. Among them, fluorescence anisotropy is a powerful technique particularly used when the fluorescence intensity of a fluorophore-labeled protein remains constant upon protein-protein interaction. In this technique, a fluorophore-labeled protein is excited with vertically polarized light of an appropriate wavelength that selectively excites a subset of the fluorophores according to their relative orientation with the incoming beam. The resulting emission also has a directionality whose relationship in the vertical and horizontal planes defines anisotropy (r) as follows: r=(IVV-IVH)/(IVV+2IVH), where IVV and IVH are the fluorescence intensities of the vertical and horizontal components, respectively. Fluorescence anisotropy is sensitive to the rotational diffusion of a fluorophore, namely the apparent molecular size of a fluorophore attached to a protein, which is altered upon protein-protein interaction. In the present text, the use of fluorescence anisotropy as a tool to study protein-protein interactions was exemplified to address the binding between the protein mutated in the Shwachman-Diamond Syndrome (SBDS) and the Elongation factor like-1 GTPase (EFL1). Conventionally, labeling of a protein with a fluorophore is carried out on the thiol groups (cysteine) or in the amino groups (the N-terminal amine or lysine) of the protein. However, SBDS possesses several cysteines and lysines that did not allow site directed labeling of it. As an alternative technique, the dye 4',5'-bis(1,3,2 dithioarsolan-2-yl) fluorescein was used to specifically label a tetracysteine motif, Cys-Cys-Pro-Gly-Cys-Cys, genetically engineered in the C-terminus of the recombinant SBDS protein. Fitting of the experimental data provided quantitative and mechanistic information on the binding mode between these proteins.
Keywords: Biochemistry, Issue 116, Fluorescence anisotropy, Fluorescence, Elongation factor-like 1, Shwachman-Diamond Syndrome protein, Protein-protein interaction, Fluorophore
Introduction
Cells contain a multitude of biomacromolecules that constantly interact with each other. This association gives rise to complexes that participate in the cellular pathways responsible for their functioning in signal transduction, regulation of gene expression and cell migration amongst others. All protein-protein interactions that occur in a cell comprise a network known as the interactome. In Saccharomyces cerevisiae more than 70% of its proteins have been shown to have interacting partners 1. Understanding the interactome of a cell and their functions provide relevant information on the complexity and diversity of living organisms. Several methodologies have been described to identify and characterize protein-protein interactions. Different high through put methods such as yeast two-hybrid 2, protein-fragment complementation assays 3, affinity purification 4 coupled to mass spectrometry and protein microarrays are used to identify an interaction 5,6. Once identified, it is necessary to validate it and this may vary on a case-by-case basis. Typically, these experiments involve disrupting the interaction itself at the level of the individual members of the interaction pair, e.g., by gene deletion or overexpression of one of the proteins, and then looking for changes in the properties or function of the other member at the cellular level. Subsequently, biophysical techniques 7 are used to characterize the protein-protein interaction at the molecular level. To this end, the structure of protein complexes are determined by X-ray crystallography, nuclear magnetic resonance and cryo-electron microscopy while calorimetry and fluorescence spectroscopy are used to quantitatively and mechanistically describe them.
In this work, fluorescence anisotropy was used as a technique to characterize the interaction between the GTPase EFL1 and the SBDS protein. These proteins participate in the synthesis of ribosomes by promoting the release of eukaryotic initiation factor 6 from the surface of the 60S ribosomal subunit 8. The SBDS protein is mutated in a disease known as the Shwachman-Diamond Syndrome 9 and acts as a guanine nucleotide exchange factor for EFL1 decreasing its affinity for guanosine diphosphate 10,11. Disease mutations in SBDS abolish the interaction with EFL1 and thus prevent its activation.
Fluorescence anisotropy is commonly used in biological applications to study protein-peptide or protein-nucleic acid interactions. It is based on the principle that a fluorophore excited with polarized light results in a partially polarized emission. Fluorescence anisotropy is defined by Equation 1:
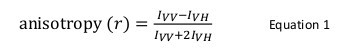
where IVV and IVH are the fluorescence intensities of the vertically (VV) and horizontally (VH) polarized emission when the sample is excited with vertically polarized light 12. Fluorescence anisotropy is sensitive to factors that affect the rate of the rotational diffusion of the fluorophore and thus depends on the temperature, the viscosity of the solution and the apparent molecular size of the fluorophore. The apparent size of a protein containing a fluorophore increases when it interacts with another protein and such change can then be evaluated as a change in anisotropy. More specifically, a fluorophore that rotates slowly in solution relative to its fluorescent lifetime will have a large IVV value and small IVH value and therefore will exhibit a relatively large anisotropy. For fluorophores that tumble rapidly relative to their fluorescent lifetime, IVV and IVH will be similar and their anisotropy value will be small 12 (Figure 1). In addition, for a good anisotropy signal to noise measurement, it is necessary to have a fluorophore with a fluorescence lifetime similar to the rotational correlation time of the molecule of interest. Otherwise, it is not possible to accurately record the difference in anisotropy between the free protein and that in the complex. For example, the anisotropy of a fluorescent probe with a lifetime close to 4 nsec such as fluorescein or rhodamine attached to a low molecular weight compound of 100 Da is 0.05. Binding to a molecule of 160 kDa will increase its anisotropy value to 0.29; a difference that can be accurately measured. In contrast, the same fluorescent probe involved in a binding reaction whose increase in molecular size varies from 65 to 1,000 kDa will only result in an anisotropy change of 0.28 to 0.3, which is too small to be accurately measured. In this scenario, a probe with a lifetime of 400 nsec would be more suitable 12.
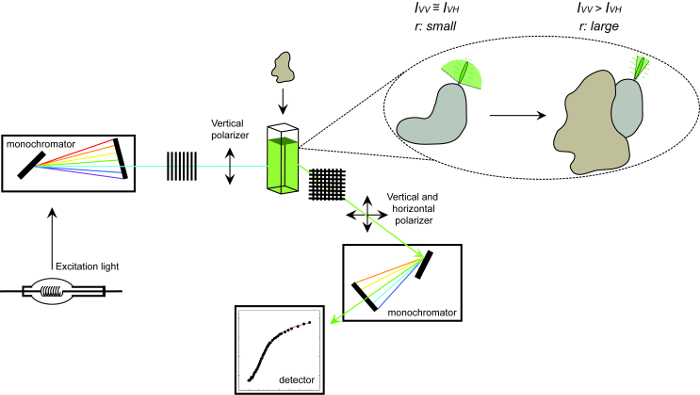 Figure 1. Schematic representation of the equipment used to measure fluorescence anisotropy and the procedure. Schematic representation of the equipment used to perform a protein-protein interaction experiment measuring fluorescence anisotropy. Fluorophores that tumble fast display small anisotropy that increases upon binding to an interaction partner. Please click here to view a larger version of this figure.
Figure 1. Schematic representation of the equipment used to measure fluorescence anisotropy and the procedure. Schematic representation of the equipment used to perform a protein-protein interaction experiment measuring fluorescence anisotropy. Fluorophores that tumble fast display small anisotropy that increases upon binding to an interaction partner. Please click here to view a larger version of this figure.
Fluorescence applications require the presence of a fluorophore in any of the molecules studied. To study protein-protein interactions there are three type of fluorophores: 1) the tryptophan residues present in the proteins, 2) chemically attached fluorophores and 3) fluorescent fusion partners such as green fluorescent protein (GFP) and its derivatives. Most proteins have tryptophan residues on its structure, thus the easiest way to measure an interaction is by monitoring the changes in the corresponding fluorescence spectra or by monitoring changes in the fluorescence intensity of the tryptophan residues. However, tryptophan residues may be present in both proteins complicating the analysis. On the other hand, for a fluorophore to change its fluorescent properties due to an interaction it needs to be located on or near the binding site and it could interfere with the interaction itself. This needs special attention when using bulky fluorophores such as GFP. If none of these fluorophores can be used for binding studies it is necessary, then, to introduce extrinsic fluorophores to the one of the proteins involved. Many chemically synthesized fluorophores exist and can be covalently attached to proteins through their reactive groups such as the amine groups (side chain of lysines or N-terminus) and the thiol groups in cysteine. Fluorophore derivatives with isothiocyanate and succinimidyl esters react with amide groups while iodoacetamide and maleimide are thiol-reactive groups 13. The most common dyes used in fluorescence applications are derivatives of the fluorescein and the rhodamine green dyes, coumarins, BODIPY fluorophores and Alexa Fluor dyes. A detailed list of commercially available fluorophores and their use can be found in references 14,15. For successful labeling, the reactive group must be exposed on the surface of the protein, but due to the large number of reactive functional groups typically present in polypeptides it is very hard to get site-specific modification. The protein of interest in this study, SBDS, contains 5 free cysteines and 33 lysines that may result in multiple site labeling. Non-uniform labeling may affect the binding and will complicate data analysis as different fluorophore molecules may elicit different fluorescent intensity signals upon binding. To overcome this problem, we used the FlAsH fluorophore, 4',5'-bis(1,3,2 dithioarsolan-2-yl) fluorescein to site-direct label the SBDS protein. This is an arsenoxide dye with a high affinity for four spaced cysteines in a motif know as FlAsH-tag consisting of the sequence CCXXCC where X is any amino acid other than cysteine 16,17. This tetracysteine motif is added to the N- or C-terminus of the protein by genetic engineering together with an appropriate linker to prevent the disruption of the overall fold of the protein. The pair consisting of FlAsH dye and FlAsH-tag was originally designed to site-specific label proteins in living cells 17 but it can also be used to label purified proteins in vitro as it is exemplified here. Additionally, enzymatic strategies have also been developed to enable site-specific functionalization of proteins 18.
In this manuscript we describe the usefulness of fluorescence anisotropy as a tool to study protein-protein interactions. Binding can be assessed by simple inspection of the binding curve shape while quantitative information can be obtained from the fit of the experimental data.
Protocol
1. SBDS-FlAsH Tag Protein Expression and Purification
NOTE: For the anisotropy experiments, a FlAsH-tag corresponding to the sequence Cys-Cys-Pro-Gly-Cys-Cys was added to the C-terminus of the human SBDS coding sequence by PCR. This construct was subcloned into the expression vector pRSET-A and transformed into Escherichia coli C41 cells to express a protein encoding a N-terminal hexahistidine tag (His-tag), the human SBDS coding sequence and a C-terminus FlAsH tag 10.
- SBDS-FlAsH protein expression
- Transform competent E. coli C41 cells with the plasmid pRSET-HisSBDS-FlAsH using a standard heat shock protocol 19. Plate the cells in solid Luria-Bertani (LB) media supplemented with 100 µg/ml ampicillin. LB solid media composition consists of 10 g NaCl, 5 g yeast extract, 10 g tryptone and 20 g agar for 1 L volume.
- Culture transformed bacteria at 37 °C until absorbance at 600 nm (A600) reaches 0.5-0.7 in 1 L of LB liquid media supplemented with 100 µg/ml ampicillin.
- Induce protein expression by adding 0.5 mM isopropyl β-D-1-thiogalactopyranoside to the culture and continue the incubation for further 5 hr.
- Collect the bacterial suspension by centrifugation at 3,800 x g for 10 min at 4 °C. Remove the supernatant. At this point, either store the cell pellet at -20 °C or use immediately for protein purification.
- SBDS-FlAsH protein purification NOTE: All chromatographic steps are performed using a fast protein liquid chromatography (FPLC) system or a peristaltic pump. Ni2+-affinity chromatography uses a 5 ml fast flow column. Anionic exchange chromatography uses a 5 ml strong sulfopropyl cation exchanger column. A flow rate of 3 ml/min was used for all the chromatographic steps.
- Resuspend the cells in 35 ml of SBDS Lysis buffer (50 mM phosphate buffer pH 7.5, 300 mM NaCl, 20 mM imidazole) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and lyse by sonication for a total time of 4 min using cycles of 10 sec ON and 30 sec OFF, at 4 °C.
- Centrifuge the sample at 9,000 x g for 50 min at 4 °C.
- Keep the supernatant and discard the pellet to remove cellular debris.
- Equilibrate the Ni2+ affinity column with 3 column volumes (CV) of SBDS Lysis buffer and introduce the whole clarified supernatant onto the column.
- Remove unbound protein by washing with 3 CV of SBDS Lysis buffer and elute with 3 CV of SBDS Elution buffer (50 mM phosphate buffer pH 7.5, 300 mM NaCl, 250 mM imidazole).
- Dilute the eluted protein 6-fold with 50 mM Phosphate buffer pH 6.5 and remove possible aggregates by filtration through a 0.22 µm cellulose membrane.
- Equilibrate the sulfopropyl cation exchanger column with 3 CV of Low salt S column buffer (50 mM phosphate buffer pH 6.5, 50 mM NaCl) and introduce the protein sample from the previous step.
- Wash unbound material with 3 CV of Low salt S column buffer and elute the protein in one step with 50 mM phosphate buffer pH 6.5, 1 M NaCl.
- Dilute the eluted protein 3.3-fold with 50 mM Phosphate buffer pH 6.5. Concentrate the protein with ultrafiltration devices by centrifugation at 3,800 x g for 15 min. Flash freeze the protein in liquid nitrogen and store it at -80 °C until further use.
- Verify the purity of the protein by SDS-PAGE analysis and Coomassie staining 20.
2. EFL1 Protein Expression and Purification
NOTE: EFL1 was expressed under the regulation of the Gal 1/10 divergent promoter 21 and the Saccharomyces cerevisiae MATA 3'UTR in the vector pRS426. The recombinant protein encodes the human EFL1 isoform 1 fused to a Tobacco Etch Virus protease (TEV) recognition site and a hexahistidine tag at the C-terminus.
- EFL1 protein expression
- Transform S. cerevisiae BCY123 cells with the plasmid pRS426-EFL1TevHis using a standard Lithium acetate protocol 22. Plate all the transformed cells in synthetic drop out media without uracil (SD-URA) supplemented with 2% (w/v) glucose. Composition of the SD-URA media consists of 8 g yeast nitrogen base without amino acids, 11 g casamino acids, 55 mg adenine sulfate, 55 mg tyrosine, 60 mg leucine and 60 mg tryptophan for 1 L volume.
- Culture transformed yeast at 30 °C until A600 reaches 1.8 in 1 L of SD-URA media supplemented with 0.5% (w/v) glucose.
- Induce protein expression by adding 2.8% (w/v) galactose to the culture and continue the incubation for further 18 hr at 30 °C.
- Collect the yeast suspension by centrifugation at 3,800 x g for 10 min at 4 °C. Remove the supernatant. At this point, either store the cell pellet at -20 °C or use immediately for protein purification.
- EFL1 protein purification NOTE: All chromatographic steps are performed using a FPLC system or a peristaltic pump. Ni2+-affinity chromatography uses a 5 ml fast flow column at a flow rate of 3 ml/min. Size exclusion chromatography uses a 125 ml column pre-packed with Superdex 200 resin at a flow rate of 1 ml/min.
- Resuspend the cells in 50 ml of EFL1 Lysis buffer (50 mM Tris-HCl pH 8, 300 mM NaCl, 20 mM imidazole, 5 mM MgCl2, 10% glycerol) supplemented with 1 mM PMSF and 1 mM benzamidine and disrupt the cells by friction on a bead beater using glass beads (Ø = 0.5 mm) for a total time of 6 min using cycles of 2 min ON and 15 min OFF, at 4 °C.
- Centrifuge the sample at 9,000 x g for 50 min at 4 °C.
- Keep the supernatant and discard the pellet to remove cellular debris.
- Equilibrate the Ni2+ affinity column with 3 CV of EFL1 Lysis buffer and introduce all the clarified supernatant onto the column.
- Remove unbound protein by washing with 3 CV of EFL1 Lysis buffer and elute with 3 CV of EFL1 Elution buffer (50 mM Tris-HCl pH 8, 300 mM NaCl, 250 mM imidazole, 5 mM MgCl2, 10% glycerol).
- Equilibrate the size exclusion column with 1.5 CV of Anisotropy buffer (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 5 mM MgCl2, 10% glycerol, 5 mM β-mercaptoethanol).
- Concentrate to 1 ml the EFL1 protein eluted from the Ni2+ affinity column with ultrafiltration devices by centrifugation at 3,800 x g to the desired volume. Introduce the sample on the size exclusion column.
- Collect the eluted protein and concentrate by ultrafiltration to a final concentration of approximately 30 µM. Flash freeze the protein in liquid nitrogen and store at -80 °C until further use. Verify the purity of the protein by SDS-PAGE analysis and Coomassie staining 20.
3. Labeling of SBDS-FlAsH with the FlAsH Fluorescent Dye 4',5'-Bis(1,3,2 dithioarsolan-2-yl) Fluorescein
Mix 3 nmol of the SBDS-FlAsH protein with 3 nmol of the 4',5'-bis(1,3,2 dithioarsolan-2-yl) fluorescein dye in 5 µl volume of Anisotropy buffer.
Let the reaction proceed for 8 hr at 4 °C. Dialyze the sample against Anisotropy buffer over night to remove the free dye.
Use the Lambert-Beer law to quantify the % of labeled protein. Measure the absorbance at 280 nm and 508 nm in a spectrophotometer using a quartz cuvette of appropriate volume. NOTE: Consider the following molar absorption coefficients (M-1 cm-1):
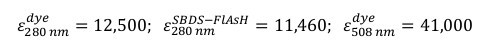
Calculate the concentration of labeled SBDS-FlAsH protein using Equation 2.

Calculate the concentration of total SBDS protein using Equation 3 by substituting the calculated CSBDS-FlAsH from previous step.
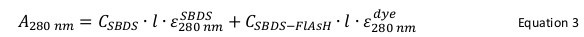
Calculate the percentage of labeled protein using Equation 4.
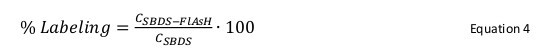
4. Fluorescence Anisotropy Experiments
NOTE: Anisotropy experiments were done in a spectrofluorometer equipped with a polarization toolbox and data collection was performed using the anisotropy program provided in the software of the equipment. The excitation wavelength was set at 494 nm with a spectral bandwidth of 8 nm and the emission was recorded using a band-pass filter of 530±25 nm. Measurements were done at 25 °C in a 200 µl cuvette with a 5 mm path length 10.
In a fluorescence cuvette, place 200 µl of 30 nM SBDS-FlAsH in anisotropy buffer and titrate 2 µl of 30 µM EFL1. Mix thoroughly and let the reaction stand for 3 min before measuring the anisotropy value.
Repeat step 4.1 until a total volume of 40 µl of EFL1 has been added.
5. Data Analysis
Fit the data to the appropriate binding model using a nonlinear least squares regression algorithm. Equations for the most common binding models are presented in Table 1.
Evaluate the best model that describes the interaction between the proteins by inspecting the residuals of the fit 23. Support the chosen model with additional experiments.
Table 1. Common protein-protein interaction binding models and the mathematical equations that describe them.
Please click here to view a larger version of this table.

Representative Results
To perform any anisotropy experiment it is important to rule out large changes in the fluorescence intensity of the fluorophore since the observed anisotropy of a mixture of species is represented by Equation 5:
![]()
where Fi represents the fractional fluorescence of each component and riis the corresponding anisotropy. The fractional fluorescence of each component depends on both, its concentration and its relative fluorescence, which is defined by Equation 6:

where Xi represents the mole fraction of the ith specie and Øi is the quantum yield of the ith specie.
Thus, if fluorescence intensity changes dramatically along the titration it is better to, either measure complex formation as a function of the change in fluorescence signal and not of the anisotropy signal, or correct the observed anisotropy value by fitting both the fluorescence intensity and the anisotropy data. The fluorescence of SBDS-FlAsH does not noticeable change upon binding to EFL1 (Figure 2) suggesting that fluorescence anisotropy is adequate for measuring the binding between the two proteins.
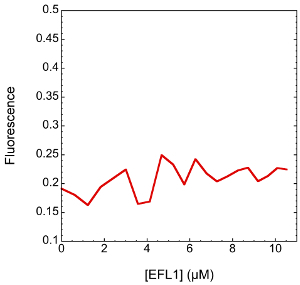 Figure 2. Fluorescence emission of SBDS-FlAsH upon titration of increasing concentrations of EFL1. Changes in the fluorescence emission of the fluorophore are negligible and random along the titration. Please click here to view a larger version of this figure.
Figure 2. Fluorescence emission of SBDS-FlAsH upon titration of increasing concentrations of EFL1. Changes in the fluorescence emission of the fluorophore are negligible and random along the titration. Please click here to view a larger version of this figure.
An example of the fluorescence anisotropy binding curve obtained from the titration of EFL1 to SBDS-FlAsH is shown in Figure 3. Qualitatively, the shape of the graph clearly shows that the two proteins interact as evidenced by an increase in the anisotropy signal upon addition of EFL1. Furthermore, anisotropy values reached a plateau suggesting that at the end of the titration all the SBDS-FlAsH protein was present in a complex with EFL1. It is possible then, to quantitatively describe the interaction between these proteins and fit the data using nonlinear least squares regression to a presumed binding model and obtain the corresponding dissociation equilibrium constant. Fitting to a one binding site model did not appropriately describe the experimental data (Figure 3A). This is evident not only from the fitting trace in the binding curve but also from the deviations of the residuals of the fit (the difference between the theoretical and experimental data) that are appreciable and nonrandom. This agrees well with the fact that a single binding site model is described mathematically by a hyperbolic curve rather than a sigmoidal curve as that observed for the experimental data presented in Figure 3. On the other hand, models such as two identical or two different binding sites describe better the experimental data and the residuals of the fit show no systematic deviation supporting a two-site association model (Figure 3B-C). In the absence of any other experimental information is difficult to establish which of the two models correspond to the binding mechanism between EFL1 and SBDS. Nevertheless, SBDS and EFL1 are both monomeric proteins, so it is difficult to envisage a two identical binding sites model.
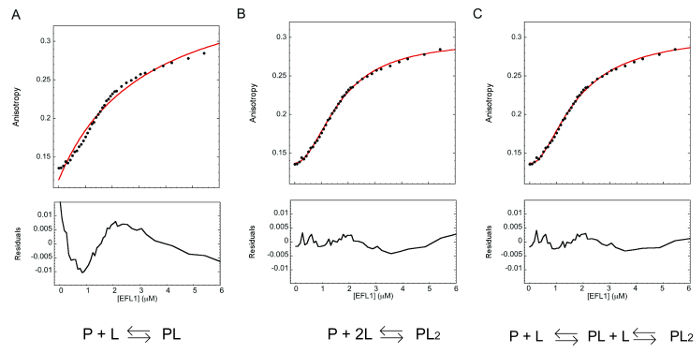 Figure 3. Fluorescence anisotropy binding isotherm of the interaction between EFL1 and SBDS-FlAsH. The continuous line in each of the upper plots corresponds to the fit of the data to different binding models: (A) single site, (B) two identical sites and (C) two non-identical sites. Lower plots represent the residuals of the fit to the corresponding model. Please click here to view a larger version of this figure.
Figure 3. Fluorescence anisotropy binding isotherm of the interaction between EFL1 and SBDS-FlAsH. The continuous line in each of the upper plots corresponds to the fit of the data to different binding models: (A) single site, (B) two identical sites and (C) two non-identical sites. Lower plots represent the residuals of the fit to the corresponding model. Please click here to view a larger version of this figure.
Discussion
Most biochemical experiments with proteins require not only pure protein but also large amounts of them, irrespective of the technique used. For this reason, the proteins used for this type of experiments are obtained by heterologous expression, as it was the case presented here. Florescence spectroscopy requires the presence of a fluorophore in the studied molecule. Aromatic residues constitute the intrinsic fluorophores of a protein, however, using their signal to study protein-protein interactions complicates the analysis since they are common to most proteins and would be present in both, the receptor protein and the ligand. Additionally, the lifetime of tryptophan is too short for fluorescence anisotropy experiments. For this reason, it is common to add extrinsic fluorophores to one of the studied proteins. To this end, a fluorophore was added to the C-terminus of SBDS through a coordination bond between the dye 4',5'-bis(1,3,2 dithioarsolan-2-yl) fluorescein with a tetracysteine motif introduced in the protein by genetic engineering. Choosing the protein that bares the fluorophore is not a random matter. Due to the size of the proteins studied here (SBDS 29 kDa and EFL1 127 kDa), we expected a larger change in anisotropy between free labeled-SBDS and that in complex with EFL1, compared to the change expected between free labeled-EFL1 and that bound to SBDS. Bearing this in mind, a FlAsH tag was introduced into SBDS rather than to EFL1. Another parameter important to consider when setting up a fluorescence anisotropy experiment is the absorption of the solutions used. The optical density of the solution in the cuvette should not be greater than 0.1 at the excitation and emission wavelength, and should remain constant along the entire titration. Otherwise, inner filter effect can interfere in the measurement and must be corrected for. In the experiment presented here, the proteins studied do not absorb at the wavelength of excitation or emission of the 4',5'-bis(1,3,2 dithioarsolan-2-yl) fluorescein dye; thus inner filter effect does not pose a problem.
Regarding data collection, and to subsequently properly fit the data, it is necessary to have well-defined lower and upper baselines with several data points through the transition along the binding curve. Otherwise, fitting of the data is inaccurate and information regarding the binding mode may be lost. To describe a decent binding curve it is necessary that the concentration of free ligand differs by two logarithmic units below and above the dissociation constant, however, experimentally this may be difficult to achieve. In the lower limit, problems with the sensitivity of the equipment may arise particularly when the affinity between the proteins is very high, while large concentrations may not be attainable due to solubility problems of the ligand. This was clearly exemplified when testing the interaction between the SBDS S143L disease mutant and EFL1. This mutant disrupted the binding to EFL1 to such an extent that it was not possible to obtain a proper binding curve since EFL1 can only be concentrated up to 70 µM and the protein gets diluted ≈ 5 fold during the titration 10. Thus, it is necessary to have a rough estimate of the affinity to optimize the titration scheme and make sure that jumps in ligand concentration do not occur at positions that would cause a less accurate fit. For example, missing the initial points of the binding curve presented in Figure 3 will make it look like a hyperbola misleading the researcher towards a single binding site model.
Once collected, data were fitted to a presumed binding model. In comparison to fluorescence data, anisotropy data can be used without further manipulation and do not need correction for dilution effects. The equations presented in Table 1 consider that [L] ≈ [L]0 at every point of the titration. This may not happen when the affinity between the proteins is very high such that in the first points of the titration ligand depletion occurs. In this scenario, more complex quadratic equations described elsewhere 23 are needed to fit the data. In the experiment described here, the ligand EFL1 was always in large excess compared to the concentration of SBDS-FlAsH and the change in EFL1 concentration at each point of the titration can be disregarded. As discussed in the results section, a two non-identical binding sites model best described the experimental data (Figure 3C). Additional biochemical information obtained in the group supports the idea that this is the correct model to describe the interaction between EFL1 and SBDS (data not shown). This mode of interaction is common in multi-domain proteins, such as EFL1 and SBDS are, and several examples exist in the literature 24-26. However, it is also important to rule out a non-specific interaction that can appear as an interaction model of distinct binding sites with different affinities for their ligand. To this end, a Scatchard plot is very useful. Compared to other techniques, fluorescence anisotropy signal reports the amount of complex formed and thus it is possible to build a Scatchard plot with the data. A convex Scatchard curve suggests a two different binding sites model with negative cooperativity or non-specific binding, while a concave Scatchard plot implies a two different binding sites model with positive cooperativity or ligand instability (Figure 4). Analysis of the experimental data showed a Scatchard plot with a concave shape supporting the model of two independent binding sites for EFL1 and SBDS (Figure 4D). Furthermore, positive cooperativity was confirmed after performing a Hill analysis that resulted in a Hill coefficient value of 1.8±0.02 (data not shown).
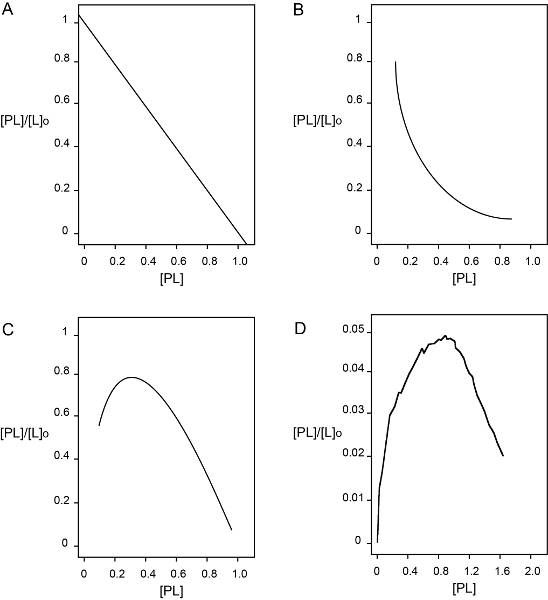 Figure 4. Scatchard plots for different protein-protein binding models. (A) Single binding site or multiple identical sites that do not interact. (B) Multiple different binding sites with negative cooperativity or non-specific binding. (C) Multiple different binding sites with positive cooperativity or ligand instability. (D) Scatchard plot resulting from analysis of the experimental anisotropy data obtained from the titration of EFL1 to SBDS-FlAsH. Please click here to view a larger version of this figure.
Figure 4. Scatchard plots for different protein-protein binding models. (A) Single binding site or multiple identical sites that do not interact. (B) Multiple different binding sites with negative cooperativity or non-specific binding. (C) Multiple different binding sites with positive cooperativity or ligand instability. (D) Scatchard plot resulting from analysis of the experimental anisotropy data obtained from the titration of EFL1 to SBDS-FlAsH. Please click here to view a larger version of this figure.
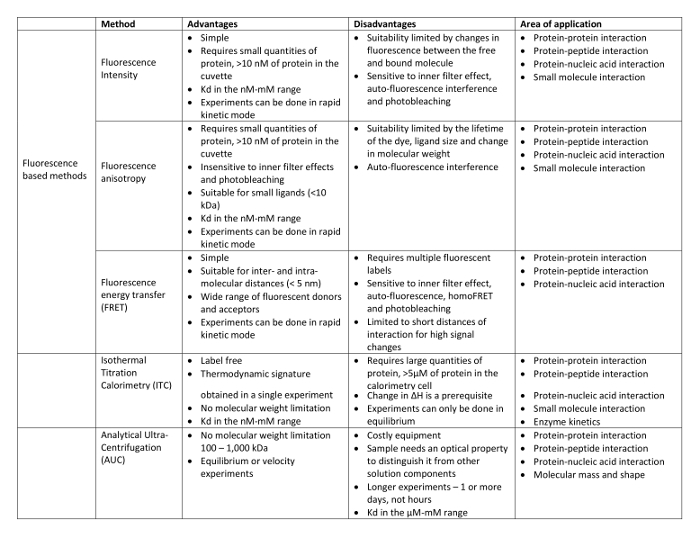
Fluorescence anisotropy is a solution-based, true-equilibrium technique that requires the studied molecules to remain in solution at the conditions tested. It can be used to study not only the interaction between full-length proteins but also the interaction between protein domains, mutant proteins or even peptides with post-translational modifications. Moreover, fluorescence anisotropy can also be used to measure the binding of proteins to lipid membranes using fluorescent sensitive membranes probes. This approach has been successfully used to study the effect that α-synuclein has on synaptic vesicle packing 27. One of the main advantages of fluorescence anisotropy is that it provides quantitative information on the binding of the studied molecules such that true dissociation constants can be obtained together with mechanistic information. Although other biophysical techniques can also be used to obtain such information, fluorescence anisotropy requires small amounts of sample and can be performed not only in equilibrium, as presented here, but also using rapid kinetics, such as stopped-flow fluorometry, to obtain the association and dissociation rate constants (kon and koff, respectively) 28. A comparative table with the benefits and disadvantages of fluorescence anisotropy with respect to other biophysical techniques is presented in Table 2. All the methods described are true-equilibrium techniques since none of them include a process of mechanical separation of the free and bound fractions. As mentioned in the results section, if the fluorescence intensity of a labeled-protein largely changes upon interaction, this property can then be used to quantify the affinity of the binding. However, changes in fluorescence intensity resulting from the interaction suggest a possible interference of the extrinsic fluorophore on the molecular interaction itself; namely a possible alteration of the dissociation constant (Kd) value of the corresponding non-labeled proteins. As such, fluorescence anisotropy can be considered a less invasive technique than changes in fluorescence intensity since the former should be applied when fluorescence intensity does not change upon interaction. In this sense, though large amounts of the protein are required, Isothermal Titration Calorimetry (ITC) can provide a true Kd value of the molecular interaction since it is a label free method. On the other hand, mechanistic information is not obtained because differences in heat only report on the enthalpy of the process and not the amount of complex formed 29.
In comparison, in vivo methods such as yeast two-hybrid or fragment complementation assays do not require purified proteins but they only provide semi-quantitative information on the mode of binding. Despite the fact that in the yeast two-hybrid technique is possible to express the strength of the binding using Miller units, this is not a real equilibrium constant and many factors out of the control of the researcher can alter it.
Table 2. Advantages and disadvantages of commonly used biophysical techniques used to study protein-protein interactions.
Please click here to view a larger version of this table.
Disclosures
The authors have nothing to disclose and they have no competing financial interests.
Acknowledgments
Authors acknowledge the financial support from CONACyT project numbers 167359 and 177138, and from DGAPA-UNAM project number IN201615.
References
- Krogan NJ, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440(7084):637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Michnick SW, Hien Ear P, Landry C, Malleshaiah MK, Messier V. A toolkit of protein-fragment complementation assays for studying and dissecting large-scale and dynamic protein-protein interactions in living cells. Methods in Enzymology. 2010;470:336–366. doi: 10.1016/S0076-6879(10)70014-8. [DOI] [PubMed] [Google Scholar]
- Puig O, et al. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Dwane S, Kiely PA. Tools used to study how protein complexes are assembled in signaling cascades. Bioeng Bugs. 2011;2(5):247–259. doi: 10.4161/bbug.2.5.17844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider J, et al. Fundamentals of protein interaction network mapping. Mol Syst Biol. 2015;11(12):848. doi: 10.15252/msb.20156351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A. In: Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. Baldwin RL, editor. W. H. Freeman and Company; 2002. pp. 191–214. [Google Scholar]
- Menne TF, et al. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;39(4):486–495. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- Boocock GR, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33(1):97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- Garcia-Marquez A, Gijsbers A, de la Mora E, Sanchez-Puig N. Defective Guanine Nucleotide Exchange in the Elongation Factor-like 1 (EFL1) GTPase by Mutations in the Shwachman-Diamond Syndrome Protein. J Biol Chem. 2015;290(29):17669–17678. doi: 10.1074/jbc.M114.626275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijsbers A, Garcia-Marquez A, Luviano A, Sanchez-Puig N. Guanine nucleotide exchange in the ribosomal GTPase EFL1 is modulated by the protein mutated in the Shwachman-Diamond syndrome. Biochem Biophys Res Commun. 2013;437(3):349–354. doi: 10.1016/j.bbrc.2013.06.077. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of fluorescence spectroscopy. Third. Springer US; 2010. [Google Scholar]
- Nishigaki T, Treviño CL. In: Tools to understand protein-protein interactions. Gòmez I, editor. Vol. 37. Transworld Research Network; 2012. pp. 1–14. [Google Scholar]
- Johnson I. The Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies. 11th. Life Technologies Corporation; 2010. [Google Scholar]
- Sabnis RW. Handbook of Fluorescent Dyes and Probes. Wiley; 2015. [Google Scholar]
- Adams SR, et al. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J Am Chem Soc. 2002;124(21):6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281(5374):269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- Rashidian M, Dozier JK, Distefano MD. Enzymatic labeling of proteins: techniques and approaches. Bioconjug Chem. 2013;24(8):1277–1294. doi: 10.1021/bc400102w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: A laboratory manual. 3rd. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Neuhoff V, Arold N, Taube D, Ehrhardt W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988;9(6):255–262. doi: 10.1002/elps.1150090603. [DOI] [PubMed] [Google Scholar]
- West RW, Jr, Chen SM, Putz H, Butler G, Banerjee M. GAL1-GAL10 divergent promoter region of Saccharomyces cerevisiae contains negative control elements in addition to functionally separate and possibly overlapping upstream activating sequences. Genes Dev. 1987;1(10):1118–1131. doi: 10.1101/gad.1.10.1118. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153(1):163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftink MR. Fluorescence methods for studying equilibrium macromolecule-ligand interactions. Methods Enzymol. 1997;278:221–257. doi: 10.1016/s0076-6879(97)78013-3. [DOI] [PubMed] [Google Scholar]
- Han H, et al. Binding of Substrates to the Central Pore of the Vps4 ATPase Is Autoinhibited by the Microtubule Interacting and Trafficking (MIT) Domain and Activated by MIT Interacting Motifs (MIMs) J Biol Chem. 2015;290(21):13490–13499. doi: 10.1074/jbc.M115.642355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Puig N, Veprintsev DB, Fersht AR. Binding of natively unfolded HIF-1alpha ODD domain to p53. Mol Cell. 2005;17(1):11–21. doi: 10.1016/j.molcel.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Trusch F, et al. The N-terminal Region of the Ubiquitin Regulatory X (UBX) Domain-containing Protein 1 (UBXD1) Modulates Interdomain Communication within the Valosin-containing Protein p97. J Biol Chem. 2015;290(49):29414–29427. doi: 10.1074/jbc.M115.680686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F, Beyer K. Binding of alpha-synuclein affects the lipid packing in bilayers of small vesicles. The Journal of Biological Chemsitry. 2006;281:9251–9259. doi: 10.1074/jbc.M512292200. [DOI] [PubMed] [Google Scholar]
- Bujalowski WM, Jezewska MJ. Fluorescence Intensity, Anisotropy, and Transient Dynamic Quenching Stopped-Flow Kinetics. Spectroscopic Methods of Analysis. 2012;875:105–133. doi: 10.1007/978-1-61779-806-1_6. [DOI] [PubMed] [Google Scholar]
- Asano N, et al. Direct interaction between EFL1 and SBDS is mediated by an intrinsically disordered insertion domain. Biochem Biophys Res Commun. 2014;443(4):1251–1256. doi: 10.1016/j.bbrc.2013.12.143. [DOI] [PubMed] [Google Scholar]