

Abstract
Many neurodegenerative diseases are still lacking effective treatments. Reliable biomarkers for identifying and classifying these diseases will be important in the development of future novel therapies. Often potential new biomarkers do not make it into the clinic due to limitations in their development and high costs. However, targeted proteomics using Multiple Reaction Monitoring Liquid Chromatography-tandem/Mass Spectrometry (MRM LC-MS/MS), specifically using triple quadrupole mass spectrometers, is one method that can be used to rapidly evaluate and validate biomarkers for clinical translation into diagnostic laboratories. Traditionally, this platform has been used extensively for measurement of small molecules in clinical laboratories, but it is the potential to analyze proteins, that makes it an attractive alternative to ELISA (Enzyme-Linked Immunosorbent Assay)-based methods. We describe here how targeted proteomics can be used to measure multiplexed markers of dementia, including the detection and quantitation of the known risk factor apolipoprotein E isoform 4 (ApoE4).
In order to make the assay suitable for translation, it is designed to be rapid, simple, highly specific and cost effective. To achieve this, every step in the development of the assay must be optimized for the individual proteins and tissues they are analyzed in. This method describes a typical workflow including various tips and tricks to developing a targeted proteomics MRM LC-MS/MS for translation.
The method development is optimized using custom synthesized versions of tryptic quantotypic peptides, which calibrate the MS for detection and then spiked into CSF to determine correct identification of the endogenous peptide in the chromatographic separation prior to analysis in the MS. To achieve absolute quantitation, stable isotope-labeled internal standard versions of the peptides with short amino acid sequence tags and containing a trypsin cleavage site, are included in the assay.
Keywords: Medicine, Issue 116, CSF, targeted proteomics, Multiple Reaction Monitoring, Apolipoprotein E isoform, dementia, biomarker, tandem mass spectrometry, multiplex
Introduction
The growing impact of neurodegenerative diseases such as Alzheimer's Disease, Lewy Body Dementia and Parkinson's Disease, is becoming a socioeconomic issue in many countries1. There is a need in additional biomarkers that can be used to identify and classify patients in the early stages of the disease, and to monitor any potential new treatments. The overall goal of this method is to create a generic pipeline for a streamlined, economic and faster way of validating potential CSF markers of neurodegeneration. The rationale is to use targeted proteomics or peptide MRM LC-MS/MS as an easily amendable method to assess multiple potential protein biomarkers from discovery experiments. These can be further multiplexed over a rapid chromatographic separation (<10 min) and assessed. Within this multiplex screen for neurodegenerative biomarkers, we have included the known dementia risk factor apolipoprotein isoform E4 (ApoE4) so we can determine simultaneously its status and level of expression, by that eliminating the need for separate genotyping tests2. LC MS/MS is routinely used as the method of choice for accurately quantitating small molecules over other methods such as ELISA or radioimmunoassay (RIA). This shift in the use of MS technology to analyzing proteins, has been driven mainly by issues with the immuno-based technologies. These include cross-specificity, batch to batch variation, limited shelf life, and high cost. Therefore, targeted proteomics is rapidly becoming a growing alternative to antibody based methods such as Western blotting, RIA and ELISA. However, the ability to multiplex many markers into one assay is the major advantage of this technique over immuno-based methods3. The technique is applicable to many tissues and has been used as a validation strategy for many proteomics studies including plasma4 and urine5,6.
The technique can be applied to any laboratory that has access and expertise in using triple quadrupole mass spectrometers. Peptide design is relatively simple with the growing use of open source databases. The competitive market of custom peptides synthesis makes them much more affordable. Heavy peptides, however, are expensive and therefore the markers should be assessed on a small cohort before moving to a larger scale. There is growing potential for the technique to be used in the clinical diagnostic settings, with most large hospitals having triple quadrupole based platforms which can easily be adapted to run targeted proteomic assays. One such application of the method, making it into the routine diagnostic setting, is its recent application to newborn blood spot screening for sickle cell anemia7.
Protocol
NOTE: A schematic of the overall protocol described here is given in Figure 1. All samples used for the development of this method are surplus clinical diagnostic samples and have ethical approval from the London Bloomsbury Ethics committee.
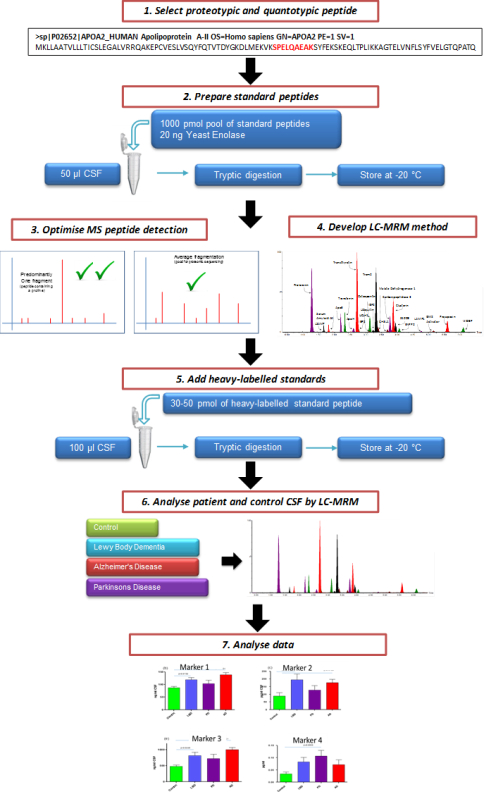 Figure 1. Schematic Illustrating the Overall Process of Creating a Targeted CSF MRM LC-MS/MS Assay. Candidate marker peptides for evaluation are selected from protein targets. Through the use of custom synthesized peptides, a targeted LC-MS/MS multiplex method is created. After evaluation, the assay can be used to assess the efficacy of potential markers of neurodegeneration.
Figure 1. Schematic Illustrating the Overall Process of Creating a Targeted CSF MRM LC-MS/MS Assay. Candidate marker peptides for evaluation are selected from protein targets. Through the use of custom synthesized peptides, a targeted LC-MS/MS multiplex method is created. After evaluation, the assay can be used to assess the efficacy of potential markers of neurodegeneration.
1. Peptide Selection and Design
NOTE: Criteria for a marker peptide is that it must be unique (proteotypic) and representing the quantitative abundance of the protein (quantotypic). To determine if a peptide is unique or not, the 'blast' search tool on the Uniprot website (http://www.uniprot.org/blast/) can be used.
Define a list of target proteins (markers), i.e., ApoE (see Table 2). NOTE: If the marker has been identified from previous proteomic profiling experiments8, then select the peptide that gives the best response from that data set.
If this information is unavailable, use open source websites, such as in silico trypsin digestion of target proteins, using a software tool such as MS-Digest.
Choose the peptide which is tryptic and not susceptible to post translational modifications. NOTE: This information can be checked on the Uniprot website www.uniprot.org. Avoid peptides prone to chemical modification during LC-MS sample preparation.
Order the custom synthesis of the chosen peptides. NOTE: Marker peptides can be custom synthesized by various commercial companies. For ApoE the quantotypic peptide used to measure total ApoE levels was determined to be AATVGSLAGQPLQER. The proteotypic peptide sequences used to determine the E2 and E4 variants are the tryptic peptides for position 158 (RLAVYQAGAR and CLAVYQAGAR) and position 112 (LGADMEDVCGR and LGADMEDVR), respectively.
2. Preparation of Standard Peptides
NOTE: In order to select the best quantitative transitions, the detection of the matrix (CSF) needs to be optimized. The most efficient way of optimizing multiplexed peptides is to create pools of the peptides at known concentrations. These pools can then be used for method development and standard curves.
Resuspend synthetic peptides (peptide details are given in Table 2) to 1 mg/ml stock concentration according to manufactures instructions. By default, if instructions are not available, resuspend peptides in 50:50 (v/v) acetonitrile (ACN)/H2O.
Prepare the 1:10 dilutions of the peptide from the stock concentration and pool 1,000 pmol of each peptide into a low binding microcentrifuge tube. Dry down in a speed-vac concentrator the final pool and store at -20 °C. Prepare several pools for future use.
Aliquot 100 µl of CSF into low binding tubes. Freeze-dry the CSF.
Resuspend an aliquot of pooled 1,000 pmol peptides in digestion buffer (100 mM Tris HCl, pH 7.8, 6 M Urea, 2 M Thiourea, 2% ASB14) to obtain concentrations of 10 and 1 pmol/µl.
Spike the pooled peptides into the freeze-dried 100 µl aliquots of CSF at 0, 1, 2, 5, 10 and 15 pM concentrations. Add 20 ng of intact unrelated protein such as yeast enolase to act as an internal standard and control for digestion efficiency of trypsin.
Top up the CSF aliquots with digest buffer to a final amount of 20 µl. Vortex.
Add 1.5 µl of dithiothreitol (30 mg in 1 ml of 100 mM Tris HCl, pH 7.8) and shake at room temperature for 1 hr.
Add 3 µl of iodoacetamide (35 mg in 1 ml of 100 mM Tris HCl, pH 7.8) and shake at room temperature for 45 min in the dark.
Add 165 µl of ddH2O.
Add 10 µl of 0.1 µg/µl sequencing grade modified trypsin solution resuspended in 50 mM ammonium bicarbonate buffer, pH 7.8. Incubate in a water bath at 37 °C O/N and stop the digestion by freezing samples. Store digests at -20 °C until ready to analyze.
3. Optimization of MS Peptide Detection
Copy and paste the sequence of the peptide to be optimized, into an appropriate software, e.g., Skyline9. Click on the peptide sequence to obtain the information of expected precursor ion mass and product ions.
Dilute the peptides from stock concentration (see section 2.1) further to 1 ng/ml concentration for MS method development.
Directly infuse the peptides in the mass spectrometer at optimized flow rate (usually 0.1 - 0.8 ml/min) and with 0 - 5% collision energy.
Acquire MS spectrum in order to identify the experimental multiply charged precursor ions8. Choose the precursor ion (m/z) that gives the most intensity (doubly- or triply-charged precursor ions are advised).
Reinfuse the peptide and apply collision energy to fragment the peptide by collision-induced-dissociation (CID). Optimize the cone and collision energies to obtain the best fragmentation pattern (fragment ions singly or doubly charged, and mass of fragment ion preferably bigger than parent ion, are advised).
Verify that the experimentally obtained transitions match transitions generated in silico (as described in step 3.1). Save the transition list in the MRM method file using at least 2 most intense transitions per precursor ion. Include 2 transitions: 1 for quantitation and 1 for confirmation in the final assay. NOTE: Some MS manufacturers have a function (i.e., Waters Intellistart) for automated MRM or SRM analysis optimization. If possible infuse peptides with combined mobile phases at 50 - 70% ACN with 0.1% FA.
4. LC-MRM Method Development
NOTE: Analyze the mixture of synthetic peptides by an Ultra Performance Liquid Chromatography (UPLC) system coupled to triple quadrupole mass spectrometer. Ensure the source is clean. Solvent A is ddH20 with 0.1% FA; Solvent B is ACN with 0.1% FA.
Use the LC-MS system equipped with a UPLC column packed with C-18 phase (1.6 µm diameter, 90 Å pores, 2.1 mm x 50 mm length) and attached to a pre-column of the same phase.
Defrost CSF digests on ice, centrifuge at 16,000 g for 10 min and transfer 60 µl into 300 µl glass insert vials and store the rest back at -20oC.
Inject the highest concentration from standard curve point using 10 min 1 - 40% ACN linear gradient (see Table 1 for gradient settings)
Open the resulting chromatogram and note retention time and top two most intense (quantitative) transitions per peptide with all transitions created in step 3.
Based on this information, update a 10 min MRM method (created in step 3.6) with timed channels to measure peptides (see Figure 2 for an example). To maintain sensitivity, keep each channel with points per peak greater than 8 and dwell time greater than 0.01 sec for at least one transition for each peptide.
Include 'solvent delays' in the MRM method: one at the beginning until 10 sec before the first peak elution and another at the end of the method, 20 sec after the last peak elution. Do this by selecting "solvent delays" in method events in MS method file.
Run the standard curve through the timed MRM method and ensure there are no interfering nonspecific peaks with transitions (generated in step 3.6) by checking for linearity.
Determine if the peptides are detectable by running non-spiked pooled control and disease CSF through the method.
Remove the peptides that are below the limit of detection from the assay.
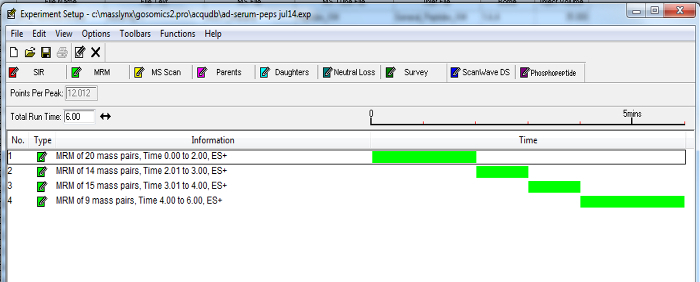 Figure 2. Example of a Dynamic MRM MS Method. Timed channels of peptide transitions can be grouped according to established retention times. Enabling MRMs to be incorporated into a timed fashion as the selected marker peptides elute from the chromatography column, minimizes the number of transitions over a time period and increases the sensitivity of the assay. Please click here to view a larger version of this figure.
Figure 2. Example of a Dynamic MRM MS Method. Timed channels of peptide transitions can be grouped according to established retention times. Enabling MRMs to be incorporated into a timed fashion as the selected marker peptides elute from the chromatography column, minimizes the number of transitions over a time period and increases the sensitivity of the assay. Please click here to view a larger version of this figure.
5. Addition of Internal Standards
NOTE: As described previously10, stable isotope-labeled internal standards can be included in the assay. Due to the expense of these standards, it is advised to first assess the peptides in the matrix.
Determine the ideal peptides optimized for detection in CSF: choose the peptides which are the most recurrent, with the highest intensity and without interference peaks.
Design corresponding peptides to include heavy 13C 15N amino acid labels that increase the mass of the peptide by at least 6 Da relative to endogenous peptide. Also, add a tag of 4 - 6 additional amino acids to either the N- or C-terminus of the heavy peptide to control for tryptic digestion.
Dilute stable isotope-labeled internal standards in digestion buffer (see step 2.4). Determine the ideal amount of stable isotope-labeled internal standard which will be spiked in CSF by spiking in various levels depending on the abundancies previously observed during development. Aim to achieve approximately 1:1 ratio of stable isotope-labeled standard to endogenous peptide. NOTE: Stable isotope-labeled internal standards can be synthesized by various commercial companies.
6. LC-MRM Assay of CSF Patient Samples
Spike optimized amount of stable isotope-labeled standards into 100 µl of CSF and freeze-dry the mixture.
Resuspend the CSF in 20 µl of digest buffer and perform the digestion as described in points 2.7 - 2.10.
Analyze the samples using LC-MRM method developed in step 4.
For quantitative analysis, run the standard peptides in concentrations of 0 -15 pmol in a standard curve. See step 2.5 for the preparation of standard curve.
7. Data Analysis
NOTE: Quantitative data is based on the intensity ratio of baseline peak internal standards (heavy labeled peptides). Detailed information regarding SRM/MRM data analysis was previously described10. Ratio data can then be used in a standard curve to determine absolute levels or calculate them from the added concentration of heavy-labeled peptide.
Analyze LC-MRM data using the mass spectrometers manufacturers' standard software10. Alternatively, use Skyline software to analyze quantitative MRM data10.
Check the sensitivity of the run by checking the response of an internal standard such as the spiked yeast enolase or a stable isotope-labeled standard in each run. Ensure that the coefficient of variation (CV) is not larger than 25%.
Manually review the data annotation to ensure accuracy. Analyze each peptide and ratio to an appropriate stable isotope-labeled internal standard, i.e., use the stable isotope-labeled version of the peptide if available.
To obtain absolute values of pmol per 100 µl of CSF, run the ratio data through appropriate standard curves that were run at the same time.
Calculate the coefficient of variation (CV). CV for each peptide should be below 25% and below 15% for high abundant peptides. NOTE: Absolute value pmol per 100 µl CSF values can used in subsequent downstream statistical analysis.
8. Apolipoprotein E Isoform Status
NOTE: To determine ApoE isoform status, the presence of the corresponding peptides can be performed by determining the presence of each isoform.
Consider the ApoE in 100 µl of CSF threshold of > 1,000 signal-to-noise as positive for that peptide. See Figure 5 for the peptides required/absent to determine a patient's isoform status. Determine the allelic expression by calculating the % of each isoform to total ApoE expression.
Representative Results
Using the method described above, a high throughput 10 min multiplex assay consisting of 74 peptides from 54 proteins was developed, as an assay for markers of the neurodegenerative disorders Alzheimer's Disease and Lewy Body Dementia (LBD)8. Figure 3 shows a multiplex chromatogram published previously8 of the significant peptide markers from the assay. The peptides included in the assay and their quantitative transitions are given in Table 2. The data generated by this method gives standardized ratios to the relevant stable isotope-labeled internal standard. These values can then be put through a standard curve to determine absolute pmol/100 µl CSF concentrations. These values can then be statistically analyzed for changes in clinical samples.
As described previously8, quantitation of the 74 peptides included in this CSF assay revealed that 25 of these markers were altered significantly in the CSF of dementia patients. To illustrate the effectiveness of this assay, results from previously described dementia markers Pro-Orexin and YKL chitinase 3-like protein (YKL-40)11,12 are given in Figure 4. The integrated ApoE assay identifies the ApoE isoform/allele status of the patient as well. Apo E4 is a known risk factor for Alzheimer's Disease, therefore integrating this into the assay will provide valuable information. The detection of the ApoE isoforms is explained in Figure 5 and is based on the detection of the corresponding peptides for the amino acid changes for isoforms E2 (R158C) and E4 (C112R). Figure 5A shows the peak pattern expected for each isoform combination and Figure 5B shows the result of a CSF tested with the assay on patient samples.
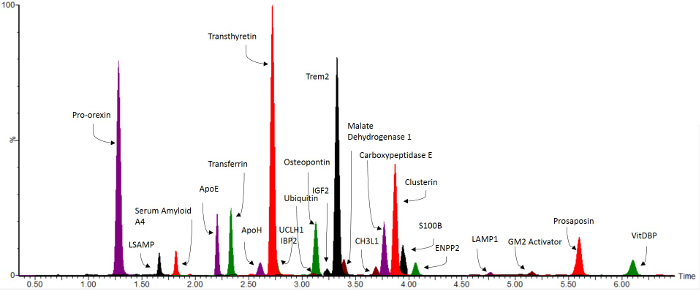 Figure 3. Representative Overlaid MRM Chromatograms. Reprint from Heywood et al.8 Biomarkers significant for the neurodegenerative disorders Lewy Body Dementia and Alzheimer's Disease8 are shown over a 10 min LC gradient. Please click here to view a larger version of this figure.
Figure 3. Representative Overlaid MRM Chromatograms. Reprint from Heywood et al.8 Biomarkers significant for the neurodegenerative disorders Lewy Body Dementia and Alzheimer's Disease8 are shown over a 10 min LC gradient. Please click here to view a larger version of this figure.
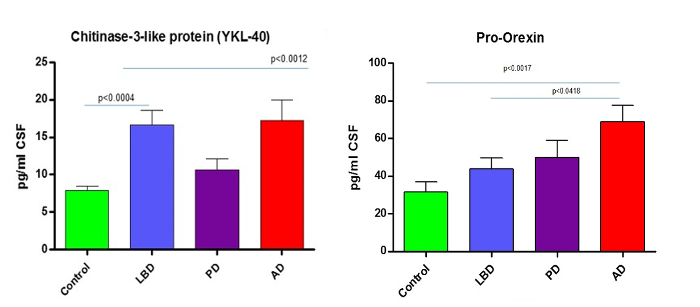 Figure 4.Example Data. Reprint from Heywood et al.8 Graphs show how the described MRM LC-MS/MS method can reliably quantitate and discriminate from controls the known neurodegenerative markers such as YKL chitinase 3-like protein (YKL-40)11,12 and the AD marker Pro-Orexin 13. AD = Alzheimer's Disease, LBD = Lewy Body Dementia and PD = Parkinson's Disease. Data previously published. Please click here to view a larger version of this figure.
Figure 4.Example Data. Reprint from Heywood et al.8 Graphs show how the described MRM LC-MS/MS method can reliably quantitate and discriminate from controls the known neurodegenerative markers such as YKL chitinase 3-like protein (YKL-40)11,12 and the AD marker Pro-Orexin 13. AD = Alzheimer's Disease, LBD = Lewy Body Dementia and PD = Parkinson's Disease. Data previously published. Please click here to view a larger version of this figure.
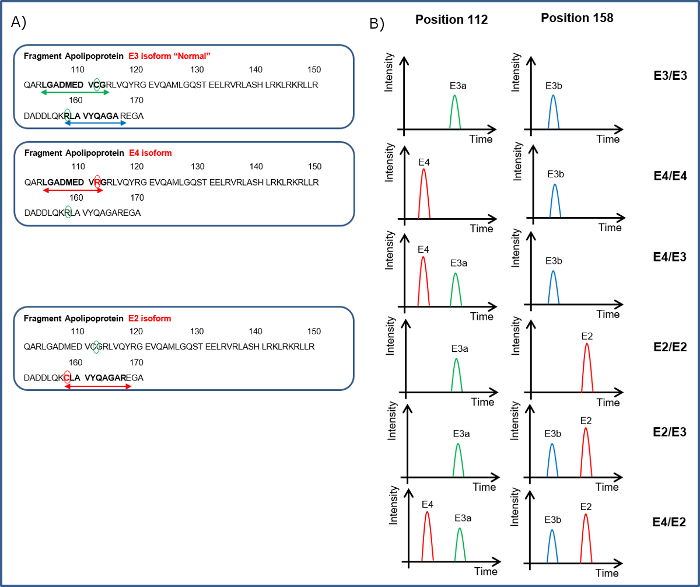 Figure 5. Illustration of How the Apo E Isoform Status of a Patient Can Be Determined.A. Indicates the peptides covering the 112 amino acid sequence LGADMEDVCGR for neutral (E3a) or LGADMEDVR for presence of E4 and for position 158 to detect RLAVYQAGAR the neutral (E3b) or CLAVYQAGAR for the E2 isoform. B. Peptides from the ApoE sequence are shown in the left hand panel. The different combinations of the peptides detected in the CSF can indicate the ApoE isoform status. Please click here to view a larger version of this figure.
Figure 5. Illustration of How the Apo E Isoform Status of a Patient Can Be Determined.A. Indicates the peptides covering the 112 amino acid sequence LGADMEDVCGR for neutral (E3a) or LGADMEDVR for presence of E4 and for position 158 to detect RLAVYQAGAR the neutral (E3b) or CLAVYQAGAR for the E2 isoform. B. Peptides from the ApoE sequence are shown in the left hand panel. The different combinations of the peptides detected in the CSF can indicate the ApoE isoform status. Please click here to view a larger version of this figure.
| Time | Flow (ml/min) | % A | % B | Curve |
| Initial | 0.8 | 97 | 3 | initial |
| 0.2 | 0.8 | 97 | 3 | 6 |
| 7 | 0.8 | 60 | 40 | 6 |
| 7.01 | 0.8 | 0.1 | 99.9 | 6 |
| 8 | 0.8 | 0.1 | 99.9 | 6 |
| 8.01 | 0.8 | 97 | 3 | 1 |
| 10 | 0.8 | 97 | 3 | 1 |
Table 1. UPLC Gradient Settings for a 10 min Method. A = ddH20 0.1% FA, B= ACN, 0.1% FA
Table 2. Peptides Included in the CSF MRM LC MS/MS Assay. Shown are all the included peptides used in the method, which are described and published 8. Indication of whether the marker was reliably detected in 100 µl of CSF is indicated. Transitions labeled in bold are the ones used for quantitative data. Please click here to download this table.
Discussion
As with all MS based assays, the critical steps in the method are the determination of the appropriate and accurate amounts of internal standards. If absolute quantitation is being used, then the correct amounts of spiked peptides in the standard curve are also critical.
Our assay does not require the precipitation of the CSF or the use of any type of clean up or desalting steps prior to MS analysis - it is an entirely one-pot reaction method. Due to the small volume of CSF and its limited complexity (compared to plasma), it has been found that these steps can be removed from the final protocol, thereby simplifying the assay and making it more suitable for translation into diagnostic setting. The inclusions of a pre-column to the main analytical column and of the solvent delays in the MS method, appear to be sufficient to maintain sensitivity during the MS run for more than 500 samples. As the assay-UPLC running time is short, it is important to unequivocally identify the correct peak in the CSF digest matrix.
This can only be achieved with the use of spiked synthesized peptides and a standard curve. If there is an uncertainty of a peptide matched to the correct chromatographic peak then it may be useful to run the sample through multiple transitions and check the transitions intensity pattern with the synthetic standards. All of our assays contain at least 2 peptides transitions to confirm the identity of the peptide.
The assay has been developed for markers detected in up to 100 µl of CSF. For some other markers of dementia, a larger volume of CSF may be needed. Another limitation of the assay is the issue of dynamic range of protein expression. Some abundant markers need to be injected on the mass spectrometer in a smaller amounts whilst some low abundant markers require a larger injection volumes. Therefore the sample may need to be injected twice.
The significance of this technique is the ability to multiplex and the increased specificity of the 3 levels of identification over antibodies (retention time, precursor and product m/z). From the perspective of clinical translation, the biggest advantage is the potential cost saving on antibodies, though an initial outlay for the mass spectrometry equipment and trained personnel is required. Another asset is the speed in which the method can be translated onto the triple quadrupole based systems. This speeds up significantly the ability to translate and validate an assay compared to all immuno-based techniques. Finally, as this platform and expertise has been routinely used to measure small molecules clinically, many large hospital centers already have this infrastructure in place. In the field of neurodegeneration there is a lot of focus and need for new biomarkers in CSF and serum. This method provides a platform for a high-throughput assay where future markers can be added (and removed) to be assessed for efficacy, etc. This technique has further application to other tissues for ApoE isoform identification, such as plasma which has already been described2 and even bloodspots.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was funded and facilitated through the GOSomics research initiative by the National Institute for Health Research, Biomedical Research Centre at Great Ormond Street Hospital and the UCL Biological Mass Spectrometry Centre at the UCL Institute of Child Health with kind donations from the Szeban Peto Foundation. The Dementia Research Centre is an Alzheimer's Research UK coordinating centre. The authors acknowledge the support of Alzheimer's Research UK, the NIHR Queen Square Dementia Biomedical Research Unit, UCL/H Biomedical Research Centre, and Leonard Wolfson Experimental Neurology Centre.
References
- Prince PM, Guerchet DM, Prina DM, et al. Policy Brief: The Global Impact of Dementia 2013-2050. Alzheimers Disease International; 2013. Available from: http://www.alz.co.uk/research/G8-policy-brief. [Google Scholar]
- Hirtz C, et al. Development of new quantitative mass spectrometry and semi-automatic isofocusing methods for the determination of Apolipoprotein E typing. Clin Chim Acta. 2015;454:33–38. doi: 10.1016/j.cca.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Marx V. Targeted proteomics. Nat Methods. 2013;10(1):19–22. doi: 10.1038/nmeth.2285. [DOI] [PubMed] [Google Scholar]
- Heywood W, et al. The development of a peptide SRM-based tandem mass spectrometry assay for prenatal screening of Down syndrome. J Proteomics. 2012;75(11):3248–3257. doi: 10.1016/j.jprot.2012.03.037. [DOI] [PubMed] [Google Scholar]
- Heywood WE, et al. Proteomic Discovery and Development of a Multiplexed Targeted MRM-LC-MS/MS Assay for Urine Biomarkers of Extracellular Matrix Disruption in Mucopolysaccharidoses I, II, and VI. Anal Chem. 2015. [DOI] [PubMed]
- Manwaring V, et al. The identification of new biomarkers for identifying and monitoring kidney disease and their translation into a rapid mass spectrometry-based test: evidence of presymptomatic kidney disease in pediatric Fabry and type-I diabetic patients. J Proteome Res. 2013;12(5):2013–2021. doi: 10.1021/pr301200e. [DOI] [PubMed] [Google Scholar]
- Moat SJ, et al. Newborn blood spot screening for sickle cell disease by using tandem mass spectrometry: implementation of a protocol to identify only the disease states of sickle cell disease. Clin Chem. 2014;60(2):373–380. doi: 10.1373/clinchem.2013.210948. [DOI] [PubMed] [Google Scholar]
- Heywood WE, et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol Neurodegener. 2015;10:64. doi: 10.1186/s13024-015-0059-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes NP, Mann JM, Nita-Lazar A. Selected Reaction Monitoring Mass Spectrometry for Absolute Protein Quantification. J Vis Exp. 2015. p. e52959. [DOI] [PMC free article] [PubMed]
- Craig-Schapiro R, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biol Psychiatry. 2010;68(10):903–912. doi: 10.1016/j.biopsych.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, et al. Identification and validation of novel cerebrospinal fluid biomarkers for staging early Alzheimer's disease. PLoS One. 2011;6(1):e16032. doi: 10.1371/journal.pone.0016032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguori C, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014;71(12):1498–1505. doi: 10.1001/jamaneurol.2014.2510. [DOI] [PubMed] [Google Scholar]