

Abstract
Green fluorescent protein (GFP) and its variants are widely used tools for studying protein localization and dynamics of events such as cytoskeletal remodeling and vesicular trafficking in living cells. Quantitative methodologies using chimeric GFP fusions have been developed for many applications; however, GFP is somewhat resistant to proteolysis, thus its fluorescence persists in the lysosome/vacuole, which can impede quantification of cargo trafficking in the endocytic pathway. An alternative method for quantifying endocytosis and post-endocytic trafficking events makes use of superecliptic pHluorin, a pH-sensitive variant of GFP that is quenched in acidic environments. Chimeric fusion of pHluorin to the cytoplasmic tail of transmembrane cargo proteins results in a dampening of fluorescence upon incorporation of the cargo into multivesicular bodies (MVBs) and delivery to the lysosome/vacuole lumen. Thus, quenching of vacuolar fluorescence facilitates quantification of endocytosis and early events in the endocytic pathway. This paper describes methods using pHluorin-tagged cargos for quantification of endocytosis via fluorescence microscopy, as well as population-based assays using flow cytometry.
Keywords: Molecular Biology, Issue 116, endocytosis, cargo sorting, quantitative microscopy, flow cytometry, live-cell imaging, lysosome, vacuole, endosome, multivesicular body
Introduction
Vesicular trafficking plays an important role in maintaining organelle identity and function in eukaryotic cells, and is a key mechanism for regulating protein and membrane composition of individual cellular compartments. At the plasma membrane, fusion of exocytic vesicles delivers new proteins and membranes to the surface of the cell, whereas vesicles generated through endocytosis remove membrane and proteins from the surface for subsequent recycling or targeting to the lysosome. Thus, endocytosis is important for nutrient uptake and for responses to the extracellular environment. Exocytosis and endocytosis are balanced to regulate plasma membrane surface area, and to allow turnover of damaged proteins.
Studies in yeast and mammalian cells have identified a large number of proteins involved in endocytosis, as well as multiple endocytic pathways that promote internalization of specific cargos or that act in response to a variety of environmental conditions. The best-studied pathway is clathrin-mediated endocytosis (CME), in which clathrin and cytosolic accessory proteins assemble into a coat structure to stabilize the nascent endocytic vesicle. Experiments in the budding yeast Saccharomyces cerevisiae have yielded key insights into the dynamics and order of recruitment for many endocytic proteins1-3. Notably, the CME machinery is highly conserved through evolution such that the majority of CME-related proteins in yeast have human orthologs; thus, budding yeast has been an important tool for understanding mechanisms of endocytosis that are conserved in higher eukaryotes. For example, studies using budding yeast determined that formation and maturation of CME structures (referred to as cortical actin patches in yeast) involves the sequential recruitment of many proteins, beginning with clathrin and cargo-binding adaptor proteins, followed by recruitment of additional endocytic accessory proteins, activation of Arp2/3-mediated actin polymerization, and recruitment of proteins involved in vesicle scission1,2. Recruitment of CME machinery proteins to cortical actin patches is a highly ordered and stereotypical process, and the precise order of recruitment for many proteins has been established with respect to other components of the CME machinery. Importantly, recent studies have confirmed a similar order of recruitment for CME proteins at clathrin-coated pits in mammalian cells4.
In addition to CME, many cell types possess one or more clathrin-independent endocytic (CIE) pathways that rely on alternative mechanisms to promote vesicle formation and internalization from the plasma membrane5-7. In yeast, we recently identified a CIE pathway that utilizes the small GTPase Rho1 and its activating guanine nucleotide exchange factor (GEF), Rom18,9. Rho1 activates the formin Bni1 to promote actin polymerization10,11, which is required for this form of clathrin-independent endocytosis in yeast12. Moreover, the α-arrestin family of proteins recruit the ubiquitin ligase Rsp5 to promote cargo ubiquitination and subsequent internalization through CME13-17, and also promote cargo internalization via the CIE pathway, possibly through direct interaction with proteins involved in CIE18. A variety of CIE pathways also exist in mammalian cells, including clathrin-independent and phagocytic pathways that rely on the Rho1 ortholog, RhoA9,19. The role of RhoA in mammalian CIE is poorly understood; thus, studies in budding yeast may provide additional mechanistic insights that are applicable to mammalian CIE.
Accurate quantification of endocytic events can provide important information about the roles of specific proteins in regulating endocytosis, and can reveal the effects of mutations on cargo internalization or progression through the endocytic pathway. For this purpose, biochemical methods can be utilized to monitor ligand uptake or to measure rates of degradation of endocytic cargo proteins. In living cells, fusion of green fluorescent protein (GFP) and its variants to cargos of interest allows direct visualization of cargo transport. However, GFP-tagged cargos are of limited use for quantification of endocytosis because GFP is resistant to degradation in the lysosome (or vacuole in yeast). Moreover, GFP fluorescence is only partially sensitive to changes in pH, and remains detectable within the vacuole lumen20,21. Consequently, fluorescence of the GFP tag persists in the vacuole long after the remainder of the cargo has been degraded, and whole cell-based quantification of cargo intensity may be inaccurate due to long-term GFP fluorescence in the vacuole.
In order to overcome the drawbacks of vacuolar GFP fluorescence, we previously made use of superecliptic pHluorin, a pH-sensitive variant of GFP that fluoresces brightly at neutral pH, but loses fluorescence in acidic environments such as the lumen of the vacuole/lysosome20,22,23. Placing a pHluorin tag on the cytoplasmic tail of endocytic cargos permits visualization of the cargos at the plasma membrane and on early endosomes, where the pHluorin tag remains exposed to the cytoplasm (Figure 1A). As early endosomes mature, the endosomal sorting complex required for transport (ESCRT) machinery packages surface-localized cargos into vesicles that bud into the lumen of the endosome, generating multivesicular bodies (MVBs)24. For cargos that have been incorporated into internal MVB vesicles, the pHluorin tag faces toward the vesicle lumen. Internal MVB vesicles are acidified; thus, pHluorin-tagged cargos lose fluorescence on early endosomes as they mature into MVBs20. Subsequent fusion of the MVB with the vacuole delivers the MVB luminal contents for degradation, and pHluorin tags remain quenched in the acidic environment of the vacuole lumen.
This paper provides detailed descriptions of quantitative endocytic assays using cargos with cytoplasmic pHluorin tags in yeast. Strains expressing pHluorin-tagged cargos can be used in kinetic and/or endpoint assays, depending on the properties and trafficking behavior of the specific cargo. In addition, some pHluorin-tagged cargos are amenable to high-throughput analysis methods, including flow cytometry. Importantly, the pHluorin tag is a versatile tool for studying endocytic events in living cells, allowing quantification of endocytosis and comparison of endocytic function in wild-type and mutant strains.
Protocol
1. Solutions and Media
- Prepare the Following Stocks, Media and Plates:
- To prepare 10x YNB, dissolve 67 g of yeast nitrogen base lacking amino acids in 1 L of water. Sterilize by filtration through a 0.22 µm nitrocellulose filter.
- Prepare YNB medium using 1x YNB (from 10x stock) with 2% dextrose (from a sterile 50% w/v stock) and 1x amino acid/nutrient mixture (from 100x stock, see step 1.1.4). For plasmid selection, omit individual amino acids or nutrients as needed. For induction of Mup1 expression, additionally omit methionine from the amino acid/nutrient mixture.
- Prepare YNB plates using 1x YNB (from 10x stock), 2% dextrose (from a sterile 50% w/v stock), 0.7 g/L amino acid/nutrient mixture (see step 1.1.5) and 2.5% agar. Prepare plate medium by autoclaving 25 g of agar and 0.7 g of amino acid/nutrient mixture per 860 ml of water. Allow the mixture to cool to 55 °C, then add 100 ml of 10x YNB and 40 ml of 50% glucose. Pour plates (approximately 30 ml of medium per 10 cm dish), allow the medium to solidify at room temperature, and store plates at 4 °C.
- Prepare 100x amino acid/nutrient stock solution (for liquid medium) by dissolving the following in 100 ml of deionized water: 0.1 g L-Methionine, 0.3 g each of L-leucine and L-lysine, and 0.2 g each of L-histidine, L-tryptophan, adenine and uracil (other amino acids and nutrients may be included as needed for specific strains). Use gentle heat if needed to dissolve, and sterilize through a 0.45 µm nitrocellulose filter. Store at room temperature with protection from light. For plasmid selection, prepare stock solutions lacking specific components as needed.
- Prepare amino acid/nutrient mixture (for plates) by combining the following: 0.5 g of adenine, 4 g of L-leucine, and 2 g each of L-histidine, L-lysine, L-methionine, L-tryptophan, L-tyrosine and uracil. Grind with a mortar and pestle, and store with protection from light. For plasmid selection, prepare mixtures lacking specific components as needed, and use 0.7 g of amino acid mixture per liter of plate medium (see step 1.1.3).
Prepare 8-well chamber slides for microscopy by coating the surface of the slide with concanavalin A (ConA). To each well of the chamber slide, add 25 µl of 2 mg/ml ConA dissolved in water. Using a pipette tip, spread the ConA across the bottom surface of the well, then allow the slide to air dry at room temperature for at least 4-8 hr. Ideally, use chamber slides within 24 hr of ConA coating.
- Generate yeast strains with in-frame fusions of GFP or pHluorin to the endocytic cargo of interest using the polymerase chain reaction (PCR)-based integration method described in Longtine et al. and Goldstein and McCusker25,26. NOTE: Plasmids containing pHluorin tagging cassettes with resistance genes for kanamycin (KANMX6) and nourseothricin (NATMX4) were described previously20.
- Amplify GFP or pHluorin cassettes containing selectable markers with primers containing additional sequences specific to the site of genomic integration20,25.
- Transform the resulting PCR product into the desired yeast strain using the lithium acetate method27.
- Select for integration of the cassette by growing cells on YPD plates containing the appropriate drug. To prepare plates, autoclave 10 g yeast extract, 20 g peptone, 0.1 g tryptophan and 25 g agar in 960 ml water. Before pouring plates, allow the mixture to cool to 55 °C, then add 40 ml of 50% glucose and either G418 to a final concentration of 200 µg/ml for KANMX6 selection, or nourseothricin to a final concentration of 100 µg/ml for NATMX4 selection.
- Confirm integration of the GFP or pHluorin tag in individual colonies by PCR and/or Western blotting using anti-GFP antibodies, as well as by detection of the tagged protein by fluorescence microscopy20. NOTE: Specific yeast strains and plasmids used in this study are listed in Tables 1 and 2, respectively.
2. Endpoint Assay for Steady-state Fluorescence of Constitutively Internalized Endocytic Cargo
Inoculate cells (~3 small colonies) in 5 ml of YNB medium lacking amino acids or nutrients as required for plasmid maintenance (if applicable). Grow cells for 16-24 hr in an orbital shaking incubator at 30 °C, with 250 rpm shaking. Alternatively, streak a ~1-2 mm colony of cells onto a YNB plate lacking amino acids or nutrients for plasmid selection, and grow cells overnight at 30 °C.
Measure the density (OD600) of each overnight culture using a spectrophotometer. Prepare a 5 ml dilution at 0.35-0.4 OD600/ml in YNB medium lacking amino acids or nutrients for plasmid maintenance, and grow for approximately 3 hr at 30 °C with shaking. If using cells streaked on a plate, skip this step and proceed with step 2.4 (see below).
Measure the density (OD600) of cells, which should now be between 0.6-1.0 OD600/ml. Transfer 1.5 ml of culture to a microfuge tube, and pellet cells at 8,000 rpm (6,800 x g) for 2 min. Remove 1,475 µl of supernatant.
Bring cells to a fluorescence microscope. Before imaging each sample, prepare a slide by resuspending the cells in the remaining medium, and mount 3 µl of cell suspension under a cover slip. Alternatively, prepare 8-well chamber slides as described below in protocol 3, using YNB medium as appropriate for plasmid selection. Ideally, image cells with a 100X, 1.4 or higher numerical aperture (NA) oil immersion objective lens, a 12- or 16-bit camera, and excitation/emission filters optimized for GFP fluorescence. NOTE: If using cells grown by streaking on a plate, prepare the slide by placing 3 µl of fresh YNB medium on a cover slip. Using a pipette tip, collect a small colony of cells from the plate, disperse the cells into the YNB medium, and mount the resulting suspension on a glass slide immediately before imaging.
Image 4-6 random fields of cells for each condition, using the same acquisition parameters (i.e., filter sets and exposure time) for all images within an experiment. NOTE: Collecting a brightfield or DIC image for each field can be helpful for cells where the pHluorin signal is quenched in MVB and vacuolar compartments.
Quantify fluorescence intensity of at least 30-50 cells for each condition (see protocol 5).
3. Kinetic Assay for Quantification of Endocytosis of Cargos that Undergo Regulated Endocytosis
Inoculate 2-3 colonies of yeast cells (approximately 2 mm in diameter per colony) in 5 ml of YNB medium lacking methionine and additional amino acids or nutrients for maintaining plasmid selection. Grow cultures for 16-24 hr at 30 °C with shaking. NOTE: In this paper, protocols for studying regulated endocytic cargo proteins will make use of the methionine permease, Mup1. Other cargos that undergo regulated endocytosis are also suitable for tagging with pHluorin (for example, the uracil permease Fur4, which accumulates at the plasma membrane in the absence of uracil, and internalizes in response to externally applied uracil); for these cargos, media conditions should be modified to reflect the specific expression, induction and internalization conditions for that cargo.
Measure the density (OD600) of each overnight culture approximately 3 hr before imaging. Prepare a 5 ml dilution at 0.35-0.4 OD600/ml in YNB medium lacking methionine and additional amino acids or nutrients for plasmid maintenance, and grow at 30 °C with shaking.
Pre-equilibrate the microscope's environmental chamber or heated stage (if available) to 30 °C during the 3 hr incubation of step 3.2.
Transfer 1 ml of cell culture to a microfuge tube, and pellet cells at 8,000 rpm (6,800 x g) for 2 min. Remove 975 µl of supernatant.
Prepare a ConA-treated, 8-well glass-bottomed chamber slide for imaging (protocol 1.2).
Add 200 µl of YNB medium lacking methionine and additional amino acids or nutrients for plasmid selection to each well. Resuspend the cell pellet from step 3.4 in the remaining supernatant, and drop 2.5 µl of cell suspension into the center of each well. Disperse cells across the surface of the well by pipetting up and down, and allow cells to settle for 5-10 min.
Place the chamber slide on an inverted fluorescence microscope equilibrated to 30 °C (see step 3.3). Locate a field of cells using brightfield illumination.
Add 50 µl of YNB medium containing 100 µg/ml methionine (pre-warmed to 30 °C) to the 200 µl of medium in the well to obtain a final methionine concentration of 20 µg/ml. Avoid disrupting the medium in the well in order to prevent the cells from floating from the bottom.
Allow the cells to equilibrate for 2 min before beginning imaging. Immediately before capturing the first image, adjust the microscope to the desired focal plane (typically, an equatorial focal plane works best).
Acquire GFP and brightfield (or DIC) images at 5 min intervals for 45-60 min, using identical acquisition parameters for all images within a time series (for example, 100X 1.4 NA oil immersion objective and 500 msec acquisition time using GFP filter, 100 msec acquisition time using DIC filter). NOTE: If the microscope's stage and imaging software do not allow for automatic correction of focal plane drift, it may be necessary to manually readjust the focus prior to each imaging time point. Moreover, acquisition times will vary between microscopes due to differences in illumination and filters, and optimal conditions should be determined manually prior to beginning the experiment. If an endocytic cargo other than Mup1 is used, it may be necessary to modify the acquisition time and imaging intervals, as well as the duration of the experiment, to reflect the internalization kinetics of that cargo.
Quantify fluorescence intensity of individual cells at each time point (see protocol 5), and express values as a percentage of the initial intensity of the first image.
4. Endpoint Assay for Quantification of Endocytic Cargos that Undergo Regulated Endocytosis
Prepare cells for imaging as described in protocol 3 (steps 3.1 through 3.6).
Image 4-5 random fields of cells for each condition immediately before adding methionine, using brightfield and GFP filter sets.
Add 50 µl of YNB medium containing 100 µg/ml methionine (pre-warmed to 30 °C) to the 200 µl of medium in the well to obtain a final methionine concentration of 20 µg/ml. Avoid disrupting the medium in the well in order to prevent the cells from floating from the bottom.
Image 4-5 random fields of cells 30 min after addition of methionine, using the same acquisition parameters as for the 0 min time point (step 4.2).
Quantify fluorescence intensity of at least 30-50 cells for each time point (see protocol 5). Express values as the percentage of Mup1-pHluorin internalized after 30 min using the formula: [Mean intensity at 0 min - Mean intensity at 30 min]/[Mean intensity at 0 min] x 100%. NOTE: It is possible to simultaneously perform up to eight endpoints assays by staggering the start time of each assay. Table 3 provides an example workflow for eight simultaneous assays.
5. Post-acquisition Analysis
Export images from acquisition software as a 16-bit tagged-image file format (.tif or .tiff format). NOTE: Other file formats may also be suitable, and should ideally use lossless compression algorithms. 8-bit images are generally not suitable for quantification purposes.
Open files using ImageJ software (freely available at 'Open' option in the 'File' menu. Use the fluorescence image for quantification, and the brightfield/DIC image as a reference to identify the location of cells and to exclude dead cells (if present), which appear darker than live cells when viewed by DIC and are autofluorescent when viewed with GFP excitation/emission filters.
- Perform a background subtraction on the fluorescence image. For images with an uneven background signal, use the 'Background Subtraction' algorithm found in the 'Process menu'. Use the default setting (rolling ball radius of 50 pixels), which is generally sufficient for quantification purposes. NOTE: An alternative background subtraction method for images with uniform background is described in step 5.3.1.-5.3.2; however, the above method also works for uniform background.
- For images with uniform background (i.e., background intensity is approximately constant at all edges and in the center of the image), perform a manual background subtraction. Click on the 'freehand selections' button found in the main ImageJ window, and select 3-5 random regions within the image that do not contain cells.
- Open the 'Set Measurements' function located in the 'Analyze' menu, select the 'Mean Gray Value' parameter, and measure intensity values using the 'Measure' function (also found in the 'Analyze' menu). Calculate the average value from the 3-5 measured regions, and subtract from all subsequent measurements in that image.
For quantification of kinetic assays (protocol 3), generate a single, stacked image for the time series. Open the fluorescence images from each time point in chronological order, and generate a stack using the 'Images to Stack' function (found in the 'Image' menu, under the Stacks sub-menu).
Outline a cell using the freehand selection tool, making sure to include the entire cell but as little area outside of the cell as possible.
Measure the following parameters: 'Area, Integrated Density' and 'Mean Gray Value'. Select parameters using the 'Set Measurements' function, located in the 'Analyze' menu. NOTE: Integrated Density corresponds to the sum of all pixel intensities within the selected region, and Mean Gray Value corresponds to [Integrated Density/Area], which corrects intensity values for cell size.
Open the 'ROI Manager' located in the 'Analyze' menu, under the 'Tools' sub-menu. In the ROI Manager window, click the 'Add' button to enter the highlighted region as a new region of interest (ROI).
Repeat steps 5.4 through 5.6 for the remaining cells in the image. Exclude any dead cells as assessed by brightfield/DIC and fluorescence (dead cells appear darker in DIC images, and have autofluorescent cytoplasmic signal when viewed with GFP excitation/emission; see Figure 2F), and any cells that touch the edge of the image. Save the selected ROIs as a .zip file by clicking the 'More' button in the 'ROI Manager' window, and export all measurements to a spreadsheet or statistical analysis software.
For quantification of kinetic assays (protocol 3), outline and measure a cell from the initial time point as described in steps 5.4 and 5.5. Use the same ROI for measurement of all subsequent time points in the experiment.
For endpoint assays (protocols 2 and 4), measure a minimum of 30-50 cells for each condition. Perform quantification for at least 2-3 images, and include all cells in each image that meet the criteria outlined in step 5.8 in the analysis.
Assess statistical significance between samples using one-way ANOVA, followed by Tukey's multiple comparison test for post hoc analysis.
6. Population Analysis of Mup1-pHluorin Endocytosis by Flow Cytometry
Inoculate 2-3 colonies of yeast cells (approximately 2 mm in diameter per colony) in 0.5 ml of YNB medium lacking methionine and additional amino acids or nutrients for maintaining plasmid selection. Grow cells in 5 ml round-bottom tubes for 16-24 hr at 30 °C on a roller drum.
Add 1.5 ml of YNB medium lacking methionine and additional amino acids or nutrients for plasmid maintenance, and grow at 30 °C for an additional 3 hr on a roller drum.
Transfer 1 ml of cells into each of two 5 ml round-bottom tubes. Add 0.25 ml of YNB -Met medium to the first tube (-Met condition), and 0.25 ml of YNB medium containing 100 µg/ml methionine to the second tube to give a final methionine concentration of 20 µg/ml (+Met condition). Incubate cells at 30 °C for 45 min on a roller drum.
Analyze cells by flow cytometry, selecting forward scatter (FS) as a measure of cell size, and Mup1-pHluorin fluorescence intensity from a fluorescein isothiocyanate (FITC) filter as a measure of cargo internalization.
Use the slider buttons to adjust the voltage of the FS and FITC channels to optimize detection for the size range and fluorescence intensity of the yeast cells being used (for these experiments, settings used were 50 V for FS and 400 V for FITC). NOTE: Voltage for both channels will vary depending on the flow cytometer being used. This step should be done before or during the 45 min treatment with methionine (step 6.3).
Measure FS and fluorescence intensity for 10,000 cells from each condition, using the high flow rate setting. Generate a scatter plot of FS against fluorescence intensity: click 'add graph', select FITC (x-axis) and FS (y-axis), and graph 1,000 points (the software will display values for 1,000 cells, even though 10,000 cells were measured). NOTE: For maximal consistency, analyze cells 45-50 min after addition of methionine; for experiments involving large numbers of samples, stagger addition of methionine to accommodate the time required for flow cytometry analysis.
Include -Met and +Met conditions for wild-type (WT) cells as a control. Using the gate function, assign a vertical gate using the WT +Met condition, such that approximately 5% of the brightest cells fall to the right of the gate. Use this gating for all other conditions in the experiment.
Representative Results
Steady-state localization and quantification of the constitutively internalized endocytic cargo protein Ste3
To demonstrate that pHluorin-tagged cargos can be utilized for quantification of endocytosis in living cells, localization of chimeric GFP and pHluorin fusions with the cytoplasmic C-terminal tail of Ste3, the a-factor pheromone receptor in yeast, were compared. Ste3 is a G protein-coupled receptor (GPCR) that is constitutively transported to the plasma membrane, internalized, and targeted to the vacuole for degradation28. Thus, under steady-state conditions, the majority of Ste3 localizes to the vacuole lumen, as seen in wild-type (WT) cells expressing Ste3-GFP (Figure 2A). In contrast, cells lacking the genes encoding the four clathrin-binding adaptor proteins Ent1, Ent2, Yap1801 and Yap1802 (ent1Δ ent2Δ yap1801Δ yap1802Δ, hereafter referred to as 4Δ) fail to stabilize clathrin at sites of endocytosis29, and are severely defective in CME30. 4Δ cells require expression of the epsin N-terminal homology (ENTH) domain of either Ent1 or Ent2 for viability30,31. The ENTH domain is not sufficient for endocytosis in 4Δ cells, as shown by retention of Ste3-GFP at the plasma membrane in 4Δ+ENTH1 cells expressing the ENTH domain of Ent1 (Figure 2A)8,30; similar plasma membrane retention in 4Δ+ENTH1 cells has been observed for other endocytic cargos, including Ste2-GFP (the α-factor pheromone receptor) and Mup1-GFP or Mup1-pHluorin (a methionine permease)8,18. In contrast, expression of full-length Ent1 from a plasmid in 4Δ cells (4Δ+Ent1) restores endocytosis and localization of Ste3-GFP to the vacuole. 4Δ+ENTH1 cells were recently used in a genetic screen to identify a CIE pathway in yeast that relies on the actin-modulating GTPase Rho1 and its GEF, Rom1, as well as the formin Bni1 and members of the α-arrestin family of proteins involved in cargo sorting8,18. Consistent with a role in CIE, expression of ROM1 or the α-arrestin LDB19 from high-copy plasmids in 4Δ+ENTH1 cells improved internalization of Ste3-GFP (Figure 2A).
Cells expressing Ste3-GFP or other GFP-tagged endocytic cargos have bright vacuoles due to accumulation and proteolytic resistance of the GFP tag. In contrast, cells expressing Ste3-pHluorin have very low levels of vacuolar fluorescence due to quenching of the pHluorin tag in acidic environments20. As seen previously, WT and 4Δ+Ent1 cells showed very little detectable Ste3-pHluorin by fluorescence microscopy (Figure 2B). In contrast, Ste3-pHluorin was readily detectable at the plasma membrane of 4Δ+ENTH1 cells, while vacuolar Ste3-pHluorin remained nearly undetectable in all cases. High-copy expression of ROM1 or LDB19 reduced the amount of detectable Ste3-pHluorin at the cell surface, similar to results with Ste3-GFP, although vacuolar Ste3-pHluorin remained nearly undetectable in all cases.
To compare the effectiveness of GFP- and pHluorin-tagged cargos as tools for quantifying changes in the endocytic capacity of cells, whole-cell fluorescence of Ste3-GFP and Ste3-pHluorin was measured in WT and 4Δ cells. Although changes in Ste3-GFP localization were readily detectable by microscopy (Figure 2A), differences in overall fluorescence intensity were not statistically significant between WT, 4Δ+Ent1 and 4Δ+ENTH1 cells transformed with empty vector (Figure 2C)20. Likewise, 4Δ+ENTH1 cells transformed with vector, ROM1 and LDB19 had similar whole-cell fluorescence intensities. Notably, 4Δ+ENTH1 cells transformed with ROM1 or LDB19 were significantly dimmer than WT cells (Figure 2C)8,18; however, Ste3-GFP protein levels were similar as assessed by immunoblotting (Figure 2E). While the difference in intensity may be due in part to changes in vacuole size or morphology, or to differences in retention of the GFP tag in the vacuole, quantification of Ste3-GFP fluorescence does not reflect the changes in localization observed by fluorescence microscopy (Figure 2A). In contrast, quantification of Ste3-pHluorin intensity demonstrated that 4Δ+ENTH1 cells transformed with vector were significantly brighter than WT or 4Δ+Ent1 cells, and high-copy expression of ROM1 or LDB19 in 4Δ+ENTH1 cells reduced Ste3-pHluorin intensity to levels that were similar to WT and 4Δ+Ent1 (Figure 2D). Thus, quantification of steady-state Ste3-pHluorin intensity accurately reflects the differences in localization observed by fluorescence microscopy.
Kinetic internalization assays using the regulated endocytic cargo, Mup1
Quantification of fluorescence intensity for constitutively internalized cargos at steady-state can reveal differences in the endocytic capacity of mutant cells compared to WT cells. However, it is possible that some cargos may reach the same steady-state distribution and intensity in WT and endocytic mutant cells, even though the mutant cells have a delay in endocytosis or slower kinetics for cargo internalization. Instead, pHluorin-tagged cargos that undergo regulated endocytosis in response to a specific stimulus or ligand can be used to monitor the kinetics of endocytosis. For this purpose, internalization of the high-affinity methionine permease, Mup1, was monitored32. In the absence of extracellular methionine, Mup1 expression is upregulated, and the permease is retained at the plasma membrane; upon addition of methionine to the medium, Mup1 undergoes rapid internalization and targeting to the vacuole13.
To monitor the kinetics of Mup1 internalization, WT strains expressing Mup1-GFP or Mup1-pHluorin from the genomic locus were grown in the absence of methionine in order to accumulate fluorescent Mup1 at the plasma membrane (Figure 3A, 0 min time point). Time-lapse imaging of cells was then performed at 5 min intervals in the absence or presence of methionine to monitor cargo internalization and changes in fluorescence. As expected, GFP- and pHluorin-tagged Mup1 imaged in the absence of methionine remained at the plasma membrane for the entire 45 min observation period20. In contrast, cells imaged in the presence of methionine showed progressive depletion of the fluorescence signal from the cell surface, consistent with cargo internalization. Notably, Mup1-GFP fluorescence accumulated in internal structures (i.e., endosomes and the vacuole), whereas Mup1-pHluorin localized to internal punctae that likely correspond to early endosomes, but was not seen in the vacuole.
When fluorescence was quantified across all time points for individual cells, Mup1-GFP and Mup1-pHluorin intensity showed little change in the absence of externally applied methionine during the course of the 45 min experiment, consistent with the protein remaining stably localized to the plasma membrane (Figure 3B). In contrast, whole-cell Mup1-pHluorin intensity rapidly decreased in the presence of methionine, such that a 50% reduction in fluorescence intensity was observed after approximately 20-25 min, and an 80% reduction was observed after approximately 40 min20. Mup1-GFP cells also showed a decrease in fluorescence intensity upon addition of methionine; however, the kinetics of fluorescence loss were delayed compared to those of Mup1-pHluorin, such that a 50% decrease in intensity was only observed after 40 min. As seen in Figure 3A, Mup1-GFP was barely detectable at the plasma membrane at this time, indicating that persistent vacuolar GFP fluorescence prevented accurate quantification of endocytic events at the cell surface.
Endpoint assays using the regulated endocytic cargo, Mup1
In addition to quantitative, kinetic assays of endocytosis as described above, regulated endocytic cargos such as Mup1 can also be used for endpoint assays, similar to quantification of steady-state Ste3-pHluorin intensity. For this purpose, the assay for quantification of Mup1-pHluorin internalization was modified to allow comparison of mean fluorescence intensities from populations of cells imaged immediately before or 30 min after addition of methionine8,18. This approach allows comparison of the percentage of Mup1 internalized between WT and endocytic mutant cells.
In agreement with the results of the kinetic assay (Figure 3), Mup1-pHluorin was rapidly depleted from the plasma membrane in WT cells (Figure 4A), such that approximately 60% of Mup1-pHluorin was internalized 30 min after addition of methionine (Figure 4B). As expected, Mup1-pHluorin internalization was similarly efficient in 4+Ent1 cells, while internalization was significantly reduced in 4Δ+ENTH1 cells, consistent with a severe defect in endocytosis. High-copy expression of ROM1 or the α-arrestin LDB19, which is specifically required for Mup1 internalization via both CME and CIE pathways13,18, improved internalization of Mup1-pHluorin, consistent with their ability to promote endocytosis in 4Δ+ENTH1 cells18. Notably, although steady-state Ste3-pHluorin intensity in 4Δ+ENTH1 cells expressing high-copy ROM1 or LDB19 was indistinguishable from WT or 4Δ+Ent1 cells, internalization of Mup1-pHluorin was only partially improved in 4Δ+ENTH1 cells expressing high-copy ROM1 or LDB19 (Figure 4C). Thus, these data indicate that kinetic and/or endpoint assays can reveal differences in endocytic rates between WT and mutant strains for some cargos, even though similar steady-state distribution of other cargos may be achieved in the same strains.
Population-based analysis of Mup1-pHluorin internalization using flow cytometry
High-throughput analysis of larger populations of cells using flow cytometry can provide an alternative to performing kinetic and endpoint assays for cargo internalization by microscopy. For this purpose, fluorescence intensity of Mup1-pHluorin was compared in WT cells from a culture grown in the absence of methionine (WT -Met, which should be "bright") or in the presence of methionine for 45 min (WT +Met, which should be "dim" compared to untreated cells based on the 80% reduction in fluorescence intensity seen in Figure 3B). After analysis by flow cytometry, a vertical gate was applied to the WT +Met condition, where ~5% of cells were contained on the right-hand side of the gate, and corresponded to the brightest cells within the population (Figure 5A and 5B, red population). When the same gate was applied to the WT -Met condition, approximately 65% of cells fell into the bright category, demonstrating that flow cytometry can be used to readily observe differences in Mup1-pHluorin fluorescence intensity between populations of cells before and after addition of methionine. In contrast, the distribution of bright versus dim cells was indistinguishable in 4Δ+ENTH1 -Met and +Met conditions using the same gate applied to the WT +Met population, consistent with defective endocytosis. Thus, flow cytometry can detect changes in the endocytic capacity of cells, and permits rapid analysis of large populations of cells. Importantly, flow cytometry can also be used in genetic screens as a tool for sorting and selection of mutant cells with defective endocytosis (K. Wrasman and B. Wendland, manuscript in preparation)33.
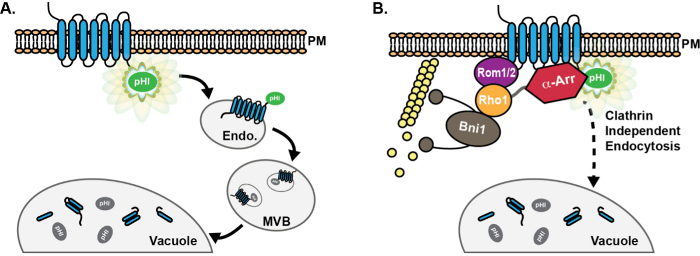 Figure 1:Schematic of pHluorin-tagged cargo progression through the endocytic pathway. (A) Chimeric fusion of a pHluorin (pHl) tag on the cytoplasmic tail of endocytic cargo proteins results in detectable fluorescence (green) of the cargo at the plasma membrane (PM). Following endocytosis, cargo proteins are delivered to the early endosome (Endo), where the pHluorin tag remains exposed to the cytoplasm, and is thus fluorescent. Subsequently, the ESCRT machinery promotes cargo sorting and incorporation into luminal vesicles of multivesicular bodies (MVB). MVBs then fuse with the vacuole to deliver proteins and membrane components for degradation. Upon incorporation into MVBs, the pHluorin tag becomes quenched (grey) due to acidification. (B) Use of pHluorin-tagged cargos has facilitated the discovery and characterization of proteins involved in a clathrin-independent endocytic (CIE) pathway in yeast. In cells with defective clathrin-mediated endocytosis, high-copy expression of the GTPase Rho1, as well as its activating GEF Rom1, promote cargo internalization via CIE8. Moreover, although the α-arrestin family (α-Arr) of proteins has known roles in promoting cargo ubiquitination and internalization via CME13-17, roles for α-arrestins in CIE were recently identified, likely due to interaction with CIE proteins rather than through recruitment of ubiquitin ligases18. Please click here to view a larger version of this figure.
Figure 1:Schematic of pHluorin-tagged cargo progression through the endocytic pathway. (A) Chimeric fusion of a pHluorin (pHl) tag on the cytoplasmic tail of endocytic cargo proteins results in detectable fluorescence (green) of the cargo at the plasma membrane (PM). Following endocytosis, cargo proteins are delivered to the early endosome (Endo), where the pHluorin tag remains exposed to the cytoplasm, and is thus fluorescent. Subsequently, the ESCRT machinery promotes cargo sorting and incorporation into luminal vesicles of multivesicular bodies (MVB). MVBs then fuse with the vacuole to deliver proteins and membrane components for degradation. Upon incorporation into MVBs, the pHluorin tag becomes quenched (grey) due to acidification. (B) Use of pHluorin-tagged cargos has facilitated the discovery and characterization of proteins involved in a clathrin-independent endocytic (CIE) pathway in yeast. In cells with defective clathrin-mediated endocytosis, high-copy expression of the GTPase Rho1, as well as its activating GEF Rom1, promote cargo internalization via CIE8. Moreover, although the α-arrestin family (α-Arr) of proteins has known roles in promoting cargo ubiquitination and internalization via CME13-17, roles for α-arrestins in CIE were recently identified, likely due to interaction with CIE proteins rather than through recruitment of ubiquitin ligases18. Please click here to view a larger version of this figure.
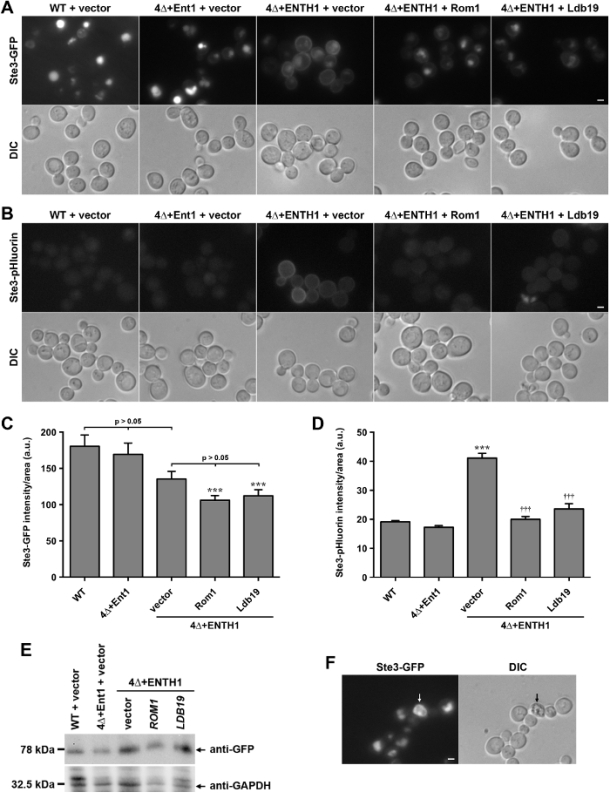 Figure 2:Localization and quantification of Ste3-GFP and Ste3-pHluorin in wild-type and endocytic mutant cells. (A) WT, 4Δ+Ent1 and 4Δ+ENTH1 cells expressing genomically encoded Ste3-GFP and transformed with vector, high-copy ROM1 or high-copy LDB19 as indicated were visualized by fluorescence microscopy (top row) or DIC (bottom row). Ste3-GFP images were adjusted to the same maximum and minimum intensities to allow direct comparison of Ste3 localization. (B) WT, 4Δ+Ent1 and 4Δ+ENTH1 cells expressing genomically encoded Ste3-pHluorin were transformed and imaged as described in panel A. Scale bar = 2 µm. (C, D) Quantification of fluorescence intensity for Ste3-GFP (C) and Ste3-pHluorin (D) strains used in panels A and B. For each condition, whole-cell fluorescence was quantified for a minimum of 40 cells. Values were corrected for cell size, and expressed in arbitrary units (a.u.) as mean ± SEM (*** p <0.001 compared to WT; ††† p <0.001 compared to 4Δ+ENTH1 + vector). (E) Relative expression of Ste3-GFP assessed in WT, 4Δ+Ent1 and 4Δ+ENTH1 cells transformed with vector, high-copy ROM1 or high-copy LDB19 as indicated. Cell extracts were prepared as described previously20, and equal amounts of each sample were resolved by SDS-PAGE. Ste3-GFP expression was assessed by immunoblotting with anti-GFP antibodies, and protein loading was assessed using anti-GAPDH. (F) Wild-type cells expressing genomically encoded Ste3-GFP viewed by fluorescence microscopy (Ste3-GFP panel) and DIC optics, with a dead cell indicated by arrows. Dead cells appear autofluorescent in the GFP filter, and are darker when viewed by DIC. Scale bar = 2 µm. Please click here to view a larger version of this figure.
Figure 2:Localization and quantification of Ste3-GFP and Ste3-pHluorin in wild-type and endocytic mutant cells. (A) WT, 4Δ+Ent1 and 4Δ+ENTH1 cells expressing genomically encoded Ste3-GFP and transformed with vector, high-copy ROM1 or high-copy LDB19 as indicated were visualized by fluorescence microscopy (top row) or DIC (bottom row). Ste3-GFP images were adjusted to the same maximum and minimum intensities to allow direct comparison of Ste3 localization. (B) WT, 4Δ+Ent1 and 4Δ+ENTH1 cells expressing genomically encoded Ste3-pHluorin were transformed and imaged as described in panel A. Scale bar = 2 µm. (C, D) Quantification of fluorescence intensity for Ste3-GFP (C) and Ste3-pHluorin (D) strains used in panels A and B. For each condition, whole-cell fluorescence was quantified for a minimum of 40 cells. Values were corrected for cell size, and expressed in arbitrary units (a.u.) as mean ± SEM (*** p <0.001 compared to WT; ††† p <0.001 compared to 4Δ+ENTH1 + vector). (E) Relative expression of Ste3-GFP assessed in WT, 4Δ+Ent1 and 4Δ+ENTH1 cells transformed with vector, high-copy ROM1 or high-copy LDB19 as indicated. Cell extracts were prepared as described previously20, and equal amounts of each sample were resolved by SDS-PAGE. Ste3-GFP expression was assessed by immunoblotting with anti-GFP antibodies, and protein loading was assessed using anti-GAPDH. (F) Wild-type cells expressing genomically encoded Ste3-GFP viewed by fluorescence microscopy (Ste3-GFP panel) and DIC optics, with a dead cell indicated by arrows. Dead cells appear autofluorescent in the GFP filter, and are darker when viewed by DIC. Scale bar = 2 µm. Please click here to view a larger version of this figure.
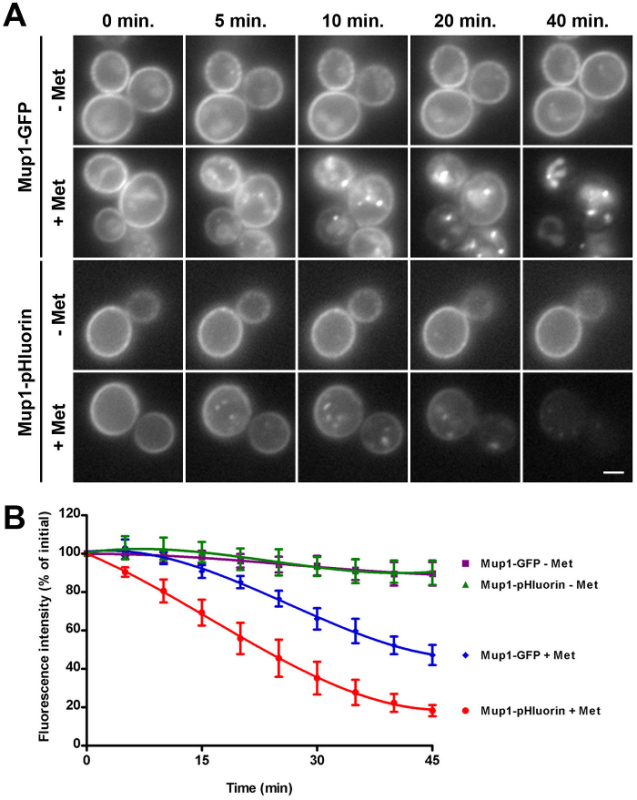 Figure 3:Kinetic assay of Mup1-GFP and Mup1-pHluorin internalization. (A) Wild-type cells expressing genomically encoded Mup1-GFP (upper two panels) or Mup1-pHluorin (lower two panels) were grown to mid-logarithmic phase in YNB medium lacking methionine to induce Mup1 expression and retention at the plasma membrane. Cells were then imaged by fluorescence microscopy every 5 min in the absence (- Met) or presence (+ Met) of 20 µg/ml methionine. For each time point within a condition, identical maximum and minimum intensity values were applied to allow comparison of fluorescence intensity and localization. 0, 5, 10, 20 and 40 min time points are shown as indicated for all conditions. Scale bar = 2 µm. (B) Quantification of Mup1-GFP and Mup1-pHluorin internalization in wild-type cells from panel A. Whole-cell fluorescence intensity of Mup1-GFP (- Met, purple squares; + Met, blue diamonds) or Mup1-pHluorin (- Met, green triangles; + Met, red circles) was measured for individual cells imaged at 5 min intervals, and expressed as a percentage of the initial fluorescence intensity (mean ± SEM, n = 6 cells per condition). Reprinted from Prosser et al. (2010)20 with permission from the publisher. Please click here to view a larger version of this figure.
Figure 3:Kinetic assay of Mup1-GFP and Mup1-pHluorin internalization. (A) Wild-type cells expressing genomically encoded Mup1-GFP (upper two panels) or Mup1-pHluorin (lower two panels) were grown to mid-logarithmic phase in YNB medium lacking methionine to induce Mup1 expression and retention at the plasma membrane. Cells were then imaged by fluorescence microscopy every 5 min in the absence (- Met) or presence (+ Met) of 20 µg/ml methionine. For each time point within a condition, identical maximum and minimum intensity values were applied to allow comparison of fluorescence intensity and localization. 0, 5, 10, 20 and 40 min time points are shown as indicated for all conditions. Scale bar = 2 µm. (B) Quantification of Mup1-GFP and Mup1-pHluorin internalization in wild-type cells from panel A. Whole-cell fluorescence intensity of Mup1-GFP (- Met, purple squares; + Met, blue diamonds) or Mup1-pHluorin (- Met, green triangles; + Met, red circles) was measured for individual cells imaged at 5 min intervals, and expressed as a percentage of the initial fluorescence intensity (mean ± SEM, n = 6 cells per condition). Reprinted from Prosser et al. (2010)20 with permission from the publisher. Please click here to view a larger version of this figure.
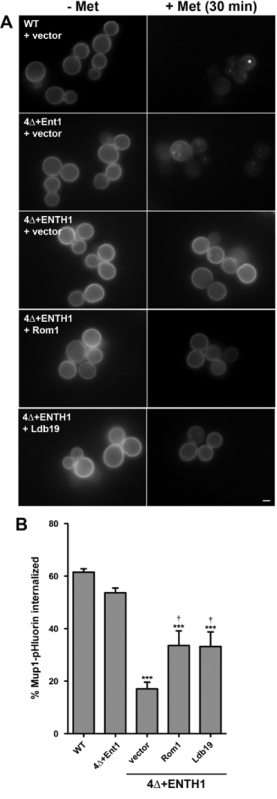 Figure 4: Endpoint assay of Mup1-pHluorin internalization. (A) WT, 4Δ+Ent1 and 4Δ+ENTH1 cells expressing genomically encoded Mup1-pHluorin and transformed with vector, high-copy ROM1 or high-copy LDB19 as indicated were grown to mid-logarithmic phase in YNB medium lacking methionine. Random fields of cells were imaged by fluorescence microscopy immediately before (- Met) or 30 min after (+ Met) addition of 20 µg/ml methionine. All images were adjusted to the same maximum and minimum intensity values. Scale bar = 2 µm. (B) Quantification of Mup1-pHluorin internalization in cells from panel A. For each condition, whole-cell fluorescence intensity was measured for a minimum of 40 cells and corrected for cell size. The percentage of Mup1-pHluorin internalized was calculated by comparing mean intensity values before and after 30 min treatment with methionine. Values are shown as mean ± SEM (n = 4; *** p <0.001 compared to WT; † p <0.05 compared to 4Δ+ENTH1 + vector). This figure has been modified from Prosser et al. (2015)18 with permission from the publisher. Please click here to view a larger version of this figure.
Figure 4: Endpoint assay of Mup1-pHluorin internalization. (A) WT, 4Δ+Ent1 and 4Δ+ENTH1 cells expressing genomically encoded Mup1-pHluorin and transformed with vector, high-copy ROM1 or high-copy LDB19 as indicated were grown to mid-logarithmic phase in YNB medium lacking methionine. Random fields of cells were imaged by fluorescence microscopy immediately before (- Met) or 30 min after (+ Met) addition of 20 µg/ml methionine. All images were adjusted to the same maximum and minimum intensity values. Scale bar = 2 µm. (B) Quantification of Mup1-pHluorin internalization in cells from panel A. For each condition, whole-cell fluorescence intensity was measured for a minimum of 40 cells and corrected for cell size. The percentage of Mup1-pHluorin internalized was calculated by comparing mean intensity values before and after 30 min treatment with methionine. Values are shown as mean ± SEM (n = 4; *** p <0.001 compared to WT; † p <0.05 compared to 4Δ+ENTH1 + vector). This figure has been modified from Prosser et al. (2015)18 with permission from the publisher. Please click here to view a larger version of this figure.
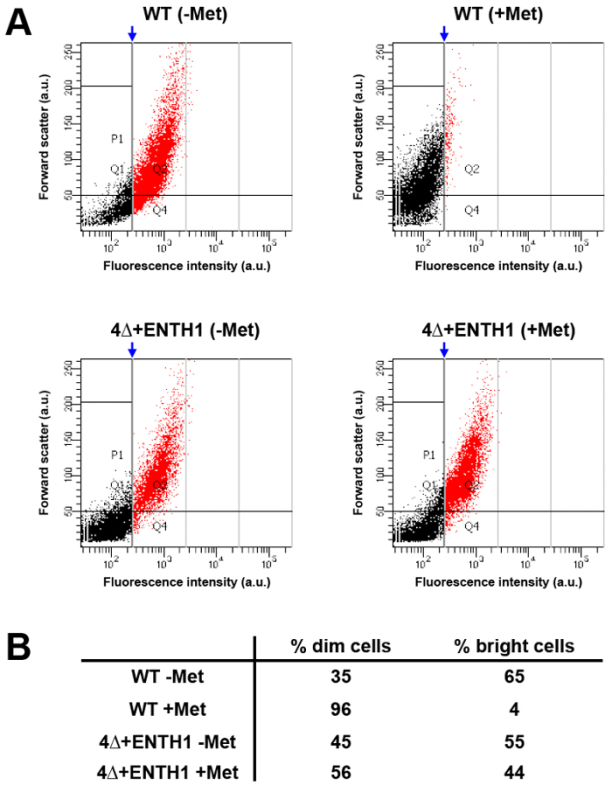 Figure 5:Population analysis of Mup1-pHuorin internalization by flow cytometry. (A) WT and 4Δ+ENTH1 cells were grown to mid-logarithmic phase in YNB medium lacking methionine. Cells were then incubated in the absence (- Met) or presence (+ Met) of 20 µg/ml methionine for 45 min prior to analysis by flow cytometry. For each condition, forward scatter (in arbitrary units, a.u.) was plotted against fluorescence intensity (a.u., using a FITC filter) for 1,000 cells out of a total of 10,000 cells measured. Populations were further analyzed by applying a vertical gate (blue arrow) to the WT + Met condition, where approximately 5% of the brightest cells (shown in red) fell on the right-hand side of the gate. The gating generated for the WT + Met condition was then applied to the remaining conditions to allow comparison of sample distributions. (B) Summary of the percentage of dim cells (black points, to the left of the gate) and bright cells (red points, to the right of the gate) for WT and 4Δ+ENTH1 ± Met trials shown in panel A. Please click here to view a larger version of this figure.
Figure 5:Population analysis of Mup1-pHuorin internalization by flow cytometry. (A) WT and 4Δ+ENTH1 cells were grown to mid-logarithmic phase in YNB medium lacking methionine. Cells were then incubated in the absence (- Met) or presence (+ Met) of 20 µg/ml methionine for 45 min prior to analysis by flow cytometry. For each condition, forward scatter (in arbitrary units, a.u.) was plotted against fluorescence intensity (a.u., using a FITC filter) for 1,000 cells out of a total of 10,000 cells measured. Populations were further analyzed by applying a vertical gate (blue arrow) to the WT + Met condition, where approximately 5% of the brightest cells (shown in red) fell on the right-hand side of the gate. The gating generated for the WT + Met condition was then applied to the remaining conditions to allow comparison of sample distributions. (B) Summary of the percentage of dim cells (black points, to the left of the gate) and bright cells (red points, to the right of the gate) for WT and 4Δ+ENTH1 ± Met trials shown in panel A. Please click here to view a larger version of this figure.
| Strain | Genotype | Source |
| SEY6210a | MAT α his3-∆200 trp1-∆901 leu2-3,112 ura3-52 lys2-801 suc2-∆9 | Laboratory plasmid |
| BWY2858 | STE3-GFP::KANMX6 | Prosser et al. (2010)16 |
| BWY2995 | STE3-pHluorin::KANMX6 | Prosser et al. (2010)16 |
| BWY3036 | ent1∆::LEU2 ent2∆::HIS3 yap1801∆::HIS3 yap1802∆::LEU2 STE3-pHluorin::KANMX6 + pEnt1 [CEN TRP1] | Prosser et al. (2010)16 |
| BWY3037 | ent1∆::LEU2 ent2∆::HIS3 yap1801∆::HIS3 yap1802∆::LEU2 STE3-pHluorin::KANMX6 + pENTH1 [CEN TRP1] | Prosser et al. (2010)16 |
| BWY3399 | ent1∆::LEU2 ent2∆::HIS3 yap1801∆::HIS3 yap1802∆::LEU2 STE3-GFP::KANMX6 + pEnt1 [CEN TRP1] | Prosser et al. (2010)16 |
| BWY3400 | ent1∆::LEU2 ent2∆::HIS3 yap1801∆::HIS3 yap1802∆::LEU2 STE3-GFP::KANMX6 + pEnt1 [CEN TRP1] | Prosser et al. (2010)16 |
| BWY3817 | MUP1-GFP::KANMX6 | Prosser et al. (2010)16 |
| BWY3818 | MUP1-pHluorin::KANMX6 | Prosser et al. (2010)16 |
| BWY4153 | ent1∆::LEU2 ent2∆::HIS3 yap1801∆::HIS3 yap1802∆::LEU2 MUP1-pHluorin::KANMX6 + pEnt1 [CEN TRP1] | Prosser et al. (2011)7 |
| BWY4154 | ent1∆::LEU2 ent2∆::HIS3 yap1801∆::HIS3 yap1802∆::LEU2 MUP1-pHluorin::KANMX6 + pENTH1 [CEN TRP1] | Prosser et al. (2011)7 |
| aAll strains used in this study are isogenic to SEY6210 except at the indicated loci. |
Table 1: Yeast strains used in this study.
| Plasmid | Description | Source |
| pRS426 | 2µ URA3 | Sikorski and Hieter, 1989 |
| pBW0768 | pRS414::ENT1 [CEN TRP1] | pEnt1, Laboratory plasmid |
| pBW0778 | pRS414::ent1(aa1-151) [CEN TRP1] | pENTH1, Laboratory plasmid |
| pBW1571 | pFA6a-pHluorin-KANMX6 | Prosser et al. (2010)16 |
| pBW2053 | YEp24::ROM1 [2µ URA3] | pROM1 (Prosser et al., 2011)7 |
| pRS426-Ldb19 | LDB19prom-LDB19 [2µ URA3] | Prosser et al. (2015)15 |
| pFA6a-GFP(S65T)-kanMX6 | pFA6a-GFP-KANMX6, PCR-based integrating plasmid for GFP-tagging in yeast | Longtine et al. (1998)21 |
Table 2: Plasmids used in this study.
| Time (min) | Action(s) |
| -5 | Image sample 1 (-Met) |
| 0 | Add methionine to sample 1 |
| Image sample 2 (-Met) | |
| 5 | Add methionine to sample 2 |
| Image sample 3 (-Met) | |
| 10 | Add methionine to sample 3 |
| Image sample 4 (-Met) | |
| 15 | Add methionine to sample 4 |
| Image sample 5 (-Met) | |
| 20 | Add methionine to sample 5 |
| Image sample 6 (-Met) | |
| 25 | Add methionine to sample 6 |
| Image sample 7 (-Met) | |
| 30 | Add methionine to sample 7 |
| Image sample 8 (-Met) | |
| Image sample 1 (+Met) | |
| 35 | Add methionine to sample 8 |
| Image sample 2 (+Met) | |
| 40 | Image sample 3 (+Met) |
| 45 | Image sample 4 (+Met) |
| 50 | Image sample 5 (+Met) |
| 55 | Image sample 6 (+Met) |
| 60 | Image sample 7 (+Met) |
| 65 | Image sample 8 (+Met) |
Table 3: Example workflow for staggering 8 Mup1-pHluorin endpoint assays.
Discussion
Applications of pHluorin for quantification of endocytic events in yeast cells makes use of transmembrane cargos in which the fluorescent tag is fused to the cytoplasmic tail of the protein (Figure 1A). For the assays described here, cargos are brightly fluorescent when the pHluorin tag is exposed to neutral environments, but become quenched when the pHluorin tag encounters acidic conditions. Thus, a pHluorin-tagged endocytic cargo is readily detected at the cytoplasmic face of the plasma membrane and on early endosomes, but becomes "dim" when incorporated into luminal MVB vesicles or upon delivery to the vacuole lumen. In general, newly synthesized endocytic cargos likely leave the endoplasmic reticulum (ER) and reach the plasma membrane before GFP and many of its variants have fully matured; thus, pHluorin-tagged cargos that have not yet reached the cell surface are predicted to remain undetectable unless exit from the ER or Golgi is impaired.
Use of pHluorin-tagged endocytic cargos such as Ste3 and Mup1 has facilitated characterization of a number of proteins involved in the yeast CIE pathway (Figure 1B)8,18. Specifically, the methods described in this paper have been used to demonstrate that increased production of the actin-modulating GTPase Rho1 and its GEF Rom1 promote endocytosis in a variety of yeast mutants with defects in CME8. In addition, pHluorin-tagged cargos were recently used to quantitatively demonstrate that α-arrestins, which have well-established roles in CME13-15,34-36, play cargo-selective roles in both CME and CIE, and have mechanistically distinct functions in the two pathways18.
For the protocols outlined in this paper, several important factors should be considered in order to obtain consistent, quantitative results. First, Mup1-GFP or Mup1-pHluorin expression and fluorescence may decrease as yeast cells approach or enter the stationary growth phase (unpublished results); thus, cells grown from an overnight culture should be diluted and re-grown for an additional 3 hr (see steps 3.1-3.2 and 6.1-6.2). In addition, when staggering multiple experimental conditions or strains in endpoint assays or for analysis by flow cytometry, it is important to collect data for all samples after the same duration of methionine treatment in order to allow comparisons between individual conditions. For post-acquisition analysis, quantification should be performed using 16-bit images, since conversion to 8-bit format causes data loss that can affect the fluorescence intensity values. Moreover, background subtraction should be performed prior to quantification (step 5.3), since the background signal can significantly occlude differences in intensity between samples.
Whereas pHluorin-tagged Ste3 and Mup1 have proven amenable to quantification, not all cargos will be well-suited to pHluorin tagging. For example, attempts to tag the α-factor receptor Ste2 have yielded inconsistent quantitative results, possibly due to lower fluorescence intensity of Ste2-pHluorin compared to that of Ste3 or Mup1 (unpublished results), although it is possible that tandem pHluorin tags could improve the signal intensity. In addition, cargos with very slow rates of ligand-induced endocytosis may not be amenable to pHluorin tagging for quantification, especially if the half-time of internalization is longer than the yeast cell cycle (approximately 90 min). For such cargos, a decrease in fluorescence could arise from endocytosis and/or from partitioning of the cargo between mother and daughter cells during division. Thus, testing individual cargos of interest is recommended in order to determine whether their expression and kinetics are suitable for pHluorin-tagging and quantification.
Although recent studies using pHluorin have focused mainly on endocytic events at the plasma membrane, pHluorin-tagged cargos can be used to study later trafficking events in the endocytic pathway. For example, Mup1-pHluorin has been successfully used in flow cytometry-based assays of ESCRT-mediated cargo sorting into MVBs37, since the pHluorin tag becomes quenched upon incorporation into MVB luminal vesicles20. This approach is similar to the more recently described luciferase reporter of intraluminal deposition (LUCID) assay, which makes use of cargos with a cytoplasmic luciferase tag to generate a bioluminescent signal in the presence of luciferin38,39. Similar to quenching of pHluorin fluorescence upon incorporation into MVB luminal vesicles, bioluminescence in the LUCID assay is attenuated upon incorporation of the luciferase tag into MVB vesicles. While pHluorin and LUCID assays are conceptually similar, pHluorin-tagged cargos can be used to study endocytosis and MVB maturation in intact cells.
Alternative methods exist for quantification of endocytosis, and have provided valuable information on rates of cargo internalization in wild-type and endocytic mutant cells. Examples include monitoring uptake of radiolabeled ligands such as α-factor (which binds to the pheromone receptor Ste2) and uracil (which is internalized by the permease Fur4)40,41, or monitoring the rate of degradation for endocytic cargos such as Ste3 as they are internalized and transported to the vacuole42. These biochemical approaches require cell lysis, and are thus not suitable for direct quantification in living cells. Alternatively, fluorescently labeled α-factor, the fluid-phase dye Lucifer Yellow, or the styryl dye FM4-64 can be used to visualize endocytosis in living cells, but do not allow direct monitoring of endogenously expressed cargo proteins40,43,44. For these approaches, persistence of fluorescence in the vacuole lumen may complicate quantification, as seen with GFP. It is also possible to use GFP-tagged cargos for quantification of endocytosis by measuring whole-cell fluorescence intensity and subtracting the signal from cytoplasmic and endosomal/vacuolar compartments; however, endosomes and vacuoles are often in close proximity to the plasma membrane. In contrast, pHluorin-tagged endocytic cargos are quenched in MVB/vacuole compartments, which can be a distinct advantage for quantification of trafficking events compared to the more widely used GFP tag. Our future studies will expand upon current knowledge of endocytic pathways and cargo sorting in yeast to identify additional factors that contribute to CME and CIE, and to understand the mechanisms that regulate cargo-sorting decisions.
Disclosures
The authors declare no competing interests.
Acknowledgments
We would like to thank Gero Miesenböck and Tim Ryan for sharing pHluorin cDNA used to generate reagents in this study, Nathan Wright, Joanna Poprawski and Lydia Nyasae for excellent technical assistance, members of the Wendland lab for helpful discussions, and Michael McCaffery and Erin Pryce at the Integrated Imaging Center (Johns Hopkins) for advice and assistance with microscopy and flow cytometry. This work was supported by grants from the National Institutes of Health (to B.W., GM60979) and the National Science Foundation (to B.W., MCB 1024818). K.W. was supported in part by a training grant from the National Institutes of Health (T32-GM007231). A.F.O. was supported by developmental funds from the Department of Biological Sciences at Duquesne University and National Science Foundation CAREER grant 553143.
References
- Kaksonen M, Sun Y, Drubin DG. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell. 2003;115(4):475–487. doi: 10.1016/s0092-8674(03)00883-3. [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Toret CP, Drubin DG. A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell. 2005;123(2):305–320. doi: 10.1016/j.cell.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Boettner DR, Chi RJ, Lemmon SK. Lessons from yeast for clathrin-mediated endocytosis. Nat Cell Biol. 2012;14(1):2–10. doi: 10.1038/ncb2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Perrais D, Merrifield CJ. A High Precision Survey of the Molecular Dynamics of Mammalian Clathrin-Mediated Endocytosis. PLoS Biol. 2011;9(3):e1000604. doi: 10.1371/journal.pbio.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CG, Nichols BJ. Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci. 2009;122(11):1713–1721. doi: 10.1242/jcs.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharanjak S, Sharma P, Parton RG, Mayor S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell. 2002;2(4):411–423. doi: 10.1016/s1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- Radhakrishna H, Klausner RD, Donaldson JG. Aluminum fluoride stimulates surface protrusions in cells overexpressing the ARF6 GTPase. J Cell Biol. 1996;134(4):935–947. doi: 10.1083/jcb.134.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser DC, Drivas TG, Maldonado-Báez L, Wendland B. Existence of a novel clathrin-independent endocytic pathway in yeast that depends on Rho1 and formin. J Cell Biol. 2011;195(4):657–671. doi: 10.1083/jcb.201104045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser DC, Wendland B. Conserved roles for yeast Rho1 and mammalian RhoA GTPases in clathrin-independent endocytosis. Small GTPases. 2012;3(4):229–235. doi: 10.4161/sgtp.21631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno H, et al. Bni1p implicated in cytoskeletal control is a putative target of Rho1p small GTP binding protein in Saccharomyces cerevisiae. EMBO J. 1996;15(22):6060–6068. [PMC free article] [PubMed] [Google Scholar]
- Evangelista M, et al. a Yeast Formin Linking Cdc42p and the Actin Cytoskeleton During Polarized Morphogenesis. Science. 1997;276(5309):118–122. doi: 10.1126/science.276.5309.118. [DOI] [PubMed] [Google Scholar]
- Kübler E, Riezman H. Actin and fimbrin are required for the internalization step of endocytosis in yeast. EMBO J. 1993;12(7):2855–2862. doi: 10.1002/j.1460-2075.1993.tb05947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, MacGurn JA, Chu T, Stefan CJ, Emr SD. Arrestin-Related Ubiquitin-Ligase Adaptors Regulate Endocytosis and Protein Turnover at the Cell Surface. Cell. 2008;135(4):714–725. doi: 10.1016/j.cell.2008.09.025. [DOI] [PubMed] [Google Scholar]
- Nikko E, Sullivan JA, Pelham HRB. Arrestin-like proteins mediate ubiquitination and endocytosis of the yeast metal transporter Smf1. EMBO Rep. 2008;9(12):1216–1221. doi: 10.1038/embor.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikko E, Pelham HRB. Arrestin-Mediated Endocytosis of Yeast Plasma Membrane Transporters. Traffic. 2009;10(12):1856–1867. doi: 10.1111/j.1600-0854.2009.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell AF, McCartney RR, Chandrashekarappa DG, Zhang BB, Thorner J, Schmidt MC. 2-Deoxyglucose impairs Saccharomyces cerevisiae growth by stimulating Snf1-regulated and α-arrestin-mediated trafficking of hexose transporters 1 and 3. Mol Cell Biol. 2015;35(6):939–955. doi: 10.1128/MCB.01183-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell AF, Huang L, Thorner J, Cyert MS. A calcineurin-dependent switch controls the trafficking function of α-arrestin Aly1/Art6. J Biol Chem. 2013;288(33):24063–24080. doi: 10.1074/jbc.M113.478511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser DC, Pannunzio AE, Brodsky JL, Thorner J, Wendland B, O'Donnell AF. α-Arrestins participate in cargo selection for both clathrin-independent and clathrin-mediated endocytosis. J Cell Sci. 2015;128(22):4220–4234. doi: 10.1242/jcs.175372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamaze C, Dujeancourt A, Baba T, Lo CG, Benmerah A, Dautry-Varsat A. Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol Cell. 2001;7(3):661–671. doi: 10.1016/s1097-2765(01)00212-x. [DOI] [PubMed] [Google Scholar]
- Prosser DC, Whitworth K, Wendland B. Quantitative analysis of endocytosis with cytoplasmic pHluorin chimeras. Traffic. 2010;11(9):1141–1150. doi: 10.1111/j.1600-0854.2010.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokman SH, Ward WW. Renaturation of green-fluorescent protein. Biochem Biophys Res Commun. 1981;101(4):1372–1380. doi: 10.1016/0006-291x(81)91599-0. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394(6689):192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The Use of pHluorins for Optical Measurements of Presynaptic Activity. Biophys J. 2000;79(4):2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex. ESCRT-I. Cell. 2001;106(2):145–155. doi: 10.1016/s0092-8674(01)00434-2. [DOI] [PubMed] [Google Scholar]
- Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14(10):953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15(14):1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Ausubel FM. Curr Prot Mol Biol. New York: John Wiley & Sons; 1991. [Google Scholar]
- Urbanowski JL, Piper RC. Ubiquitin sorts proteins into the intralumenal degradative compartment of the late-endosome/vacuole. Traffic. 2001;2(9):622–630. doi: 10.1034/j.1600-0854.2001.20905.x. [DOI] [PubMed] [Google Scholar]
- Newpher TM, Smith RP, Lemmon V, Lemmon SK. In Vivo Dynamics of Clathrin and Its Adaptor-Dependent Recruitment to the Actin-Based Endocytic Machinery in Yeast. Dev Cell. 2005;9(1):87–98. doi: 10.1016/j.devcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Maldonado-Báez L, Dores MR, Perkins EM, Drivas TG, Hicke L, Wendland B. Interaction between Epsin/Yap180 adaptors and the scaffolds Ede1/Pan1 is required for endocytosis. Mol Biol Cell. 2008;19(7):2936–2948. doi: 10.1091/mbc.E07-10-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar RC, et al. Epsin N-terminal homology domains perform an essential function regulating Cdc42 through binding Cdc42 GTPase-activating proteins. Proc Natl Acad Sci U.S.A. 2006;103(11):4116–4121. doi: 10.1073/pnas.0510513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isnard AD, Thomas D, Surdin-Kerjan Y. The study of methionine uptake in Saccharomyces cerevisiae reveals a new family of amino acid permeases. J Mol Biol. 1996;262(4):473–484. doi: 10.1006/jmbi.1996.0529. [DOI] [PubMed] [Google Scholar]
- Wendland B, McCaffery JM, Xiao Q, Emr SD. A novel fluorescence-activated cell sorter-based screen for yeast endocytosis mutants identifies a yeast homologue of mammalian eps15. J Cell Biol. 1996;135(6):1485–1500. doi: 10.1083/jcb.135.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvaro CG, et al. Specific Alpha-Arrestins Negatively Regulate Saccharomyces cerevisiae Pheromone Response by Down-Modulating the G-Protein-Coupled Receptor Ste2. Mol Cell Biol. 2014;34(14):2660–2681. doi: 10.1128/MCB.00230-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaddar K, Merhi A, Saliba E, Krammer EM, Prevost M, Andre B. Substrate-Induced Ubiquitylation and Endocytosis of Yeast Amino Acid Permeases. Mol Cell Biol. 2014;34(24):4447–4463. doi: 10.1128/MCB.00699-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becuwe M, Léon S. Integrated control of transporter endocytosis and recycling by the arrestin-related protein Rod1 and the ubiquitin ligase Rsp5. eLife. 2014;3 doi: 10.7554/eLife.03307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Buchkovich NJ, Zhao Y, Emr SD. The Endosomal Sorting Complex ESCRT-II Mediates the Assembly and Architecture of ESCRT-III Helices. Cell. 2012;151(2):356–371. doi: 10.1016/j.cell.2012.08.039. [DOI] [PubMed] [Google Scholar]
- Nickerson DP, Russell MRG, Lo S-Y, Chapin HC, Milnes JM, Merz AJ. Termination of Isoform-Selective Vps21/Rab5 Signaling at Endolysosomal Organelles by Msb3/Gyp3. Traffic. 2012;13(10):1411–1428. doi: 10.1111/j.1600-0854.2012.01390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson DP, Merz AJ. LUCID: A Quantitative Assay of ESCRT-Mediated Cargo Sorting into Multivesicular Bodies. Traffic. 2015;16(12):1318–1329. doi: 10.1111/tra.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic V, Egerton M, Elguindi I, Raths S, Singer B, Riezman H. Yeast endocytosis assays. Methods Enzymol. 1991;194:697–710. doi: 10.1016/0076-6879(91)94051-d. [DOI] [PubMed] [Google Scholar]
- Silve S, Volland C, Garnier C, Jund R, Chevallier MR, Haguenauer-Tsapis R. Membrane insertion of uracil permease, a polytopic yeast plasma membrane protein. Mol Cell Biol. 1991;11(2):1114–1124. doi: 10.1128/mcb.11.2.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miliaras NB, Park J-H, Wendland B. The Function of the Endocytic Scaffold Protein Pan1p Depends on Multiple Domains. Traffic. 2004;5(12):963–978. doi: 10.1111/j.1600-0854.2004.00238.x. [DOI] [PubMed] [Google Scholar]
- Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128(5):779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima JY, Toshima J, Kaksonen M, Martin AC, King DS, Drubin DG. Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent alpha-factor derivatives. Proc Natl Acad Sci U.S.A. 2006;103(15):5793–5798. doi: 10.1073/pnas.0601042103. [DOI] [PMC free article] [PubMed] [Google Scholar]