

Abstract
We have determined the distribution of each of the 65,536 DNA sequences that are eight bases long (8-mer) in a set of 13,010 human genomic promoter sequences aligned relative to the putative transcription start site (TSS). A limited number of 8-mers have peaks in their distribution (cluster), and most cluster within 100 bp of the TSS. The 156 DNA sequences exhibiting the greatest statistically significant clustering near the TSS can be placed into nine groups of related sequences. Each group is defined by a consensus sequence, and seven of these consensus sequences are known binding sites for the transcription factors (TFs) SP1, NF-Y, ETS, CREB, TBP, USF, and NRF-1. One sequence, which we named Clus1, is not a known TF binding site. The ninth sequence group is composed of the strand-specific Kozak sequence that clusters downstream of the TSS. An examination of the co-occurrence of these TF consensus sequences indicates a positive correlation for most of them except for sequences bound by TBP (the TATA box). Human mRNA expression data from 29 tissues indicate that the ETS, NRF-1, and Clus1 sequences that cluster are predominantly found in the promoters of housekeeping genes (e.g., ribosomal genes). In contrast, TATA is more abundant in the promoters of tissue-specific genes. This analysis identified eight DNA sequences in 5082 promoters that we suggest are important for regulating gene expression.
Vertebrate gene expression is often regulated by the basal promoter, which traditionally is defined as being between –200 bp and the transcription start site (TSS). The DNA sequence properties of basal promoters are poorly described because it is difficult to identify the TSS. Two recent results have helped to resolve this problem: (1) RefSeq (Maglott et al. 2000; Pruitt et al. 2000; Pruitt and Maglott 2001) sequences have been mapped to their location in the complete human genome sequence, and (2) TSSs have been experimentally verified for 7889 genes by using cDNA synthesis methods that identify the 5′ CAP site (Suzuki et al. 2002). We have combined these data to assemble genomic DNA sequences that are putative promoter regions for 13,010 genes aligned relative to the putative TSS. We have examined these aligned sequences for 8-mers that are preferentially localized relative to the TSS, namely, clusters.
A fundamental question in gene expression studies is to determine which DNA sequences that are bound by TFs are biologically relevant. Often, the same DNA sequence is functional in one context but not in another. We reasoned that if a DNA sequence clusters relative to the TSS, the DNA sequences that are in the cluster have a high likelihood of being biologically significant. In human promoters the CAAT box, SP1, and TATA box are recognized by the constitutive transcription factors NF-Y, SP1, and TBP, respectively, and are thought to be localized near the TSS (Breathnach and Chambon 1981). Recently, a genome-wide analysis has demonstrated that the CRE sequence clusters in human promoters (Conkright et al. 2003).
To identify additional DNA sequences that localize near the TSS and thus may be biologically important, we determined the distribution of each of the 65,536 8-mer DNA sequences in 13,010 human promoters sequences from –2500 to 500 bp relative to the TSS. A detailed analysis of the 8-mers with the most significant clustering indicates that they primarily represent variations of only nine DNA consensus sequences. Eight motifs cluster between –100 and the TSS. They include (1) TF binding sites that have been previously suggested to cluster within the promoter (CAAT, SP1, CREB, and TATA); (2) TF binding sites that were not known to localize in the core promoter region, ETS, NRF-1, and USF; and (3) a single DNA sequence, designated Clus1, that is not a known TF binding site. The ninth motif is the Kozak sequence that clusters downstream of the TSS. We observe correlations between the presence of DNA sequences that cluster in promoters and the mRNA expression properties and function of genes.
RESULTS
We combined the cDNA data for RefSeq genes (Maglott et al. 2000; Pruitt et al. 2000; Pruitt and Maglott 2001) with TSS data (Suzuki et al. 2002) and mapped the 5′ end of these cDNA sequences onto assembled human genomic DNA sequences (Lander et al. 2001) for 13,010 genes. For this study, we aligned these human promoter genomic sequences relative to the putative TSS. We then analyzed the distribution of DNA sequences (2-mers to 8-mers) between –1000 and 500 bp relative to the putative TSS and, as a control set, the region between –2500 and –1000 bp. The 1500-bp regions were divided into 75 bins. Each bin contains 20 bp, where bin 1 is from –1000 to –981 bp, and bin 51 is from 1 bp to 20 bp. We determined the number of times that a particular DNA sequence occurred in each 20-bp bin for all 13,010 promoter sequences.
Distribution of Dinucleotide Pairs in Promoters
Initially, we determined the distributions of each dinucleotide (2-mer) in the set of 13,010 human promoter sequences. We determined the position of each 2-mer on the DNA coding strand across the 1500 bp, between –1000 and 500 bp, and plotted the results as a frequency histogram. Three general distributions are observed: (1) a peak near the TSS for the 2-mers containing G and/or C (GC, CG, GG, and CC), (2) a valley near the TSS for the 2-mers containing A and/or T (AT, TA, TT, and AA), and (3) no preference for the remaining 2-mers (Fig. 1). Although peaking around the TSS, the CG sequence, which can be methylated on the C base, is the least abundant outside the promoter region, as is observed in genomic DNA (Hapgood et al. 2001).
Figure 1.

Distribution of the dinucleotides (CG, GC, TT, CA) from –1000 to 500 bp in 13,010 human promoters.
Distribution of All 8-mer DNA Sequences
To identify DNA sequences that cluster relative to the TSS, we determined the distribution of all sequences ranging from 2-mers to 8-mers in this set of 13,010 promoter sequences. As the length of the DNA sequence increased, we identified sequences that clustered more dramatically. This manuscript will focus on the distribution of 8-mers. Because both strands of complementary DNA were examined, the number of independent 8-mers was reduced from 65,536 to 32,896 (32,640 nonpalindromic 8-mers + 256 palindromic 8-mers).
To determine if a DNA sequence clustered, the mean (x̄) and standard deviation (σ) were determined based on its abundance in each of the 75 bins. Those bin values that were ≥2 SD above the mean were considered to be part of the cluster and a new mean (x̄′) and standard deviation (σ′) were calculated excluding these bin values. A clustering factor (CF) was then calculated based on this corrected mean and standard deviation,  CF values for all 32,896 8-mers were plotted against the bin with the maximum value (Fig. 2A). There is a clear preference for the CF to be higher near the putative TSS. Limiting the analysis to the 8687 8-mers with at least 20 members in the most abundant bin gave a more prominent peak near the TSS (Fig. 2B). The distribution pattern for each 8-mer, along with the identity of the promoters containing each 8-mer, is available at http://genome.nci.nih.gov/publications/promoters.
CF values for all 32,896 8-mers were plotted against the bin with the maximum value (Fig. 2A). There is a clear preference for the CF to be higher near the putative TSS. Limiting the analysis to the 8687 8-mers with at least 20 members in the most abundant bin gave a more prominent peak near the TSS (Fig. 2B). The distribution pattern for each 8-mer, along with the identity of the promoters containing each 8-mer, is available at http://genome.nci.nih.gov/publications/promoters.
Figure 2.

Clustering factor of each 8-mer DNA sequence plotted at the position of the most populated bin: All 32,896 8-mers (A); 8687 8-mers with a maximum bin containing ≥20 members (B); clustering factor from –1000 bp to 500 bp for the 6838 8-mers that contain a maximum bin with ≥20 members from one of the 1000 random seventh-order Markov model data sets (C); clustering factor for the 7471 8-mers that contain a maximum bin with ≥20 members from –2500 to –1000 bp (D); and clustering factor values from –1000 to 500 bp for the 8687 8-mers from Figure 2B based on a randomized translocation of the TSS of between 0 and 500 bp (E).
Three controls evaluated the significance of the observed localization of particular 8-mers near the TSS. The distribution plot of a seventh-order Markov random data set (see below) shows a complete lack of clustering for any of the 8-mers (Fig. 2C). Figure 2D presents the CF values between –2500 and –1000 bp for the 13,010 putative promoter sequences in which no preferential localization is observed for the 7471 8-mers that contain at least 20 members in the most abundant bin. To further characterize the unique nature of the distribution of 8-mers around the TSS, we performed an experiment in which we aligned the 13,010 sequences based on a translocation of the putative TSS within a random distance between 0 and 500 bp upstream or downstream (Fig. 2E). The CF distribution for this data set does not identify sequences that cluster.
To determine the statistical significance of the CF values, we converted the CF into a probability term. One thousand random data sets, each containing 13,010 sequences that are 1500 bp long, were generated by using the 8-mer frequencies observed in the original data set. For each of the 1000 data sets, the distribution and CFexpt value for all 32,896 8-mers were determined. From the 1000 separate CFexpt values for each 8-mer, the frequency distribution was plotted, and then a mean (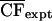 ) and standard deviation (σexpt) were computed. The probability term, P, represents, –log10(1 – p), where p is the area that lies under the normalized curve of the distribution of CFexpt. Thus, the greater the P value, the more nonrandom the result. Figure 3 shows a plot of the P values versus the maximum bin number for all 8-mers with at least 20 members in the most abundant bin. There is a group of 8-mers near the TSS with clustering that is statistically nonrandom. Of the 120 sequences with the greatest CF, 111 were observed in the 120 sequences with the greatest P values.
) and standard deviation (σexpt) were computed. The probability term, P, represents, –log10(1 – p), where p is the area that lies under the normalized curve of the distribution of CFexpt. Thus, the greater the P value, the more nonrandom the result. Figure 3 shows a plot of the P values versus the maximum bin number for all 8-mers with at least 20 members in the most abundant bin. There is a group of 8-mers near the TSS with clustering that is statistically nonrandom. Of the 120 sequences with the greatest CF, 111 were observed in the 120 sequences with the greatest P values.
Figure 3.

The probability term P = [–log10(1 – p)] for the 8687 8-mers with a maximum bin containing ≥20 members. The 159 DNA sequences above the line at P = 7, a one in 10 million (single sampling) chance of being random, were manually annotated.
To determine if the 8-mers with the highest CF values are also the most abundant sequences, we compared the abundance of these sequences in the 13,010 promoters between –1000 and 500 bp to the abundance of all 32,896 8-mers (Fig. 4). To determine the abundance of a sequence, we counted the total numbers of occurrence of each 8-mer between –1000 and 500 bp in the set of 13,010 genes. The overall prevalence of the different 8-mers is very variable. On average, we expect 590 occurrences of an 8-mer across the whole promoter region in 13,010 sequences. The observed occurrences, however, are scattered in a very wide range: from minimum of 12 for the palindrome TCGTACGA to maximum of 43,517 for TTTTTTTT. Although the 156 8-mers that showed the most significant clustering in bins 45 through 56 are not the rarest sequences in promoters, they do not appear to represent only the abundant 8-mers. Although they are frequently ignored (masked) in promoter analyses, we have not excluded repetitive sequences in this study. Our rationale is that such sequences may actually contain active control elements by virtue of their specific location.
Figure 4.

The number of occurrences of each 32,896 DNA sequence in the 13,010 promoter sequences is plotted as a gray dot. The abundance of all 159 sequences with P ≥ 7 is plotted as black triangles.
The 159 8-mers with a P value ≥7, a one in 10 million single sampling probability of occurring by chance, were examined in more detail. One hundred fifty-six of these sequences had peaks between bin 45 and bin 56. Those sequences were found to be composed of overlapping sequences that could be grouped into nine distinct classes (Table 1). The manual placement of an 8-mer into a particular group was guided by (1) the similarity among DNA sequences, (2) the shapes of the distribution histogram, and (3) the peak position relative to the TSS. Seven of the DNA sequences that cluster are known TF binding sites, listed in the order as they appear in promoters, starting with the most 5′ member: CCAAT, SP1, USF, CREB, TATA, NRF-1, and ETS (Table 1). One sequence, TCTCGCGA that we name Clus1, did not resemble any known TF DNA-binding site. The final sequence is the Kozak sequence that is 3′of the TSS, and thus is transcribed into mRNA, and encodes the initiating methionine of the protein. The distribution of the 8-mer with the greatest probability of having a nonrandom distribution (P = 40.6) is shown in Figure 5A, and the distribution of the 159th 8-mer with a P = 7.0 is shown in Figure 5B.
Table 1.
DNA 8-mer Sequences That Cluster

One hundred fifty-six DNA sequences are grouped into related sequences and arranged by their peak position relative to the TSS. From the left the table contains the most abundant bin, the number of times the sequence occurs in the distribution, the 8-mer sequence, and finally the P value (see text). The end of the table contains consensus sequences. Here the leftmost numbers are the bins defining the peak, followed by the clustering factor (CF), the consensus sequence, and finally the number of occurrences of the sequence in the bins that comprise the peak. Exclamation point (!) denotes sequences that are at least threefold more abundant in the maximum bin on the DNA strand presented in the table than on the opposite strand. The asterisk (*) denotes sequences used in Tables 2 and 3. IUPAC letters used to represent degenerate bases are R (G,A), W (A,T), Y (T,C), K (G,T), V (G, C, A), D (G,A,T), and N (A,T,G,C).
Figure 5.

Distribution (number of occurrences per bin as a function of position relative to the TSS) of the DNA 8-mer (ACCGGAAG) that shows the greatest clustering (A) and the 159th 8-mer (CCGCCTCC; B).
Extending DNA Sequences That Cluster
For each of the eight groups in Table 1 that are 5′of the TSS, we manually constructed a consensus sequence, some of which extended 5′and 3′ beyond the central core of identity that initially guided the formation of the groups. For example, Figure 6A shows the result of expanding the 5-mer CCAAT sequence to the degenerate 9-mer (RRCCAATSR). The background is dramatically reduced, whereas the peak height is not. The longer consensus sequences provide greater confidence that the sequences within the peak may be functional TF binding sites in their promoters because the occurrence of the sequence outside the peak is so low.
Figure 6.

Distribution of the 5-mer CCAAT and the 9-mer RRCCAATSR (A) and the CCAAT consensus RRCCAATSR and the 15 single base variants of the central CCAAT (B).
For each consensus, we also varied the identity of each base to determine if the related DNA sequences also clustered. The general result was that related sequences do not cluster.
Eight Clustering Sequences Upstream of the TSS
The eight clustering DNA sequences upstream of the TSS are found in 5082, or 39%, of the 13,010 promoters analyzed and are discussed in the order they occur in promoters, starting with the most upstream element.
CAAT
Thirty-two 8-mers contain an invariant 4-mer CAAT, and 27 of them contain the invariant 5-mer CCAAT. Neighboring DNA sequences are constrained resulting in the degenerate consensus 9-mer (RRCCAATSR; Fig. 6A) found in 994, or 7.6%, of the promoters examined. The CCAAT sequence (Dynan and Tjian 1985; Mantovani 1999) cluster is located the furthest upstream from the TSS. The CCAAT sequence binds NF-Y (Sinha et al. 1995), a trimer protein complex that bends DNA by using the histone fold motif (Romier et al. 2003). We individually varied each of the five invariant bases (CCAAT) and did not observe any clustering of these 15 related sequences (Fig. 6B).
SP1
Twenty-seven sequences have been grouped into binding sites for the SP1 family of three-zinc finger motif proteins. We highlight three sequences that cluster, the 7-mer CCCGCCC, the 8-mer CCCCGCCC, and the 9-mer CCCCGCCCC; the first two are found in 2696, or 20.7%, of promoters (Fig. 7A). The central G is critical for the sequences to cluster, changing it to C results in a sequence that does not cluster (Fig. 7A). Two additional 8-mers (CCCCTCCC and CCTCCCTC) also cluster, suggesting that they either are degenerate sequences or are bound by different members of the SP1 family of proteins.
Figure 7.

Distribution of selected sequences (8-mers and consensus patterns). (A) Three SP1 (CCCGCCC, CCCCGCCC, CCCCGCCCC) sequences and a nonpeaking single base variation (CCCCCCCC). (B) Clus1 (TCTCGCGA) sequence. (C) Two USF (TCACGTGG, TCACGTGA) sequences. (D) Three (TGACGTCA, TGATGTCA, TTGCGTCA) CREB like sequences. (E) Strand-specific localization of the TATAAAD sequence. (F) Two variants (TATATAD and TATAAGD) of TATA, plus strand (+) only. (G) Three NRF-1 (CGCCTGCG, CGCGTGCG, CGCATGCG) sequences. (H) ETS core (CCGGAA), consensus sequence (VCCGGAARY), and a peaking (VGCGGAARY) and nonpeaking VCCGGAAYR variant.
Clus1
The 8-mer sequence TCTCGCGA that we termed Clus1 is found in 140, or 1.1%, of promoters. No related sequence is observed to cluster when each of the 8 bp is varied (Fig. 7B). No described TF is known to bind this sequence.
USF
The palindromic 8-mer TCACGTGA and the related TCACGTGG sequence cluster in 191, or 1.5%, of promoters examined (Fig. 7C). These sequences are bound by the USF family of dimeric B-HLH-ZIP proteins (Bendall and Molloy 1994; Boyd and Farnham 1999). The core of this sequence is the E box 6-mer CANNTG that is bound by B-HLH-ZIP proteins (Ferre-D'Amare et al. 1993). Varying each base in this consensus or keeping one-half of the palindrome constant and varying the other half (NNNNGTGA) does not identify additional clustering DNA sequences.
CRE
The palindromic 8-mer TGACGTCA sequence is known as the cAMP responsive element (CRE; Shaywitz and Greenberg 1999; Mayr and Montminy 2001). The CRE is bound by a variety of B-ZIP proteins, including CREB, ATF1, and Oasis homodimers, and FOS|JUN and ATF2|JUN heterodimers (Vinson et al. 2002). Varying each of the nucleotides in the CRE consensus revealed a related sequence that also clusters (TGATGTCA), which is not one of the 159 most clustering sequences. These two sequences occur in 310, or 2.4%, of promoters examined. To identify related DNA sequences that may cluster, we maintained one-half of the CRE palindrome constant and allowed the second half to vary (NNNNGTCA). This identified an additional clustering sequence (TTGCGTCA) that consists of a C/EBP and a CREB half site (Moll et al. 2002) that can be bound by a C/EBP|ATF4 (Vinson et al. 1993) or a C/EBP|ATF2 heterodimer (Fig. 7D; Shuman et al. 1997).
Eleven 8-mers contain the six-base sequence GTGACG. These appear to be of two classes. One class may be degenerate CRE and/or USF sites or a binding site for an unknown TF. The second class, for example, the complement of GAAGTGAC (GT CACTTC), can be extended to the clustering 11-mer CCG GAAGTGAC, which is the juxtaposition of an ETS sequence and half of a CRE sequence (data not shown).
TATA
Nine 8-mers have been grouped together and are predicted to be bound by the TATA binding protein (TBP; Kim et al. 1993) that recruits the basal machinery to initiate transcription (Geiger et al. 1996). The consensus 7-mer TATAAA is found in 335, or 2.6%, of all promoters examined. The TATA sequence shows the sharpest peak but also has the highest background. This is the only clustering TF binding site that occurs in a DNA strand–specific manner (Fig. 7E). Two variants of the TATA sequence (TATATAD and TATAAGD) show weaker clustering and are found in a total of 259, or 2.0%, of promoters (Fig. 7F).
NRF-1
Eleven GC-rich 8-mers are the most divergent set that we grouped. We highlight the sequence GCGCATGC because it shows the greatest CF of these 11 sequences. Varying each base identified one additional sequence that was not found in the group of 156 most significant clustering sequences, resulting in the consensus CGCVTGCG that is found in 776, or 6.0%, of promoters (Fig. 7G). This sequence is bound by the transcription factor NRF-1 that regulates expression of nuclear-encoded mitochondrial genes (Scarpulla 2002).
ETS
Twenty-seven 8-mers have been grouped into binding sites for the ETS family of transcription factors (Graves and Petersen 1998; Sharrocks 2001). The extended consensus is the 9-mer VCCG GAARY found in 897, or 6.9%, of promoters. This extended consensus was identified in in vitro DNA-binding site selection experiments by using ETS proteins (Graves and Petersen 1998). DNA sequences with the two variable bases RY changed to YR do not peak, showing the importance of the extended sequence for clustering (Fig. 7H).
Four 8-mers contain a 1-bp variant of the ETS sequence, the 6-mer GCGGAA. The extension of this sequence is the 9-mer RGCGGAAGY found in 243, or 1.9%, of promoters. DNA-binding site selection experiments indicate that this ETS site variant is bound by the PEA-3 subfamily of ETS proteins (Brown and McKnight 1992).
Kozak Sequence
The Kozak sequence is the only sequence to cluster 3′ of the start site. It is found in 32 of the DNA 8-mers examined. This sequence includes the initiating ATG, which encodes the amino-terminal methionine found in all proteins, and surrounding nucleotides important for ribosome binding to the mRNA. These sequences are preferentially found on one strand of DNA as is expected if they function in the mRNA to initiate protein translation (Fig. 8). These DNA sequences have the same general distribution, a peak that abruptly decreases going 5′ and more slowly trails going 3′. This shape is the opposite of what we observe for transcription factor binding sites. Several of these sequences extend to the 3′ end.
Figure 8.

Distribution of the Kozak octamer AGATGGCG on the plus strand (+) and minus strand (–).
Transcription Factor DNA-Binding Sites That Do Not Cluster
To determine if clustering is a property exhibited by all TF binding sites, we determined the distribution of 193 DNA sequences reported to be TF binding sites found in the TRANSFAC Database, version 3.4 (http://transfac.gbf.de/TRANSFAC; Matys et al. 2003). This set of DNA sequences ranges in length from 5- to 20-mers. Three general distribution patterns were observed: (1) 15 DNA sequences formed peaks between –120 and the TSS (these represent variations of the six consensus DNA sequences we identified); (2) two sequences are severely underrepresented near the TSS, the zinc finger protein LYF1 (Ikaros) TTTGGGAGR, and the HMG protein SRY (sex-determining region Y gene product; WWAACAAWA; Fig. 9A); and (3) the majority of TF binding sites are uniformly distributed from –1000 to 500 bp including Myb (AACKGNC), HSF2 (GAANNWTCK), and TRE (TGAGTCA; Fig. 9B).
Figure 9.

Distribution of selected sequences from the TRANSFAC database that are underrepresented near the TSS, SRY (WWAACAAWA), and LYF1 (TTTGGGAGR; Ikaros; A); and uniformly distributed, Myb (AACKGNC), HSF2 (GAANNWTCK), and TRE (TGAGTCA; B). (C) The core promoter element Initiator, Inr (YYANWYY). (D) The core promoter element downstream promoter element, DPE (RGWCGTG).
A couple of DNA sequences have been implicated in the initiation of polymerase II transcription. These include the initiator (YYANWYY; Lo and Smale 1996) and the downstream promoter element (RGWCGTG; Kutach and Kadonaga 2000). However, an analysis of the 13,010 promoters does not indicate that these sequences are uniquely positioned relative to the TSS (Fig. 9C).
DNA Sequences That Cluster Occur Together in Promoters
The identified DNA sequences that cluster in promoters have a complex pattern of interdependence within the same promoter (Table 2). In general, the presence of a clustered DNA sequence positively correlates with other clustered DNA sequences in the same promoter. The only exception is the TATA sequence that negatively correlates with all sequences except CCAAT and CREB. Most notably, TATA is totally absent in promoters containing the ETS sequence, an estimated random probability of 10–12.4. The positive correlations are most pronounced for a sequence predicting the presence of additional copies of the same sequence in a single promoter. This is true for all the sequences except for CREB. Notably, Clus1 is found in 1.1% of promoters, but 45% of these promoters have more than one site in the cluster.
Table 2.
The Number of Promoters That Contain One DNA Consensus Sequence Also Containing a Second DNA Consensus Sequence

To the left are the eight consensus sequences followed by the number of their occurrences in the peak, and the percentage of promoters containing this sequence. Across the top is the same set of consensus sequences. The intersection is the number of promoters containing both sequences in the peak, followed by the percentage of the promoters containing the top sequence that also contain the sequence from the side, and the probability of having the number of elements in the intersection more dramatic than given. For example, 20.7% of the 13,010 promoters contain the SP1 sequence in the peak (2696), but 33.5% of promoters that contain a USF sequence (191) in the peak also contain the SP1 sequence in the peak. The probability of this positive correlation between these two DNA sequences is P = 4.7. Those correlations that are greater than P = 5 are shown in black, a positive correlation has an asterisk in the probability column.
Clustering DNA Sequences Correlate With Biological Activity
We examined whether the presence of clustered DNA sequences in promoters predicts their mRNA expression properties. Initially, we divided genes into two groups depending on whether or not they had a GO ontology annotation that was indicative of some biochemical insight into the function of the gene (Ashburner and Lewis 2002). These two groups are of similar size and have a similar frequency of DNA sequences that cluster in their promoters, indicating a lack of bias toward well-characterized genes (Table 3).
Table 3.
Functional Properties of Genes With Promoters That Contain Consensus DNA Sequences That Are in the Peak

A variety of functional characteristics was examined for each gene, including if they had a GO ontology annotation, were involved in related biological processes (e.g. ribosomal, proteasomal, or channel) and mRNA expression properties (housekeeping or tissue specific). For each criterion, we present the total number of genes in the group. We next present the three numbers for each consensus sequence: (1) the absolute number of promoters in the group with the consensus sequence in the peak, (2) the fraction of genes in the group that have this consensus, and (3) a statistical measure of the correlation between these two terms. For example, 7.6% of the 13,010 promoters contain the CCAAT sequence in the peak, but only 2% of the 147 channel genes contain the CCAAT sequence in the peak. Those correlations that are greater than P = 3 are shown in black, a positive correlation has an asterisk in the probability column.
We next examined if clustering sequences were found in the promoters of genes with a related function (Table 3). The most general observation is that proteins involved in essential cellular functions, for example, translation (ribosome) and degradation (proteosome) often have ETS sequences in their promoters. For example, the ETS sequence clusters in 8% of promoters but is observed in 23% of ribosomal genes, 43% of mitochondrial ribosomal genes, and 42% of proteosomal genes. NRF-1 and Clus1 are preferentially observed in the ribosomal genes, but unlike ETS sequences, they are not observed in proteosomal genes. This suggests a combinatorial system of DNA sequences is used to regulate expression of functionally related genes. These data are in sharp contrast to the 147 channel-related genes that do not have a single ETS or Clus1 sequence in their promoters.
We also determined whether clustering of DNA in promoters correlates with tissue-specific mRNA expression (Table 3). The Web site (http://expression.gnf.org) contains mRNA expression data for 29 human tissues derived from microarray data (Su et al. 2002); 6744 genes are in common with the set of 13,010 promoters we examined. From these 6744 genes, we extracted two subsets based on their differential mRNA expression levels in tissues: We defined tissue-specific genes to be highly expressed in one or two tissues and housekeeping genes to be expressed in 62 or more of the 63 samples. Based on this classification, 12.6% of the genes are housekeeping genes and 9.2% are tissue-specific genes. Table 3 presents the tissue-specific mRNA expression properties of genes with specific DNA sequences in their promoters. ETS, NRF-1, and Clus1 are preferentially observed in the housekeeping genes, whereas only TATA is preferentially observed in tissue-specific genes. For example, 8.2% of genes have promoters that contain an ETS sequence in the peak, but 14.6% of housekeeping genes have promoters that contain an ETS sequence in the peak.
To ascertain if the sequence within the peak had different properties than the same sequence outside the peak, we examined the mRNA expression profile for promoters that contained ETS, NRF-1, or Clus1 sequences outside the clustering peak. These DNA sequences outside the peak do not correlate with housekeeping genes (Table 4). In addition, promoters containing the ETS sequences in the peak contain 4.5 times more mitochondrial ribosomal genes than expected. In contrast, promoters containing the ETS sequences outside the peak contain 0.8 times the number of mitochondrial ribosomal genes as expected. This type of analysis provides greater assurance that individual ETS sequences that occur in the peak are biologically important. A similar analysis of TATA sequences indicates that the observation that TATA correlates with tissue-specific gene expression is only true for those TATA sequences under the peak.
Table 4.
Functional Properties of Consensus Sequences Outside of the Peak

The genes with promoters that contain the ETS, NRF-1, Clus1, and TATA are divided into two groups, those in which the consensus sequence is in the peak, and those in which the consensus sequence is not in the peak. The same set of parameters as for Table 3 for each functional criteria and consensus sequence is presented. For example, 124 of the 850 housekeeping genes contain the ETS sequence in the peak. This is 2.0 times more than the expected frequency: (124/850)/(1072/13,010). Those correlations that are greater than P = 3 are shown in black, a positive correlation has an asterisk in the probability column.
DISCUSSION
We determined the distribution of all 65,536 8-mer DNA sequences in 13,010 human promoters relative to the TSS. One hundred fifty-nine sequences clustered relative to TSS with a random single sampling probability of less than one in 10 million (P ≥ 7). One hundred fifty-six of the 159 sequences clustered near the TSS and were variants of nine sequences, eight were 5′ to the TSS, and one (Kozak) was 3′. Seven of the eight DNA sequences that cluster upstream of the TSS are known TF binding sites. The distribution of the TF binding sites relative to the TSS is different for each sequence. The CAAT and SP1 sequences cluster at around –100 bp, whereas the other sequences cluster closer to the TSS. Additional sequences may also cluster but were not identified because our analysis was limited to those 8-mers that occurred frequently enough in 13,010 promoters to allow reliable analysis.
An enigma in eukaryotic promoter analysis is that not all DNA sequences that can be bound by a TF are biologically relevant. We suggest, however, that if a particular DNA sequence is observed in the same position relative to the TSS, it is likely that the individual DNA sequences that comprise the cluster are important for regulating gene expression of their promoters. We identify 5082 promoters that contain one or more of eight DNA sequences that cluster. For each of these eight DNA sequence families, we generated a consensus sequence. However, although our approach permits us to identify sequences that are likely to be biologically relevant, it does not necessarily imply that related DNA sequences are not important. It could simply be that the related sequences are not sufficiently abundant to form a peak.
A prevailing theme in gene expression studies is that TFs bind a variety of related DNA sequences to regulate gene expression. To determine if variants of the DNA sequence we identified also clustered, we systematically varied each base in the consensus DNA sequences. Different results are obtained for each consensus sequence. For example, when the five invariant bases of the RRCCAATSR consensus are individually varied, none of the related 15 DNA sequences cluster. However, for the ETS consensus sequences, the variants VCCGGAARY and VGCGGAARY both form peaks. In vitro DNA-binding selection experiments have shown that different ETS family members preferentially bind one or the other of these two sequences (Brown and McKnight 1992; Graves and Petersen 1998; Sharrocks 2001). However, it remains to be determined if the variant sequences for SP1, CREB, and USF are bound by different TFs, as is observed for ETS, or are bound by the same TF. One of the sequences that clusters, Clus1, is not a known TF binding site.
Three of the nine sequences highlighted in this analysis are palindromic (CREB, USF, and NRF-1), although only 0.3% (256/65,536) of all octamers are palindromic. Two properties of palindromes may explain their predominance as important TF binding sites. First, palindromes can be bound on either strand of DNA, thus doubling their concentration and increasing the number of productive encounters between the TF and the DNA. Second, palindromes can be bound by dimeric proteins, as is known to be the case for the CREB and USF sites. Monomers dimerize to double their local concentration and now bind palindromic DNA that, again, is in a higher concentration because it can be “viewed” on both strands of DNA. Both of these effects make palindromic sequences “attractive” structures for TFs to bind.
Coordinate gene expression is a hallmark of genetic regulation and may be mediated by TFs that bind in the promoter of coordinately regulated genes. We have addressed this issue by determining if correlations exist between the presence of a particular DNA sequence in a cluster and the mRNA expression properties and/or function of the gene. In general, at the mRNA expression level, promoters containing ETS, NRF-1, and Clus1 tend to be housekeeping genes. Looking at gene function, the ancient classes of essential housekeeping genes, for example, the ribosomal and proteosome genes, have the ETS, NRF-1, and Clus1 sequence in their promoters. The significance of these results is bolstered by the observation that the promoter of mitochondrial ribosomal genes has NRF-1 sites (Scarpulla 2002). Thus, these sequences may be an ancient regulatory element that has been conserved throughout metazoan evolution.
TATA is often considered the prototypical DNA promoter element, however, in this analysis TATA is an exception. It is the only TF binding site to show strand-specific distribution, it has the sharpest peak, it has the highest background, it has the most variant sequences that cluster, and it is the only TF that positively correlates with tissue-specific gene expression. In the Eukaryotic Promoter Database (EPD), 51% of the genes contain a TATA (Davuluri et al. 2000) [T/A] element. Recently, it was found that the same TATA consensus occurs in 27% of 10,276 putative promoters (600 bp) derived from the Mammalian Gene Collection (Trinklein et al. 2003). However, the study presented here indicates that only 2.6% of promoters contain a TATA site in the peak. This does not suggest that TATA sequences outside of the peak are not important, only that those TATA sequences in the peak are likely important and might more accurately reflect functional TATA sequences. The traditional experimental focus on TATA containing promoters probably reflects the fact that these promoters can dramatically alter their transcriptional activity, making them ideal experimental systems, as is expected for tissue-specific genes (Smale 1997).
This study did not identify a single DNA sequence that clustered relative to the TSS for the majority of promoters. Thus, if such a sequence exists, it is sufficiently degenerate to be missed by this analysis. Previous studies of eukaryotic promoters have identified the initiator element as a DNA sequence that can act with or without TATA to direct accurate transcription initiation by RNA polymerase II (Smale 1997). However, the initiator element (Inr) has a degenerate sequence YYANWYY that does not cluster at the TSS. Instead of a universal DNA sequence that defines the TSS, the transcription factors that bind the eight clustering DNA sequences may themselves be involved in recruiting the basal machinery and regulating the position of transcriptional initiation. In support of this conjecture is the observation that CREB (Ferreri et al. 1994) and USF (Sawadogo and Roeder 1985) interact with components of the TFIID complex.
The observation that many clustering sequences positively correlate with the presence of additional clustering sequences, including themselves, suggests that promoters tend to contain multiple TF binding sites.
This analysis has identified key DNA sequences in 5082 promoters that cluster relative to the TSS and thus may be important for regulating gene expression. We expect that analyses using less stringent parameters may identify additional DNA sequences that are critical for gene expression.
METHODS
Data Set Generation
We combined the DNA sequence data for the annotated RefSeq genes in the Golden Path Human Genome Assembly (version December 2001; Kent et al. 2002; http://genome.ucsc.edu/), with data from the database of TSS (DBTSS; Suzuki et al. 2002; http://dbtss.hgc.jp/index.html) and generated two data sets: the control set representing sequences from –2,500 to –1000 bp relative to the TSS, and the experimental set containing sequences –1000 to 500 bp relative to the TSS. Both data sets contain sequences from 13,010 RefSeq genes. The BLAST and BLAT programs were used to align the DBTSS and Golden Path data, and sequences that showed multiple homologies were discarded. These data sets were queried with the program fuzznuc from the EMBOSS suite of software (Rice et al. 2000) or tacg (Mangalam 2002) to locate the occurrence and position of different DNA sequence motifs.
8-mer Analysis
There are 65,536 possible octameric sequences, and of these 256 are palindromic. Among these 65,536 possible octameric sequences, each sequence and its complement is represented. Thus, the number of possible comparisons can be reduced from 65,536 to 32,896 (32,640 nonpalindromic + 256 palindromic sequences) when both strands of the target sequence are examined. All 32,896 8-mers used in this analysis were automatically generated by custom software and were searched against all 13,010 promoter sequence in batches of ∼3800. The raw data was then processed by a combination of scripts and programs to generate the final binned distribution for each 8-mer. To analyze the data, we divided the 1500 bp into 75 bins with each bin containing 20 bp. For the data set –1000 to 500 bp, the numbering for bin 1 is –1000 to –981, thus bin 51 is form 1 to 20 bp. We determined the number of times a particular DNA sequence occurred in each 20-bp bin.
CF Calculation
To determine if a DNA sequence forms a peak in its distribution (i.e., clustered), we used an automated method of detecting and quantifying peak height. For the 75 bins in each frequency distribution, a mean (x̄) and standard deviation (σ) were determined. Those points, which were ≥2 SD above the mean, were considered to be part of the peak and a new mean (x̄′) and standard deviation (σ′) were calculated excluding these points. The CF was then calculated based on the maximum bin value (xmax) and the corrected mean and standard deviation CF = (xmax – x̄′)/(σ′).
Calculation of P Value for Distribution
To evaluate the probability that the results were obtained by chance, we converted the CF values into probability terms based on the analysis of the occurrence of each 8-mer in 1000 random data sets. One thousand random data sets, each containing 13,010 sequences 1500 bp long, were generated by using the 8-mer frequencies observed in the original data set. To populate each 1500-bp promoter, initially an 8-mer was chosen at random. To determine each next base, the preceding 7-mer was identified. The frequency of the four 8-mers starting with this 7-mer was determined, and the next bp was chosen by chance maintaining this frequency. This process was continued until the entire 1500-bp sequence was determined (seventh-order Markov model). For each of the 1000 data sets, the distribution of all 32,896 8-mers was determined, and the CF determined, as above. From the 1000 separate CF values (CFexpt) for each octamer, a mean (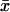 ) and standard deviation (σexpt) were computed. Finally, we calculated the probability term, P, that represents –log10(1 – p), where p is the area that lies under the normalized curve of the distribution of CFexpt. Thus, the greater the P value, the more nonrandom the result.
) and standard deviation (σexpt) were computed. Finally, we calculated the probability term, P, that represents –log10(1 – p), where p is the area that lies under the normalized curve of the distribution of CFexpt. Thus, the greater the P value, the more nonrandom the result.
The clustering and graphing of the data was performed using the programs Excel (Microsoft) and/or Grace (http://plasma-gate.weizmann.ac.il/Grace/).
A collection of 193 transcription motifs were selected from the TRANSFAC database (version 3.4) for analysis of the distribution of TF binding sites across the 1500 bp.
Tissue Specificity Classification
Based on the mRNA expression data for 63 samples representing 29 human tissues (http://expression.gnf.org), from the set of 6744 genes, which is in common with these data and 13,010 promoters that were examined, we extracted two subsets of genes: tissue-specific genes and housekeeping genes.
To classify those genes we defined two floating cutoffs, the values of which were individual for each given gene and depended on the maximum expression value of that gene: (1) high expression cutoff, the value that is 70% of the maximum expression level for the given gene always staying within the limits of ≥250 and ≤400; and (2) middle expression cutoff, the value that is 40% of the maximum expression level always staying within the limits of ≥130 and ≤200.
Those genes, the expression level of which is greater than high expression cutoff in one or two samples, and at the same time the expression level is greater than middle expression cutoff in four or fewer samples, were classified as tissue-specific genes (12.6% of the 6744 genes). The genes with an expression level that was greater than middle expression cutoff or high expression cutoff at least in 62 of the 63 samples were classified as housekeeping genes (9.2% of the 6744 genes).
Calculation of P Value for Subsets in a Set
To determine the significance of the numbers presented in Tables 2,3,4, we introduced a “probability” parameter, which is the normalized probability that the results observed occurred at a higher (or lower) frequency than would be expected by random chance. We calculated this parameter based on the standard P value for a two-tailed distribution. For each set containing S members, we calculated the number of possible combinations in which the two subsets containing s1 and s2 members have m members in common
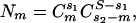 |
the total number of combinations
 |
and the probability of having m members in the intersection
 |
where 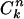 is combinatorial combination.
is combinatorial combination.
Then we calculated the integrated probability that our observed value (m*) occurred at greater than expected frequency, if m* is greater than the most probable value of m
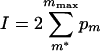 |
or our observed value (m*) occurred at lower frequency than expected when m* is less than the most probable value of m
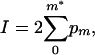 |
where mmax = min(s1,s2). We doubled the result so integrated probability, I value, should be varied in a range from zero to one, and took the logarithm P(m*) = –log10(I).
The value of P indicates the statistical probability of numbers being nonrandom: The greater the number, the more statistically nonrandom the result. For instance, in Table 2, in the CCAAT–SP1 intersection: there are 13,010 genes in total, 994 of them have a CCAAT site (7.6%), 2696 have a SP1 site (20.7%), and 288 have both sites. Thus, of CCAAT genes 29.1% have SP1, which is ∼1.5-fold greater than expected. The probability to have 288 (or more) members that have both CCAAT and SP1 sites is P = 10.2, a big number, which means that the number of elements in the intersection is not a result of random fluctuation but is determined by the nature of interdependence of the presence of CCAAT and SP1 sites under the peaks in the promoter region.
Acknowledgments
We thank Barbara Graves for conversations about ETS DNA binding, Robert Perry for conversations about ribosomal gene promoters, and David FitzGerald for comments on the manuscript. This study used the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, Maryland (http://biowulf.nih.gov).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1953904. Article published online before print in July 2004.
References
- Ashburner, M. and Lewis, S. 2002. On ontologies for biologists: The Gene Ontology: Untangling the web. Novartis Found. Symp. 247: 66–80. [PubMed] [Google Scholar]
- Bendall, A.J. and Molloy, P.L. 1994. Base preferences for DNA binding by the bHLH-Zip protein USF: Effects of MgCl2 on specificity and comparison with binding of Myc family members. Nucleic Acids Res. 22: 2801–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd, K.E. and Farnham, P.J. 1999. Coexamination of site-specific transcription factor binding and promoter activity in living cells. Mol. Cell. Biol. 19: 8393–8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breathnach, R. and Chambon, P. 1981. Organization and expression of eucaryotic split genes coding for proteins. Annu. Rev. Biochem. 50: 349–383. [DOI] [PubMed] [Google Scholar]
- Brown, T.A. and McKnight, S.L. 1992. Specificities of protein–protein and protein–DNA interaction of GABP α and two newly defined ets-related proteins. Genes & Dev. 6: 2502–2512. [DOI] [PubMed] [Google Scholar]
- Conkright, M.D., Guzman, E., Flechner, L., Su, A.I., Hogenesch, J.B., and Montminy, M. 2003. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol. Cell 11: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Davuluri, R.V., Suzuki, Y., Sugano, S., and Zhang, M.Q. 2000. CART classification of human 5′ UTR sequences. Genome Res. 10: 1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynan, W.S. and Tjian, R. 1985. Control of eukaryotic messenger RNA synthesis by sequence-specific DNA-binding proteins. Nature 316: 774–778. [DOI] [PubMed] [Google Scholar]
- Ferre-D'Amare, A.R., Prendergast, G.C., Ziff, E.B., and Burley, S.K. 1993. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 363: 38–45. [DOI] [PubMed] [Google Scholar]
- Ferreri, K., Gill, G., and Montminy, M. 1994. The cAMP-regulated transcription factor CREB interacts with a component of the TFIID complex. Proc. Natl. Acad. Sci. 91: 1210–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger, J.H., Hahn, S., Lee, S., and Sigler, P.B. 1996. Crystal structure of the yeast TFIIA/TBP/DNA complex. Science 272: 830–836. [DOI] [PubMed] [Google Scholar]
- Graves, B.J. and Petersen, J.M. 1998. Specificity within the ets family of transcription factors. Adv. Cancer Res. 75: 1–55. [DOI] [PubMed] [Google Scholar]
- Hapgood, J.P., Riedemann, J., and Scherer, S.D. 2001. Regulation of gene expression by GC-rich DNA cis-elements. Cell. Biol. Int. 25: 17–31. [DOI] [PubMed] [Google Scholar]
- Kent, W.J., Sugnet, C.W., Furey, T.S., Roskin, K.M., Pringle, T.H., Zahler, A.M., and Haussler, D. 2002. The human genome browser at UCSC. Genome Res. 12: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., Geiger, J.H., Hahn, S., and Sigler, P.B. 1993. Crystal structure of a yeast TBP/TATA-box complex. Nature 365: 512–520. [DOI] [PubMed] [Google Scholar]
- Kutach, A.K. and Kadonaga, J.T. 2000. The downstream promoter element DPE appears to be as widely used as the TATA box in Drosophila core promoters. Mol. Cell. Biol. 20: 4754–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Lo, K. and Smale, S.T. 1996. Generality of a functional initiator consensus sequence. Gene 182: 13–22. [DOI] [PubMed] [Google Scholar]
- Maglott, D.R., Katz, K.S., Sicotte, H., and Pruitt, K.D. 2000. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 28: 126–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangalam, H.J. 2002. tacg: A grep for DNA. BMC Bioinformatics 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani, R. 1999. The molecular biology of the CCAAT-binding factor NF-Y. Gene 239: 15–27. [DOI] [PubMed] [Google Scholar]
- Matys, V., Fricke, E., Geffers, R., Gossling, E., Haubrock, M., Hehl, R., Hornischer, K., Karas, D., Kel, A.E., Kel-Margoulis, O.V., et al. 2003. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 31: 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr, B. and Montminy, M. 2001. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell. Biol. 2: 599–609. [DOI] [PubMed] [Google Scholar]
- Moll, J.R., Acharya, A., Gal, J., Mir, A.A., and Vinson, C. 2002. Magnesium is required for specific DNA binding of the CREB B-ZIP domain. Nucleic Acids Res. 30: 1240–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt, K.D. and Maglott, D.R. 2001. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 29: 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt, K.D., Katz, K.S., Sicotte, H., and Maglott, D.R. 2000. Introducing RefSeq and LocusLink: Curated human genome resources at the NCBI. Trends Genet. 16: 44–47. [DOI] [PubMed] [Google Scholar]
- Rice, P., Longden, I., and Bleasby, A. 2000. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 16: 276–277. [DOI] [PubMed] [Google Scholar]
- Romier, C., Cocchiarella, F., Mantovani, R., and Moras, D. 2003. The NF-YB/NF-YC structure gives insight into DNA binding and transcription regulation by CCAAT factor NF-Y. J. Biol. Chem. 278: 1336–1345. [DOI] [PubMed] [Google Scholar]
- Sawadogo, M. and Roeder, R.G. 1985. Interaction of a gene-specific transcription factor with the adenovirus major late promoter upstream of the TATA box region. Cell 43: 165–175. [DOI] [PubMed] [Google Scholar]
- Scarpulla, R.C. 2002. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene 286: 81–89. [DOI] [PubMed] [Google Scholar]
- Sharrocks, A.D. 2001. The ETS-domain transcription factor family. Nat. Rev. Mol. Cell. Biol. 2: 827–837. [DOI] [PubMed] [Google Scholar]
- Shaywitz, A.J. and Greenberg, M.E. 1999. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68: 821–861. [DOI] [PubMed] [Google Scholar]
- Shuman, J.D., Cheong, J., and Coligan, J.E. 1997. ATF-2 and C/EBPα can form a heterodimeric DNA binding complex in vitro: Functional implications for transcriptional regulation. J. Biol. Chem. 272: 12793–12800. [DOI] [PubMed] [Google Scholar]
- Sinha, S., Maity, S.N., Lu, J., and de Crombrugghe, B. 1995. Recombinant rat CBF-C, the third subunit of CBF/NFY, allows formation of a protein-DNA complex with CBF-A and CBF-B and with yeast HAP2 and HAP3. Proc. Natl. Acad. Sci. 92: 1624–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale, S.T. 1997. Transcription initiation from TATA-less promoters within eukaryotic protein-coding genes. Biochim. Biophys. Acta 1351: 73–88. [DOI] [PubMed] [Google Scholar]
- Su, A.I., Cooke, M.P., Ching, K.A., Hakak, Y., Walker, J.R., Wiltshire, T., Orth, A.P., Vega, R.G., Sapinoso, L.M., Moqrich, A., et al. 2002. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. 99: 4465–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, Y., Yamashita, R., Nakai, K., and Sugano, S. 2002. DBTSS: DataBase of human Transcriptional start sites and full-length cDNAs. Nucleic Acids Res. 30: 328–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinklein, N.D., Aldred, S.J., Saldanha, A.J., and Myers, R.M. 2003. Identification and functional analysis of human transcriptional promoters. Genome Res. 13: 308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson, C.R., Hai, T., and Boyd, S.M. 1993. Dimerization specificity of the leucine zipper-containing bZIP motif on DNA binding: Prediction and rational design. Genes & Dev. 7: 1047–1058. [DOI] [PubMed] [Google Scholar]
- Vinson, C., Myakishev, M., Acharya, A., Mir, A.A., Moll, J.R., and Bonovich, M. 2002. Classification of human B-ZIP proteins based on dimerization properties. Mol. Cell. Biol. 22: 6321–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://genome.nci.nih.gov/publications/promoters; Supplemental data for this paper.
- http://transfac.gbf.de/TRANSFAC; the Transcription Factor Database.
- http://expression.gnf.org; GNF Gene Expression Atlas.
- http://genome.ucsc.edu/; UCSC Genome Bioinformatics site.
- http://dbtss.hgc.jp/index.html; database of TSS (DBTSS).
- http://plasma-gate.weizmann.ac.il/Grace/; Grace Graphing Software.
- http://biowulf.nih.gov; NIH Biowulf cluster.