

Abstract
We present the first analysis of stress-induced DNA duplex destabilization (SIDD) in a complete chromosome, the Escherichia coli K12 genome. We used a newly developed method to calculate the locations and extents of stress-induced destabilization to single-base resolution at superhelix density σ = –0.06. We find that SIDD sites in this genome show a statistically highly significant tendency to avoid coding regions. And among intergenic regions, those that either contain documented promoters or occur between divergently transcribing coding regions, and hence may be inferred to contain promoters, are associated with strong SIDD sites in a statistically highly significant manner. Intergenic regions located between convergently transcribing genes, which are inferred not to contain promoters, are not significantly enriched for destabilized sites. Statistical analysis shows that a strongly destabilized intergenic region has an 80% chance of containing a promoter, whereas an intergenic region that does not contain a strong SIDD site has only a 24% chance. We describe how these observations may illuminate specific mechanisms of regulation, and assist in the computational identification of promoter locations in prokaryotes.
Because the initiation of transcription and the initiation of replication both require local separation of the DNA duplex, the locations and occasions where this transition occurs in vivo must be stringently controlled. One biologically important way in which local DNA stability is regulated is through superhelical stresses imposed on the duplex (Benham 1979). Negative DNA superhelicity exerts untwisting torsional stresses on the base pairs that experience it. When these stresses are sufficiently large, they can drive local structural transitions to conformations whose right-handed helicities are less than that of the B-form (Benham 1981). Because strand separation locally unwinds the DNA, it localizes some of the imposed superhelicity, which relaxes by a corresponding amount the level of stress on the rest of the domain. Thus the free energy cost of this transition is partially or fully offset by the free energy returned from the fractional relaxation it provides. When this return exceeds its cost, a transition will be favored at equilibrium.
Local sites of strand separation (also called local denaturation, duplex opening, or unwinding), the most extreme form of duplex destabilization, can be induced by moderate levels of negative superhelicity (Benham 1980; Kowalski et al. 1988; Lyubchenko and Shlyakhtenko 1988; Voloshin et al. 1989). Partial destabilizations also can occur, in which the imposed superhelical stresses fractionally decrease the free energy needed to separate the duplex. Partial destabilization can be biologically important, as it decreases the amount of free energy other molecules must provide to drive separation at the site involved, and hence can facilitate regulatory events.
The relaxation induced by strand separation at any one site is felt by all other base pairs, and their propensities to separate change accordingly. Thus, transitions that involve only near-neighbor interactions when they occur in linear or nicked DNA will in stressed DNA be coupled to the conformational states of all other base pairs that experience the stress. Whether transition occurs at a given site thus depends not just on its local properties, such as thermodynamic stability, but also on how that site competes with all others in the domain. In consequence of this coupling, stress-induced transitions have a large repertoire of highly intricate, nonlinear, and interactive behaviors that far transcend what is possible for thermally driven transitions in unconstrained molecules (Benham 1996). For this reason, the thermodynamic stability profile of a region does not adequately reflect its stress-induced destabilization properties.
Superhelical stresses are modulated in vivo by a variety of processes, including topoisomerase enzyme activity, translocation of RNA polymerase during transcription, changes of nucleosome binding patterns, histone acetylation, helicase activity, and constraints imposed by other DNA-binding events. In prokaryotes, the basal level of superhelical stress is known to vary with the energy charge, both between stationary and growth phases, and in response to environmental changes such as altered osmolarity, hydrogen peroxide, or thermal stress (Rohde et al. 1994; Lopez-Garcia and Forterre 2000; Weinstein-Fischer et al. 2000; Cheung et al. 2003). The patterns of gene expression also change to suit these altered conditions. Superhelically induced DNA duplex destabilization has been shown to be involved in the mechanisms regulating specific promoters (Sheridan et al. 1998, 1999). This has led to the proposal that chromosomal superhelicity may be a global regulator of gene expression in Escherichia coli (Hatfield and Benham 2002).
Strand separation in most DNA regulatory events is not mediated by superhelical stresses alone, but instead usually involves interactions between the DNA and other molecules, commonly proteins. However, stress-induced changes in the local stability of the DNA duplex can strongly affect these events. The ease with which a DNA region can be opened by a reversible intermolecular process depends exponentially on the energy required; destabilizing the duplex at the site by 3 kcal/mole, much less than what is required for its opening, will shift the equilibrium more than 100-fold toward the denatured state, other factors remaining unchanged. Even relatively small amounts of stress-induced duplex destabilization (SIDD), therefore, well below what would be required to drive full strand separation, can greatly facilitate opening reactions that are mediated by other molecules. In this way, even fractional changes of stability at a regulatory site can drastically affect its activity.
SIDD has been implicated in the mechanisms of activity governing a wide variety of biological processes. One mechanism of transcriptional regulation known to occur in E. coli involves the binding-induced transmission of superhelical destabilization. This mechanism has been implicated in the activation of the ilvPG promoter by ihf protein binding, and in the fis binding-mediated regulation of the leuV promoter (Sheridan et al. 1998, 1999; Hatfield and Benham 2002). Initiation of transcription from the human MYC proto-oncogene is regulated by the binding of FBP to the single-stranded DNA regulatory element FUSE (He et al. 2000). The minimal conditions for in vitro transcriptional initiation from the yeast CUP1 (YHR053C) gene promoter are a negatively superhelical DNA substrate plus RNA polymerase (RNAP); no other regulatory molecules are required (Leblanc et al. 2000). SIDD also has been implicated in transcriptional termination in yeast (Benham 1996; Aranda et al. 1997). The initiation of replication in both prokaryotes and yeast has been shown to require the presence at a precise position of a site that is susceptible to superhelical strand separation (Kowalski and Eddy 1989; Huang and Kowalski 1993). Recent work has shown that SIDD sites created by mutations can function as replication origins (Potaman et al. 2003). This has been proposed as the mechanism of expansion of the pentameric repeat responsible for spinocerebellar ataxia type 10. Sites susceptible to stress-induced duplex destabilization also characterize a variety of chromosomal attachment regions. Examples include yeast centromeres (Tal et al. 1994) and matrix attachment regions (MARs), which are positions where the eukaryotic chromosome putatively attaches to the interphase nuclear matrix (Benham et al. 1997).
These and other results show that stress-induced duplex destabilization is an essential component of regulatory mechanisms governing a wide range of normal and pathological events. This makes it essential to have computational methods that accurately analyze the SIDD properties of DNA sequences. This research group has developed three such methods, all based in statistical mechanics, to calculate the pattern of duplex destabilization experienced by a short DNA sequence in response to negative superhelicity (Benham 1990; Sun et al. 1995; Fye and Benham 1999). These methods all evaluate the equilibrium distribution among states of denaturation of a DNA molecule of specified base sequence, on which a defined level of superhelicity has been imposed. All conformational and energy parameters are given their experimentally measured values, thus there are no free parameters to be fit. Yet in all cases where experiments have been performed, the computations are found to make accurate predictions of the locations and extents of strand separation as functions of base sequence and imposed superhelicity (Benham 1992; He et al. 2000). Moreover, many of the sites that were predicted to separate under stress have subsequently been experimentally shown to open, both in vitro and in vivo (Aranda et al. 1997; Benham et al. 1997; Sheridan et al. 1998; Fye and Benham 1999; He et al. 2000; Potaman et al. 2003). The demonstrated quantitative accuracy of these methods allows one to have confidence in their predictions of SIDD sites in other sequences, on which experiments have not been performed. Indeed, computational analyses of the SIDD properties of specific DNA sequences have played central roles in the collaborations that have elucidated many of the biological roles of stress-induced duplex destabilization.
These methods have recently been extended to enable the analysis of long DNA sequences, including complete genomes (Benham and Bi 2004). As a first application of this method, we present here the results of the analysis of the complete E. coli chromosome. We document a strong tendency for the stress-destabilized sites to avoid coding sequences. And among the noncoding, intergenic regions, we show that sites that are known or inferred to contain promoters are highly enriched for destabilized sites, whereas sites that are inferred not to contain promoters are not thusly enriched. We also present and apply a Monte Carlo method for assessing the statistical significance of the associations found between SIDD sites and biological markers.
RESULTS
Here we report the results of the SIDD analysis of the E. coli K12 chromosomal sequence (version M54, accession number NC000193; Blattner et al. 1997), performed as described in the Methods section below. This circular molecule contains 4,639,221 bp, of which 4,117,360 bp, or 88.75% of the genome, occur within its 4395 annotated coding regions (i.e., open reading frames [ORFs]). The other 521,861 bp occur in noncoding, intergenic regions. Because there are 811 cases in which neighbor coding regions either overlap or abut, this molecule has 3584 annotated intergenic regions. These may be classified according to the directions of transcription of their bounding coding regions as tandem (TAN), divergent (DIV), or convergent (CON). There are 624 DIV regions, 2405 TAN regions, and 555 CON regions.
The Distribution of SIDD Sites in the E. coli K12 Chromosome
The most informative parameter for describing destabilization is the incremental energy G(x) needed to guarantee separation of the base pair at position x (Benham 1993, 1996). More strongly destabilized sites have lower values of G(x), whereas positions that remain stable have high values. A value of G(x) near 10.2 kcal/mole indicates full stability. Such a region is as stable as it would be in a relaxed or unconstrained molecule.
Figure 1 presents three basic properties of the distribution of SIDD sites in the E. coli genome. The top left graph gives the cumulative distribution of destabilization levels expressed in terms of the destabilization energy G(x). For each value G on the horizontal axis, the curve plots the total number of base pairs for which G(x) ≤ G. Thus, 51,215 bp, just 1.1% of the genome, are strongly destabilized at the level G(x) ≤ 0.0. A total of 153,994 bp, comprising 3.32% of the genome, have G(x) ≤ 2.0 kcal/mole indicative of substantial destabilization, whereas 556,036 bp (11.98%) show moderate destabilization at the level G(x) ≤ 6.0. Conversely, >75% of the genome is not significantly destabilized (G(x) > 8.0), despite the imposed stress. Destabilization clearly is not distributed uniformly throughout this genome. Significant destabilization is calculated to be confined to a small fraction of sites, with the large majority of base pairs remaining virtually fully stable despite the imposed stress.
Figure 1.

The top left graph plots the cumulative G(x) distribution. For each value of G on the horizontal axis, the curve plots the number of base pairs destabilized below that value, that is, G(x) ≤ G. The top right graph shows the number of SIDD regions (sets of consecutive base pairs, all of which are) destabilized below the indicated level. The graph on the bottom shows the distributions of lengths of these SIDD regions.
The top right graph of Figure 1 shows the number of SIDD regions that are destabilized below a threshold G, that is, G(x) ≤ G for G = 0, 1, 2, 3, 4, 5, and 6. This is the number of runs of contiguous base pairs that satisfy this inequality. Finally, the graph at the bottom of the figure gives the distribution of lengths of the SIDD sites that satisfy this condition for each integer threshold value G = 0,..., 6. Taken together, these last two graphs show that destabilization tends to occur in a relatively small number of reasonably long sites. For example, there are 692 sites of strong destabilization, satisfying G(x) ≤ 0.0, whose average length is 74.0 bp. Similarly, 1448 runs have G(x) ≤ 2.0, with an average length of 106.3 bp. This gives an average density of approximately one site destabilized to this level per 3 kb.
The SIDD profile of a representative 5-kb region of the E. coli genome is shown in Figure 2, annotated with the coding regions and promoters known to be present. In this example, the sites of significant destabilization are largely confined to intergenic regions, whereas coding regions are not significantly destabilized. Indeed, the stabilities within coding regions remain around G(x) = 9–10 kcal/mole, essentially unchanged from what they would be in a relaxed or unconstrained molecule. The strongest destabilization occurs in the intergenic region separating the dnaK gene from the divergently oriented yaa1 ORF. This region contains two documented promoters, both regulating dnaK, whose locations are annotated in the figure. Indeed, the three most strongly destabilized sites in this profile all occur at intergenic positions located in the upstream, 5′-flanks of coding regions. All of these sites are destabilized below G(x) ≤ 5, and the 5′-flank of each ORF in this region is located at one of these three sites. In contrast, the 3′-flanks of all ORFs in this profile are either less destabilized than this or not destabilized. Specifically, there are two intergenic regions located between convergently oriented ORFs, which are unlikely to contain promoters. One of these is moderately destabilized, and the other is not destabilized.
Figure 2.

The SIDD profile of a representative 5-kb region of the E. coli genome. ORF locations are given by the bars above the graph; the upper level of bars are ORFs that transcribe directly; the lower level are ORFs that transcribe in reverse. The documented dnaK promoters also are shown. Destabilization is largely confined to the 5′-flanks of coding regions. The strongest SIDD site is between the divergently oriented dnaK and yaaI ORFs, a region that contains both dnaK promoters. The convergent terminal region at 11,300 is destabilized below G(x) < 5, but the other such region at position 9900 is not.
In summary, this SIDD profile shows a distinctive pattern in which significant destabilization is concentrated at noncoding regions. And among these, strong destabilization appears to occur primarily at those intergenic regions that contain promoters. Visual inspection shows that this pattern of SIDD distribution occurs frequently in the E. coli genome. We next perform a variety of analyses to determine precisely how representative this pattern is of the arrangement of SIDD sites throughout this genome.
Correlations Between SIDD Sites and Promoters, Terminators, and Coding Regions
We investigate the association between SIDD sites and promoter-containing regions in greater detail. For this purpose, we examine the differences in SIDD properties between coding regions and intergenic regions of various types. Divergent intergenic regions (DIV, 624 instances) may be inferred to contain promoters, whereas convergent regions (CON, 555 instances) may similarly be inferred not to contain promoters. Tandem regions (TAN, 2405 instances) either may or may not contain promoters.
We also examine the set of intergenic regions that contain experimentally characterized promoters, noting that the number of known promoters is small. For this purpose, we have used two sets of documented promoters—those that are so annotated in the GenBank entry for this sequence, and those compiled in the PromEC database (Hershberg et al. 2001). The results we find are entirely equivalent for these two sets, as described below. There are 305 documented promoters annotated in GenBank. Of these, 262 occur in 189 distinct intergenic regions, all of which are either divergent or tandem. No documented promoter in either of these data sets is found in a convergent intergenic region. We call this set of intergenic regions that contain GenBank-annotated documented promoters DTP.
In the analyses that follow, we define an SIDD site to be a collection of consecutive base pairs whose G(x) values all are <8.0 kcal/mole. This energy level is thereby regarded as the threshold for regarding a region as being destabilized. We denote the minimum value of G(x) within such an SIDD site by Gm. This value is used to classify the degree of destabilization within a site. (We note that this definition differs from the one used to compile the information in Figure 1. There we counted the numbers of base pairs, and the numbers and lengths of regions, where G(x) falls below each value G. Here an SIDD site is a region where G(x) falls below 8 kcal/mole. Such an SIDD site could contain multiple minima, and hence may have several distinct internal regions where G(x) falls below a given level G.) We sort the SIDD sites according to their levels of destabilization in two ways, either cumulatively or into disjoint bins. The cumulative sets are those satisfying Gm ≤ Go with Go = 0, 1, 2, 3, 4, 5, or 6. In this case, the SIDD locations determined by the choice Go will be a subset of the sites having threshold G′o ≥ Go. In the binned case, the lowest bin is determined by Gm ≤ 0, and the other bins contain the SIDD sites satisfying i – 1 < Gm ≤ i, for i = 1,..., 6. These binned sets are disjoint; each SIDD site with Gm ≤ 6.0 belongs to exactly one bin.
The Fraction of SIDD Sites That Overlap Intergenic Regions
In the following analysis, we use the binned sets of SIDD sites. We first determine the fraction of SIDD sites in each set that overlap intergenic regions. This is done for the complete set of 3584 intergenic regions, and for each of the three subtypes DIV, TAN, and CON. These results are shown in Figure 3. For example, 89% of the SIDD sites satisfying Gm ≤ 0 are found to overlap intergenic regions. Thus, only 11% of the SIDD sites destabilized to this level are internal to coding regions. As 88.75% of this genome is coding and only 11.25% is noncoding, the density of these strongest SIDD sites is more than 60 times greater in intergenic regions than in coding regions! This represents an extremely pronounced clustering of SIDD sites within intergenic regions, far beyond what would occur if they were randomly located. Even the least destabilized sites we examined, those having 5 < Gm ≤ 6, are enriched more than threefold within intergenic regions relative to coding regions. These results demonstrate that the pattern shown in Figure 2, whereby SIDD sites in E. coli show a strong preference for noncoding, intergenic regions, in fact prevails throughout the genome.
Figure 3.

This figure plots the percentage of binned SIDD sites that overlap the various types of intergenic regions. The top line (circles) is for all intergenic regions, which are partitioned into the disjoint sets TAN, DIV, and CON. The values given by squares give the fraction of SIDD sites at each level that overlap regions in TAN; diamonds give the same information for DIV, and triangles for CON.
The most strongly destabilized site in the SIDD profile in Figure 2 coincides with the intergenic region containing the documented promoters dnaKp1 and dnaKp2. The results shown in Figure 3 demonstrate that this pattern is representative of the genome-wide SIDD distribution. Although 89% of the sites having Gm ≤ 0 colocate with noncoding regions, only 3% occur at CON sites that are unlikely to contain promoters. In contrast, 31% occur at DIV sites, which may be inferred to contain promoters. The remaining 55% occur at tandem regions, which either may or may not contain promoters. There are almost four times as many tandem regions as divergent ones, thus this represents a twofold enrichment of SIDD sites in DIV over TAN. This clearly shows that strongly destabilized sites are most closely associated with promoter-containing intergenic regions. Next we examine this association in greater detail.
The Fractions of Intergenic Regions That Overlap SIDD Sites
Here we consider the converse question: How frequently do intergenic regions that are known or inferred either to contain promoters (DTP and DIV), or not to contain promoters (CON), overlap sites that are destabilized to a specific level? We also determine the percentage of tandem regions (TAN) that are destabilized to that level, and the percentage of coding regions that have internal SIDD sites. In this analysis we use the cumulative sets of SIDD sites. The complete results are shown in Figure 4. Consider, for example, the results at the moderate destabilization level of Gm ≤ 4.0. We find that 137 of the 189 DTP regions (72%) intersect SIDD sites that are destabilized at this level. Similarly, 422 of the 624 DIV regions (68%) achieve this level of destabilization. However, only 101 of the 555 CON regions (18%) and 714 of the 4395 coding regions (16%) are destabilized to this level. The last number is especially remarkable, as coding regions constitute almost 90% of this genome. Clearly SIDD sites show a strong tendency to cluster specifically at promoter-containing intergenic regions, and to avoid coding sequences. Similar clusterings are found when any destabilization threshold is used, from Gm ≤ 6 to Gm ≤ 0.
Figure 4.

The fractions of sites of the four types DIV, DTP, CON, and TAN that overlap SIDD regions are shown at each level of destabilization. The fraction of coding regions that have internal SIDD sites at this level also are shown. Here the cumulative sets of SIDD sites are used.
These results show that the strongest association with SIDD sites occurs for intergenic regions that are known (DTP) or inferred (DIV) to contain promoters. Tandem intergenic regions (TAN), which may or may not contain promoters, have an intermediate level of SIDD association, whereas CON sites (which are inferred not to contain promoters) and coding regions have the lowest levels of association. The fact that the fractions of SIDD-associated documented promoters (DTP) and divergent intergenic regions (DIV) are closely similar at each destabilization level lends further support to the inference that promoters are present within DIV regions.
The Statistical Significance of These Associations
Next we assess the statistical significance of the associations we have documented between SIDD sites and the categories DTP, DIV, TAN, CON, and coding regions. For each category, we compare its level of association with actual SIDD sites to the level it would have with a matched set of randomly located sites. We use a Monte Carlo procedure to choose these matched sets of sites. We first perform this analysis at each cumulative SIDD level as follows. For each value Go = 0,..., 6, we count the number of SIDD sites throughout the genome with Gm ≤ Go, and determine the length of each such site. Then we use a random number generator to select a set of randomly located, nonoverlapping and nonabutting sites, equal in number and having the same lengths as this set of actual SIDD sites. We determine how many of these randomly located sites overlap regions in each of the categories DTP, DIV, CON, and TAN, and how many are internal to coding regions. We do this 1000 times in each case, and determine the distribution that arises for each category. This is done for each cumulative SIDD level Gm ≤ Go, Go = 0, 1,..., 6. The results for Gm ≤ 2 are shown in Figure 5, along with the observed values for the actual distribution of SIDD sites, for the four sets DIV, CON, DTP, and coding. In each case, the blue distribution is the number of genomic regions that colocate with SIDD sites, and the orange distribution is the number of SIDD sites that colocate with the particular type of genomic region. Because coding regions can be long, a single ORF may contain more than one SIDD site at random, thus in this case the blue and orange curves are displaced. Because the other types of regions are shorter, these curves overlap. In all cases, the random distributions are seen to be approximately normal, thus we may evaluate their means and standard deviations. We then determine how many standard deviations away from this mean one finds the actual distribution. (Here the number of genomic regions that are actually colocated with SIDD sites is given as the red square, and the number of SIDD sites that colocate with these genomic regions is denoted with a pink X.) This gives the statistical significance of whatever associations or avoidances there may be between SIDD sites and each of these five classes of transcriptional regions. As shown in Figure 5, the actual level of association between SIDD sites and either DTP or DIV exceeds that expected at random by many standard deviations. This is also true for TAN (data not shown). SIDD sites also are highly statistically significantly under-represented within coding regions. They are somewhat overrepresented at CON, but with at most a moderate level of statistical significance.
Figure 5.

The calculated distributions are shown of the numbers of sites of each of the four types (DIV, DTP, CON, and coding) that would overlap SIDD sites with Gm ≤ 2 if these 1448 SIDD sites were distributed at random. The results for DTP are shown in the upper left, for DIV in the upper right, for CON in the lower left, and for coding regions in the lower right. Blue distributions give the number of genomic regions that colocate at random with SIDD sites, whereas orange distributions show the number of SIDD sites that colocate at random with genomic regions. In each case, the number of genomic regions that actually colocate with SIDD sites is given as the red square, and the number of actual SIDD sites that colocate with the genomic regions is denoted with a pink X.
Next we investigate in greater detail how the statistical significance of each type of association varies with SIDD level. For this purpose, we repeat the Monte Carlo analysis at each SIDD level, with the SIDD sites now partitioned into seven disjoint bins according to their minimum G(x) value, Gm. Sites with Gm ≤ 0 are placed in bin 0, whereas sites with i – 1 < Gm ≤ i are placed in bin i, 1 ≤ i ≤ 6. In all cases, the random distributions by inspection are seen to be approximately normal (data not shown), as was observed above for the cumulative distributions. We calculate their means and standard deviations of these distributions, and from them the z-scores of the actual SIDD associations. (The z-score is the number of standard deviations that the observed value is away from the mean of the random distribution. Positive z-scores correspond to values above the mean, negative z-scores to values below the mean.) The statistical significances of associations between these SIDD sites and the five classes DTP, DIV, CON, TAN, and internal coding regions are plotted in Figure 6 for each destabilization level (i.e., bin) as the z-scores found by this Monte Carlo procedure. A z-score of ±3.5 corresponds to a probability of slightly less than 0.001 that this value occurs by chance. We see that for values Gm ≤ 0, 1, or 2, the SIDD sites are statistically highly significantly associated with both promoter-containing sets DTP and DIV. The association between SIDD sites and CON, which are regions that are inferred not to contain promoters, is never more than marginally significant at the 0.001 level. At all destabilization levels, SIDD sites show a statistically highly significant propensity to avoid coding regions, and to associate with TANs. The statistical significances found for the four categories DTP, DIV, TAN, and coding regions are greatest for the highest levels of destabilization. For the most destabilized sites, those satisfying Gm ≤ 0, we find they cluster at DIV regions at rates >27 standard deviations above random, and avoid coding regions at rates that are >30 standard deviations below random.
Figure 6.

The statistical significance of the associations of SIDD sites with the five classes of genomic regions DTP, DIV, TAN, CON, and coding region are given as z-scores, the number of standard deviations by which the given value varies from what would be expected if SIDD sites were randomly located. Here binned sets of SIDD sites are used, bin 0 corresponding to SIDD sites with Gm ≤ 0, and bin i to sites with i – 1 < Gm ≤ i. A positive z-score indicates that the number of SIDD sites at the given type of region exceeds the value expected at random, whereas a negative z-score indicates that the actual number is smaller than what would be expected at random. The gray band is the threshold for statistical significance at the 0.001 level. Sites outside this band are significant at this level.
A second set of 472 documented promoters has been compiled in the PromEC database (Hershberg et al. 2001). As some intergenic regions contain multiple promoters, this database documents 285 distinct intergenic regions that together contain 399 experimentally characterized promoters. Of these 285 promoter-containing intergenic regions, 151 TAN (tandem) regions contain a total of 195 documented promoters, and 134 regions in DIV contain 204 documented promoters. The family CON contains no documented promoters. These observations justify our inference that regions in CON, by virtue of the convergent orientations of their bounding coding regions, are expected not to contain promoters. The analysis of documented promoters using this data set yields results that are entirely equivalent to those found above from the GenBank data set. For example, 71 of the 189 GenBank-documented promoter-containing intergenic sites (37.6%) are destabilized below Gm ≤ 0, whereas 105 of the 285 PromEC-documented sites (36.8%) are destabilized to this level.
SIDD Sites at Promoters in E. coli
These results show that SIDD sites strongly avoid coding regions. And among intergenic regions, destabilization at or below the level Gm ≤ 2.0 kcal/mole is highly statistically significantly associated with the presence of promoters. Here we investigate the implications of these results regarding the numbers and locations of promoters in E. coli.
We have estimated the probabilities that an intergenic region either is (D) or is not (D̃) destabilized to the specified level, given that it either does (P) or does not (P̃) contain a promoter. In symbols, these are the conditional probabilities p(D|P), p(D̃|P) = 1 – p(D|P), p(D|P̃), and p(D̃|P̃) = 1 – p(D|P̃). This information can be used to estimate the probability that an intergenic region contains a promoter, given that it either is or is not destabilized. This involves finding the reversed conditional probabilities p(P|D) and p(P|D̃), which may be done using Bayes's theorem:
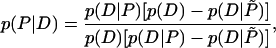 |
(1) |
and
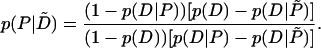 |
(2) |
In this analysis we regard all intergenic regions as a priori identical, and assess the probabilities that they contain promoters given that they either are or are not destabilized at the level Gm ≤ 2. The probabilities required in these formulas are found as follows. Of the 555 regions in CON, which are inferred not to contain promoters, only 62 are destabilized at this level, so p(D|P̃) = 62/555 = 0.112. We first estimate p(D|P) from the 189 intergenic regions in DTP, which contain experimentally characterized promoters. We find that 107 of these DTP sites are destabilized at this level, giving p(D|P) = 107/189 = 0.566. Finally, of the 3584 intergenic regions, we find that 1032 are destabilized at the level Gm ≤ 2.0, thus the overall probability of destabilization is p(D) = 0.288. Substituting these values into the above formulas, we find that p(P|D) = 0.763, and p(P|D̃) = 0.236. If we use DIV instead of DTP as our promoter-containing data set, we get p(P|D) = 0.81, and p(P|D̃) = 0.249. This analysis indicates that SIDD properties alone are anticipated to be highly accurate predictors of the presence of promoters within strongly destabilized intergenic regions.
We may use these results to make a rough estimate of the number n(P) of promoter-containing intergenic regions in E. coli. Using the set of SIDD sites satisfying Gm ≤ 2.0 and the results from the documented promoters DTP, we find
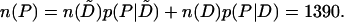 |
Those intergenic regions that contain promoters as documented in the PromEC database have an average of 1.4 promoters each (Hershberg et al. 2001). If this average is accurate on the genome-wide scale, these 1390 regions would contain ∼1940 promoters. This database also finds that 15% of the documented promoters overlap or are internal to coding regions. If this is representative of the genome-wide average, there would be an additional 350 promoters, for an estimated total of 2290 promoters in this genome.
DISCUSSION
This paper presents the results of the first analysis of the stress-induced duplex destabilization properties of a complete genome, that of E. coli K12. This is the first application to a complete chromosome of our newly developed computational method for analyzing the SIDD properties of long DNA sequences (Benham and Bi 2004). We have investigated the distribution of predicted SIDD sites in this genome, and shown that they have a specific and statistically highly significant pattern of association with transcriptional regions. We document a very strong association of highly destabilized sites with promoter-containing intergenic regions, but not with other intergenic regions. SIDD sites also are found in coding sequences at frequencies far below what would occur at random.
The statistics of this association are indeed extreme. The most strongly destabilized sites are present in intergenic regions that contain either documented (DTP) or inferred (DIV) promoters at rates that are >20 standard deviations above random. And they occur within coding regions at rates >30 standard deviations below random. The p-values associated with these z-scores are infinitesimal. Stated otherwise, the density of the strongest SIDD sites is >85 times greater in promoter-containing intergenic regions than in coding regions. We are unaware of any other single attribute that shows such a high degree of clustering at promoters. Such extreme density differences would be very unlikely to persist within a population without strong selection pressure to conserve them.
The results presented here suggest that SIDD attributes may be useful for finding promoter locations in prokaryotes, a problem that has proven surprisingly difficult to resolve using string-based methods alone (Hertz and Stormo 1996; Vanet et al. 1999; Eskin et al. 2003). One of the latest and allegedly most accurate of such methods is based on the hexameric sequence properties of putative –10 and –35 regions (Huerta and Collado-Vides 2003). The best implementation of this method achieves an overall accuracy of 53%. We contrast this with our results, which are based on SIDD properties alone. If an SIDD site having Gm ≤ 2.0 kcal/mole overlaps an intergenic region, the analysis presented in the previous section estimates that the probability of this region containing a promoter is ∼80%. If an intergenic region does not overlap such an SIDD site, the probability that it contains a promoter falls below 25%. Suppose one were to predict the presence of a promoter within an intergenic region based exclusively on whether or not it is destabilized at this level. Using the known number of intergenic regions that contain such SIDD sites, we estimate that a 67% statistical accuracy would be achieved by such a predictor. Thus, predictions that are based exclusively on one structural attribute—SIDD properties—would substantially outperform even the best sequence-based predictor.
We anticipate that improved promoter prediction algorithms may be achievable by including both the SIDD properties and the known sequence attributes of promoters. (In addition to the sequence attributes used in previous predictors, there is statistical evidence, for example, that TAN regions that do not contain promoters are usually quite short, commonly ∼10 bp in length; Salgado et al. 2000.) We are currently working to develop and test alternative promoter prediction strategies based on this approach. We anticipate that the accuracy achievable by inclusion of SIDD properties will be a substantial improvement over all existing methods. This work will be the subject of a future paper.
One might anticipate that specific structural and physico-chemical attributes could be closely associated with particular types of regulatory regions. After all, the mechanisms by which regulation is effected involve interactions with other molecules that depend on the structures and physical–chemical properties of all the participants, including the DNA. Thus, it should not be surprising that the propensity of sites to become destabilized under the types of stresses that occur in vivo should correlate with the locations of regulatory regions governing processes in which duplex opening is required.
Stress-induced destabilization could be expected to be associated specifically with promoters because strand separation is a necessary step in the initiation of transcription. Although this opening is mediated by the polymerase holoenzyme, even fractional destabilization can greatly assist this process. For example, destabilization of the DNA duplex at the –10 region by 2.8 kcal/mole (from 10.2 kcal/mole to 7.4 kcal/mole) would shift the equilibrium of the opening reaction by two orders of magnitude toward the open state. This is a direct mechanism by which SIDD could alter the transcriptional activities of promoters. However, specific cases have shown that destabilization events also can play other specific roles in transcriptional regulatory mechanisms.
A strong SIDD site has been shown to be present 90 bp upstream of the ilvPG promoter of E. coli (Sheridan et al. 1998, 1999). This region coincides with an IHF-binding site. IHF binding at this position is known to activate transcription in a superhelix-dependent manner. Experiments have shown that in the absence of IHF, this site denatures when the superhelix density satisfies σ ≤ –0.03. But IHF binding causes this site to reanneal back to B-form. This causes the stress-induced destabilization to be transferred to the most susceptible remaining site, which is in the –10 region of this promoter. The predicted SIDD site in this intergenic region has been shown experimentally to actually denature under negative superhelical stress, and to participate in the transcriptional regulation of this promoter by this IHF-binding-induced transmission of stress-induced destabilization from a remote site into the –10 region.
We note that the strong SIDD site involved in this mechanism coincides with the binding site for a regulatory molecule, not with the location of the promoter itself. This shows the possible functional importance of SIDD sites located at any positions within intergenic regions, not just at the promoter. It also shows that regulatory mechanisms can involve interactions between destabilization and binding events. It is reasonable to suppose that SIDD sites near other promoters in E. coli also could be involved in the specific mechanisms of their regulation. These issues, and many others arising from the analysis of this data set, will be carefully addressed in future publications.
An initial analysis of several yeast genes has shown that the strongest SIDD sites in that primitive eukaryote are found in the 3′-terminal flanks of genes, not in their 5′-flanks (Benham 1996). Yeast promoters commonly are destabilized, but to a much smaller degree than are terminators, whereas coding regions are not significantly destabilized. The preferential occurrence of strong SIDD sites in the 3′ gene flanks of yeast genes stands in stark contrast to the association with promoters that has been documented here for E. coli. The significance of these species- or kingdom-specific differences in SIDD patterns will be a matter for future investigation.
We currently are developing a Web site to provide access to the results of our SIDD analyses at the address http://www.genomecenter.ucdavis.edu/benham. There one can get SIDD profiles of any regions of interest in any complete chromosome that has been analyzed. To date this is just E. coli K12, although yeast results will be added soon.
We do not at present intend our analysis to exactly reflect in vivo conditions. Indeed, one anticipates these will vary in complex ways according to the precise manner in which stresses are imposed, how the DNA is constrained, binding events, and many other specific effects. The present calculations are intended to illuminate a relatively simple physical–chemical attribute of the DNA duplex—its propensity to become locally destabilized in response to the superhelical stresses that are imposed on it in vivo. Although the assumptions implicit in this approach are much simpler than is the in vivo situation, their results already have illuminated attributes of the DNA that have been implicated in a variety of important regulatory processes. Accordingly, we designed our analysis procedure to most effectively illuminate the SIDD behavior of genomic sequences, and thereby enable correlations of SIDD sites with regulatory and other regions to be evaluated. Correlations that are found can suggest possible roles of SIDD in mechanisms of regulation, and experimental tests thereof. But calculations that accurately reflect in vivo conditions—where the basal level of superhelicity can vary in complex ways with growth state and environmental parameters, transient supercoiling is driven by replication and transcription, and both domain boundaries and susceptibilities to transition vary with protein-binding events—must await a fuller understanding of these conditions.
METHODS
The Analysis of SIDD in Long DNA Sequences
The Equilibrium Thermodynamics of Superhelical DNA Denaturation
In a superhelical domain, the DNA is constrained so that its linking number Lk is fixed. (Lk is the total number of helical turns in the DNA within a domain when its central axis is held planar.) When the domain is relaxed, it has linking number Lk0. But DNA in vivo is commonly constrained in a negatively superhelical state, in which Lk < Lk0. This results in a (negative) linking difference α = Lk – Lk0 < 0, which exerts untwisting torsional stresses on the base pairs within the domain. The superhelix density σ is σ = α/Lko.
A given linking difference (i.e., superhelicity) α imposed on a DNA molecule can be accommodated by many combinations of supercoiling deformations and conformational transitions. In particular, there are 2N states of strand separation possible in a domain containing N base pairs. Denaturation of n base pairs decreases their total unstressed twist by n/A turns, where A = 10.5 bp per turn is the sequence-averaged helicity (twist per length) of unstressed B-form DNA (Wang 1979). Torsional stresses will remain unless n has the precise value that exactly relaxes the domain, which is n = –αA. Because single-stranded DNA is much more flexible than is the B-form duplex (Bloomfield et al. 1974), the separated strands in a denatured region will tend to twist around each other in response to these residual stresses. We denote the total twist of the denatured regions by I . Finally, the residual linking difference αr is the component of α that is not accommodated by either of these two deformations. The superhelical constraint coupling together these modes of deformation is expressed by the conservation equation:
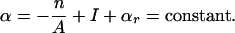 |
(3) |
At thermodynamic equilibrium, a population of identical superhelical DNA molecules will be distributed among its available states according to Boltzmann's law (Landau and Lifshitz 1969). If the set 𝒢 of available states is indexed by i, and if the free energy of state i is Gi, then the fraction of a population of identical molecules that is in state i at equilibrium will be
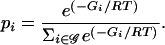 |
(4) |
where R is the gas constant and T is the absolute temperature. (For simplicity, all states are denoted here as though they are discrete in character. For parameters that vary continuously, it is understood that relevant summations actually involve integrals.) Thus the fractional occupancies of individual states decrease exponentially as their free energies increase. If a parameter ζ has value ζi in state i, then its population average (i.e., expected) value 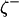 at equilibrium is
at equilibrium is
 |
(5) |
To apply this analysis strategy to superhelical DNA, we must identify the states available to the molecule, and associate a free energy to each.
To specify a state of strand separation in a DNA molecule of specified base sequence and imposed superhelicity α, we first identify its open base pairs. We construct N binary variables nj, 1 ≤ j ≤ N, one for each base pair, having the value nj = 1 if base pair j is open in the state, and nj = 0 otherwise. Next, we specify the torsional deformation τj of each open base pair. This will determine the residual superhelicity through the conservation equation 3 above. The total free energy ascribed to this state is the sum of the free energies associated to each of these deformations:
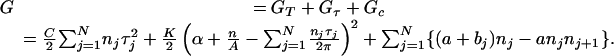 |
(6) |
A complete derivation of this equation is given in Fye and Benham (1999).
Equations 4 and 5 may be used to evaluate the equilibrium value of any property of interest, once the states and their energies have been specified. The method by which this is done has been presented elsewhere (Benham 1992; Fye and Benham 1999; Benham and Bi 2004). We commonly calculate two quantities of interest—the ensemble average probability p(x) of denaturation of the base pair at each position x along the DNA sequence, and the incremental free energy G(x) needed to guarantee separation of that base pair (Benham 1993, 1996). Strong destabilization is indicated by low values of G(x), whereas positions that remain stable have high values of G(x). Plots of G(x) versus x are called SIDD profiles. (An example is shown in Fig. 2.) SIDD profiles are more informative than transition probability profiles because they also depict sites where the amount of free energy needed to induce denaturation is decreased relative to background, but not necessarily to levels where opening is favored at equilibrium. Such regions could be biologically important as sites that stresses render vulnerable to or abet opening by enzymatic or other processes. In E. coli, for example, promoter opening at the –10 region is driven by the activity of the σ-factor within the polymerase complex. Even a fractional destabilization at this site will drive the equilibrium exponentially toward the open complex, as indicated by equation 4. A destabilization of 4.2 kcal/mole, well below what is required for strand opening, will still favor open complex formation by a factor of 1000.
The Approximate Method of SIDD Analysis
We use the following strategy to analyze the SIDD properties of a single, relatively short DNA sequence (Benham 1990, 1992). We first find the state of absolute lowest free energy, and denote its energy by Gmin. Then an energy threshold θ is specified, and the set  of all states s is found whose free energies Gs exceed Gmin by no more than θ:
of all states s is found whose free energies Gs exceed Gmin by no more than θ:
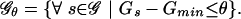 |
Approximate ensemble average values of the partition function and of all the sums needed to calculate the quantities of interest are evaluated using only this subset of states. Comparison with the results of exact (but very slow) calculations performed at T = 37°C and midphysiological superhelix densities (viz., –0.6 ≤ σ ≤ –0.04) show that the approximate method achieves four to five significant figures of accuracy in all calculated parameters when a threshold of θ = 12 kcal/mole is used (Fye and Benham 1999). Calculations performed on sequences of length N ≈ 5000 bp under these conditions commonly will have somewhere between 106 and 109 states that satisfy this threshold, a number that is small enough to execute efficiently.
Long DNA sequences, including complete chromosomes, are analyzed by partitioning them into windows and analyzing each window separately (Benham and Bi 2004). All windows are chosen to have the same length N. Successive windows are offset by a distance do, with do|N so that each internal base pair appears in w = N/do windows. Each window is analyzed individually using the approximate method described above. The final values of the opening probability p(x), the destabilization energy G(x), and any other parameters of interest for the base pair at position x are calculated as weighted averages of the values computed for the windows containing that base pair:
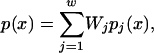 |
(7) |
and
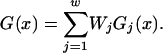 |
(8) |
Here we use do = N/10 so that each base pair appears within 10 windows that span a total of 9500 bp. We assign weights to the windows in which a particular base pair appears using a strategy whereby large weights are given to windows in which the base pair is central, and successively smaller weights to windows where it is increasingly peripheral. Here we assign exponential relative weights of 1, 2, 4, 8, 16, 16, 8, 4, 2, 1 as the windows proceed from left to right across the base pair of interest. Because they must add to unity, the actual weights Wi are found by dividing these numbers by their sum, which is 62. These choices have the effect of directly coupling each base pair strongly to its nearer neighbors, with the strength of this coupling falling off approximately sigmoidally with distance. We note, however, that every base pair is indirectly coupled to every other base pair by a chain of direct couplings.
Computational Implementation
The windowing algorithm has been implemented in both FORTRAN and C++ with default values N = 5000 bp, do = 500 bp, and θ = 12 kcal/mole. Circular DNA molecules, such as the E. coli chromosomes, are accommodated by a simple sequence wrap-around boundary condition. The precision of the floating point arithmetic needed for implementation depends on the value of the threshold θ. When θ = 12 kcal/mole, double precision is required on a 32-bit processor, but only single precision is needed in a fully 64-bit implementation.
All energy parameters used in these calculations are given the values that have been experimentally determined to occur at an ionic strength of 0.01 M and a temperature of 37°C (Benham 1992). These are the conditions used in the mung bean nuclease digestion procedure by which superhelical denaturation has been most accurately assessed in vitro (Kowalski et al. 1988). The opening energies are regarded as copolymeric (one value for every AT base pair and a different value for every GC base pair), although near-neighbor energetics also can be used.
Previous calculations performed on a wide variety of short (i.e., 3–8 kb length) DNA sequences have shown that this approach, with these energy parameters, provides highly accurate predictions of stress-induced transition behavior, both in vitro and in vivo. For sequences in which the locations and extents of superhelically driven duplex opening have been measured using the mung bean digestion procedure, our computational predictions of the positions of opening and the relative amounts of opening at each position, both as functions of imposed superhelicity, have been shown to be quantitatively accurate to within experimental precision (Benham 1992, 1993, 1996). In cases in which superhelical destabilization was followed either by other methods (viz., S1 nuclease digestion or the single-strand-specific binding of small molecules such as KMnO4, chloracetaldehyde, or OsO4) or under other conditions, in every case the sites that were predicted to open were, in fact, found experimentally to open (Benham et al. 1997; Sheridan et al. 1998; Aranda et al. 1997; Fye and Benham 1999). Even in cases in which the experimental conditions did not correspond to those assumed in the calculations, their results still accurately predicted the observed details of the transition. Thus, for example, the opening transition at the IgH enhancer region was assessed by chloracetaldehyde binding at fixed superhelicity but varying ionic conditions. The sites of opening and the sequence of events that occurred as opening proceeded were both accurately reflected in the results of our computations, although they were done at fixed ionic strength and varying superhelicity (Benham et al. 1997). Even in vivo, where the exact superhelical and environmental conditions are not fully understood, predicted SIDD sites have been experimentally found to open (Aranda et al. 1997; He et al. 2000). Indeed, in the latter case, even the fine details of how opening progressed through the site were accurately predicted. These examples indicate that the results of the present theory are quite robust as to locations of destabilized sites and the progression of the transition. This gives confidence in the accuracy of our predictions of these transition properties when applied to other sequences, on which experiments have not been performed.
We note, however, that the precise level of superhelicity needed to drive a given extent of transition does depend on environmental conditions of temperature and ionic strength. Our results are quantitatively highly accurate when compared with experiments performed under the environmental conditions of the nuclease digestion procedure of Kowalski, as these are the conditions in which the energy parameters we use pertain. One may modify our calculations to suit other conditions by simply using the energy parameters that are appropriate to those conditions. However, at present the experimental information available regarding superhelical transitions under other conditions is not sufficiently detailed to allow a rigorous assessment of the quantitative accuracy of our predictions of the superhelical dependence of transition properties.
We use a window size of 5000 bp for our calculations because previous analyses of sequences of this size have proven to be highly accurate and informative, as described above. As there is no information available regarding the distances over which superhelical stresses propagate in vivo, there is at present no biological basis for selecting any specific length scale. Indeed, one may speculate that the sizes and boundaries of topological domains may change dynamically with protein-binding events, translocation of polymerases, reptation through constraints, and perhaps other effects. If experiments suggest that a particular length scale is relevant to a specific phenomenon, the calculations can be easily modified to use that scale.
Analysis of the E. coli Genome
The E. coli K12 chromosome is the first complete genome to be analyzed using this SIDD windowing strategy. The analyzed sequence is version M54, accession number NC000193. This circular molecule contains 4,639,221 bp. The complete calculation on this sequence required 9290 windows using the default window size N and offset distance do. The results reported here assume superhelix density α/Lko = σ = –0.06, which corresponds to an intermediate physiological value. The complete analysis required 40.15 CPU h to run on a 1-GHz Pentium III processor using the GNU C++ compiler at optimization level 2, with double precision floating point arithmetic. It found 81,101,140,512 states in total. The number of states and the execution time both vary dramatically with the level of superhelicity, decreasing by a factor of 4 when σ = –0.055.
The results of these calculations may be accessed at http://www.genomecenter.ucdavis.edu/benham. There the user may request the G(x) values for any specified region of any chromosome for which the calculation has been performed. The SIDD and probability profiles of regions 5 kb in length can be graphed, or tabulated output for specified regions can be sent by e-mail. The output for the entire chromosome can be provided on request.
Acknowledgments
We are grateful for the assistance of Peter Morrison in writing programs implementing our early algorithmic strategies for analyzing the SIDD profiles of E. coli. The work reported here was supported in part by grants DBI 99-04549 from the National Science Foundation and RO1-HG01973 from the National Institutes of Health, and by additional support from the Diversa Corporation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2080004.
References
- Aranda, A., Perez-Ortin, J., Benham, C.J., and del Olmo, M. 1997. Analysis of the in vivo structure of a natural alternating d(AT)n sequence in yeast. Yeast 13: 313–326. [DOI] [PubMed] [Google Scholar]
- Benham, C.J. 1979. Torsional stress and local denaturation in supercoiled DNA. Proc. Natl. Acad. Sci. 76: 3870–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham, C.J. 1980. The equilibrium statistical mechanics of the helix-coil transition in torsionally stressed DNA. J. Chem. Phys. 72: 3633–3639. [Google Scholar]
- Benham, C.J. 1981. A theoretical analysis of competing conformational transitions in torsionally stressed DNA. J. Mol. Biol. 150: 43–68. [DOI] [PubMed] [Google Scholar]
- Benham, C.J. 1990. Theoretical analysis of heteropolymeric transitions in superhelical DNA molecules of specified sequence. J. Chem. Phys. 92: 6294–6305. [Google Scholar]
- Benham, C.J. 1992. The energetics of the strand separation transition in superhelical DNA. J. Mol. Biol. 225: 835–847. [DOI] [PubMed] [Google Scholar]
- Benham, C.J. 1993. Sites of predicted stress-induced DNA duplex destabilization occur preferentially at regulatory regions. Proc. Natl. Acad. Sci. 90: 2999–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham, C.J. 1996. Duplex destabilization in superhelical DNA is predicted to occur at specific transcriptional regulatory regions. J. Mol. Biol. 255: 425–434. [DOI] [PubMed] [Google Scholar]
- Benham, C.J. and Bi, C.-P. 2004. The analysis of stress-induced duplex destabilization in long genomic DNA sequences. J. Comp. Biol. (in press). [DOI] [PubMed]
- Benham, C.J., Kohwi-Shigematsu, T., and Bode, J. 1997. Stress-induced duplex destabilization in chromosomal scaffold/matrix attachment regions. J. Mol. Biol. 274: 181–196. [DOI] [PubMed] [Google Scholar]
- Blattner, F., Plunkett, G., Bloch, C.A., Perna, N.T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J.D., Rode, C.K., Mayhew, G.F., et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277: 1453–1474. [DOI] [PubMed] [Google Scholar]
- Bloomfield, V., Crothers, D., and Tinoco, I. 1974. The physical chemistry of nucleic acids. Harper & Row, New York.
- Cheung, K.J., Badarinarayana, V., Selinger, D.W., Janse, D., and Church, G.M. 2003. A microarray-based antibiotic screen identifies a regulatory role for supercoiling in the osmotic stress response of Eschicheria coli. Genome Res. 13: 206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskin, E., Keich, U., Gelfand, M.S., and Pevzner, P. 2003. Genome-wide analysis of bacterial promoter regions. 2003 Pac. Symp. Biocomp. 29–40. [PubMed]
- Fye, R.M. and Benham, C.J. 1999. Exact method for numerically analyzing a model of local denaturation in superhelically stressed DNA. Phys. Rev. E 59: 3408–3426. [Google Scholar]
- Hatfield, G.W and Benham, C.J. 2002. DNA topology-mediated control of global gene expression in Escherichia coli. Ann. Rev. Genet. 36: 175–203. [DOI] [PubMed] [Google Scholar]
- He, L., Liu, J., Collins, I., Sanford, S., O'Connell, B., Benham, C.J., and Levens, D. 2000. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 19: 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg, R., Bejerano, G., Santos-Zavaleta, A., and Margalit, H. 2001. PromEC: An updated database of Escherichia coli mRNA promoters with experimentally identified transcriptional start sites. Nucleic Acids Res. 29: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz, G.Z. and Stormo, G.D. 1996. Escherichia coli promoter sequences: Analysis and prediction. Methods Enzymol. 273: 30–42. [DOI] [PubMed] [Google Scholar]
- Huang, R.Y. and Kowalski, D. 1993. A DNA unwinding element and an ARS consensus comprise a replication origin within a yeast chromosome. EMBO J. 12: 4521–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta, A.M. and Collado-Vides, J. 2003. σ70 promoters in Escherichia coli: Specific transcription in dense regions of overlapping promoter-like signals. J. Mol. Biol. 333: 261–278. [DOI] [PubMed] [Google Scholar]
- Kowalski, D. and Eddy, M.J. 1989. The DNA unwinding element: A novel, cis-acting component that facilitates opening of the E. coli replication origin. EMBO J. 8: 4335–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski, D., Natale, D., and Eddy, M. 1988. Stable DNA unwinding, not breathing, accounts for single-strand specific nuclease hypersensitivity of specific A+T-rich regions. Proc. Natl. Acad. Sci. 85: 9464–9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau, L.D. and Lifshitz, E.M. 1969. Statistical physics. Pergamon Press, New York.
- Leblanc, B., Benham, C.J., and Clark, D. 2000. An initiation element in the yeast CUP1 promoter is recognized by RNA polymerase II in the absence of TATA box-binding protein if the DNA is negatively supercoiled. Proc. Natl. Acad. Sci. 97: 10745–10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Garcia, P. and Forterre, P. 2000. DNA topology and the thermal stress response, a tale from mesophiles and hyperthermophiles. BioEssays 22: 736–746. [DOI] [PubMed] [Google Scholar]
- Lyubchenko, Y.L. and Shlyakhtenko, L.S. 1988. Early melting of supercoiled DNA. Nucleic Acids Res. 16: 3269–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potaman, V., Bissler, J., Hashem, V., Oussatcheva, E., Lu, L., Shlyakhtenko, L., Lybuchenko, Y., Matsuura, T., Ashizawa, T., Leffak, M., et al. 2003. Unwound structures in SCA10 (ATTCT)n · (ATTCT)n repeats. J. Mol. Biol. 326: 1095–1111. [DOI] [PubMed] [Google Scholar]
- Rohde, J.R., Fox, J.M., and Minnich, S.A. 1994. Thermoregulation in Yersinia enterocolitica is coincident with changes in DNA supercoiling. Mol. Microbiol. 12: 187–199. [DOI] [PubMed] [Google Scholar]
- Salgado, H., Moreno-Hagelsieb, G., Smith, T.F., and Collado-Vides, J. 2000. Operons in Escherichia coli: Genomic analysis and predictions. Proc. Natl. Acad. Sci. 97: 6652–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan, S., Benham, C.J., and Hatfield, G.W. 1998. Activation of gene expression by a novel DNA structural transmission mechanism that requires supercoil-induced DNA duplex destabilization in an upstream activating sequence. J. Biol. Chem. 273: 21298–21308. [DOI] [PubMed] [Google Scholar]
- Sheridan, S., Benham, C.J., and Hatfield, G.W. 1999. Inhibition of DNA supercoiling-dependent transcriptional activation by a distant B-DNA to Z-DNA transition. J. Biol. Chem. 274: 8169–8174. [DOI] [PubMed] [Google Scholar]
- Sun, H.-Z., Mezei, M., Fye, R.M., and Benham, C.J. 1995. Monte Carlo analysis of conformational transitions in superhelical DNA. J. Chem. Phys. 103: 8653–8665. [Google Scholar]
- Tal, M., Shimron, F., and Yagil, G. 1994. Unwound regions in yeast centromere IV DNA. J. Mol. Biol. 243: 179–189. [DOI] [PubMed] [Google Scholar]
- Vanet, A., Marsan, L., and Sagot, M. 1999. Promoter sequences and algorithmical methods for identifying them. Res. Microbiol. 150: 779–799. [DOI] [PubMed] [Google Scholar]
- Voloshin, O.N., Shlyakhtenko, L.S., and Lyubchenko, Y.L. 1989. Localization of melted regions in supercoiled DNA. FEBS Lett. 243: 377–380. [DOI] [PubMed] [Google Scholar]
- Wang, J.C. 1979. Helical repeat of DNA in solution. Proc. Natl.Acad. Sci. 76: 200–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein-Fischer, D., Elgrably-Weiss, M., and Altuvia, S. 2000. Escherichia coli response to hydrogen peroxide: A role for DNA supercoiling, Topoisomerase I and FIS. Mol. Microbiol. 35: 1413–1420. [DOI] [PubMed] [Google Scholar]