

Abstract
Determining linkage phase from population samples with statistical methods is accurate only within regions of high linkage disequilibrium (LD). Yet, affected individuals in a genetic mapping study, including those involving cases and controls, may share sequences identical-by-descent stretching on the order of 10s to 100s of kilobases, quite possibly over regions of low LD in the population. At the same time, inferring phase from nuclear families may be hampered by missing family members, missing genotypes, and the noninformativity of certain genotype patterns. In this study, we reformulate our previous haplotype reconstruction algorithm, and its associated computer program, to phase parents with information derived from population samples as well as from their offspring. In applications of our algorithm to 100-kb stretches, simulated in accordance to a Wright-Fisher model with typical levels of LD in humans, we find that phase reconstruction for 160 trios with 10% missing data is highly accurate (>90%) over the entire length. Furthermore, our algorithm can estimate allelic status for missing data at high accuracy (>95%). Finally, the input capacity of the program is vast, easily handling thousands of segregating sites in ≥1000 chromosomes.
The field of elucidating complex diseases by using genetic methods is still without a generalized methodology. Association studies hold great promise in overcoming remaining challenges (Lander 1996; Risch and Merikangas 1996; Collins et al. 1997). Aside from whole genome-wide association studies to be undertaken in the future, these techniques have already proven useful in the fine mapping of regions identified by linkage and candidate gene studies.
Single nucleotide polymorphisms (SNPs) have come into the fore as the marker of choice for association studies. Recent articles have clarified their various properties in the context of gene mapping. Individually, a SNP has the greatest power for detection of association to disease when it is the actual susceptibility mutation. Otherwise, even when high linkage disequilibrium (LD) exists between the two, the power of a SNP to detect a disease variant diminishes as its allele frequency differs from that of the disease mutation (Muller-Myhsok and Abel 1997). The presence of multiple disease susceptibility alleles at a given locus has the same effect (Slager et al. 2000). It has been shown that tests in which association is sought to haplotypes rather than single SNPs may in certain situations increase statistical power (Akey et al. 2001). This result holds even for loci harboring multiple disease alleles (Morris and Kaplan 2002).
Unfortunately, phase information is not readily discernible from standard DNA molecular methodologies for diploid organisms. Three methods exist by which this information can be obtained: experiments, statistical algorithms exploiting population LD, and inference in pedigrees.
Experimental methods of haplotyping include diploid-to-haploid conversion (Papadopoulos et al. 1995), allele-specific PCR, and cloning. Although these techniques have been shown to impart more information over certain statistical methods (Douglas et al. 2001; Schaid 2002), they are time-consuming and/or expensive and are thereby inapplicable to large-scale applications.
With regard to computational approaches, there exist three main methods of reconstructing the haplotypes of population samples. First, Clark's parsimony method (Clark 1990) seeks to minimize the number of distinct haplotypes reconstructed in a sample. Second, the EM algorithm (Excoffier and Slatkin 1995) uses the expectation maximization method on a likelihood assuming only Hardy-Weinberg equilibrium. Third, the Stephens-Smith-Donnelly (SSD) method (Stephens et al. 2001; Stephens and Donnelly 2003) uses a Bayesian framework to compute the haplotype distribution based on a population genetic (infinite-sites coalescent) model with the Hardy-Weinberg assumption. Derivative methods exist as well. One example illustrates the distinction between the statistical methodology and the underlying model; the PL algorithm (Niu et al. 2002) assumes only Hardy-Weinberg equilibrium as with the EM algorithm but uses Bayesian statistics. Another derivative method was our previous algorithm (Lin et al. 2002), a variant of algorithm 2 of Stephens et al. (2001), which assumes an infinite-alleles, coalescent model calculated within a Bayesian framework.
In Lin et al. (2002), as well as Stephens and Donnelly's (2003) recent work, haplotype reconstruction for long sequences (∼100 to 300 kb) as a whole, as adjudged by individual error rate, was reportedly poor for the various computational methods examined. With regard to our particular program, we found that many of the mistakes occurred in phase relations straddling regions of low LD (for any two heterozygous sites, a phase relation is defined to be the information that would resolve these sites into two haplotypes). We remarked that there may be little information inherent in population samples to correctly reconstruct these sites. The unreliability in reconstructing haplotypes spanning regions of low LD limits the utility of in silico methods for disease gene mapping. Indeed, correct haplotype reconstruction across blocks of low LD may be crucial in some cases. We often imagine that the genome exhibits monolithic patterns of LD, with many regions showing relatively small blocks of high LD (Wall and Pritchard 2003) intermixed with rare regions showing longer LD (Dawson et al. 2002). However, LD is a property not of a region of the genome but instead of the sites of variation within that region. Sites that are young will necessarily have more LD than older sites (Hudson and Kaplan 1985). If we posit that common diseases are at least sometimes caused by collections of recent, individually rare, mutations (Pritchard 2001), it is quite possible that disease alleles will show greater LD over longer ranges than that exhibited by surrounding sites with each other. We posit that construction of haplotypes on the order of 10s to 100s of kilobytes may allow the full exploitation of such a feature (Zwick et al. 2001).
The final method of discerning phase is to genotype related individuals and deduce haplotypes within families. This approach is not without its own complexities, as evidenced by the plethora of strategies put forth to construct haplotypes in the context of linkage mapping (Kruglyak et al. 1996; Sobel and Lange 1996; Sobel et al. 1996). Nevertheless, inferring haplotypes within families does provide substantial phase information for tightly linked SNPs (Schaid 2002). Even within this context, however, unavailable family members, missing genotype calls, and the noninformativity of certain genotype patterns may obscure phase relations. In certain situations, these ambiguities may lead to completely spurious haplotype/disease associations (Schaid et al. 2002). Even though the output of programs implementing the aforementioned strategies may represent the most probable haplotype reconstructions, they may be only one of several global maxima, a situation especially likely for small pedigrees. It is not difficult to understand that making inferences in such a context leads to erroneous conclusions.
As pointed out by Schaid et al. (2002), LD information from the population sample may potentially be used to differentiate these maxima from a flat likelihood. Indeed, simultaneous exploitation of both statistical methods applied to population samples as well as genotypes of additional family members would compensate to some degree the respective shortcomings of each method alone. More specifically, the tightly linked genotypes of children could, say, be used to aid the phasing of a sample of parents across regions of low LD. Reciprocally, the characteristics of a population sample of haplotypes could be used to resolve probabilistic ambiguities remaining after familial inference. Already, a method incorporating these properties has been devised by Rohde and Fuerst (2001) and has been shown to be significantly more accurate than methods using either population or familial sources of information singly.
However, because the Rohde-Fuerst program (http://www.bioinf.mdc-berlin.de/∼rob/) is based on the EM algorithm, its input capacity is 30 sites (although their Web site recommends 12). This limitation may be suitable for candidate gene approaches but is inadequate for fine mapping of putative loci implicated in linkage studies, which stretch for megabases, let alone genome-wide association studies.
There are also front-end programs to existing phasing algorithms, for example, PHamily (http://archimedes.well.ox.ac.uk/pise). This program determines parental sequences rendered completely unambiguous by their children's haplotypes. Users then input these haplotypes with remaining ambiguous parental diplotypes into PHASE. Unfortunately, because the downstream analysis program (PHASE) is unaware of the upstream family structure (PHamily), this two-step approach, when applied to large data sets, often results in reconstructions that require inordinate numbers of double cross-over events within individual families. As a result their utility in post-haplotype reconstruction analyses, such as in transmission disequilibrium tests (Spielman et al. 1993), is diminished.
In this study, we reformulate our previous algorithm and its associated computer program (Lin et al. 2002), implementing the infinite-alleles model and adding procedures to phase between blocks of high LD. We apply the program to the same empirically derived, phase-known sequences we have generated and used before. The sequences comprise eight X-linked regions from a sampling of 40 males (Cutler et al. 2001) and range from 87 to 327 kb in length, with 45 to 165 segregating sites per locus. We find a modest improvement in accuracy (86.2% versus the previously reported 83.2%). Given the reformulation, we are able to expand the program to incorporate genotype data from nuclear families while retaining its vast input capacity. When couples are simulated to have zero to three children, the accuracy improves markedly to 95.3%. Moreover, we find that our program reconstructs phase and fills in missing data at a high accuracy rate for simulated sequences.
RESULTS
The performance of our previous and current methods on the eight X-linked genomic regions is shown in Table 1 (of note, males in these simulations were given two X-chromosomes). Comparison of our previous and current programs on single individuals reveals a modest but statistically significant improvement of 3.0% (P < 0.0001). As our current program has the capacity to handle nuclear family data, it was tested on data in which zero to three children were assigned randomly to couples. This accuracy increased more markedly by 12.1% (P < 0.0001) to 95.3% over the original result.
Table 1.
Accuracy of Haplotype Inference
|
Accuracy (fraction correct)
|
||||
|---|---|---|---|---|
| Locus | Symbol | Lin et al. 2002 | Infinite allele (no children) | Infinite allele (0-3 children) |
| Glycine receptor, α2 (89 kb, 102)a | GLRA2 | 0.863 | 0.900 | 0.962 |
| Monoamine oxidase A (102 kb, 97) | MAOA | 0.904 | 0.921 | 0.983 |
| Potassium voltage-gated channel, Shal-related family, member 1 (122 kb, 60) | KCND1 | 0.781 | 0.824 | 0.945 |
| α thalassemia/mental retardation syndrome X-linked (RAD54 [S. cerevisiae] homolog) (151 kb, 45) | ATR | 0.713 | 0.750 | 0.911 |
| α-Galacosidase (102 kb, 116) | GLA | 0.781 | 0.855 | 0.953 |
| Transient receptor potential channel 5 (327 kb, 165) | TRPC5 | 0.872 | 0.782 | 0.918 |
| Bombesin-like receptor 3 (87 kb, 66) | BRS3 | 0.860 | 0.901 | 0.969 |
| Methyl CpG binding protein 2 (106 kb, 103) | MECP2 | 0.773 | 0.815 | 0.927 |
| Average | 0.832 | 0.862 | 0.953 | |
Genomic length, number of SNPs.
Comparing switch accuracy of our program to that of Rohde and Fuerst (2001) reveals that our program phased haplotypes more accurately by 2.2% (0.966 versus 0.944, respectively; P = 0.0048). It should be noted that roughly half of the 100 sets of input data incurred a time-out after 5 min on our internet browser when we attempted to reconstruct haplotypes at the Fuerst group's Web site. The reported values and statistical analysis are based on only those data sets of 12 contiguous sites that were successfully completed without a timeout.
The real boon of incorporating family information is the ability to reconstruct accurately haplotypes over long distances, namely, over multiple regions of low LD. This feature is, of course, unappreciable by metrics such as switch accuracy. To evaluate phase reconstruction beyond neighboring phase relations, pairwise accuracies of reconstructed haplotypes were plotted against bins of 5 kb. Figure 1 shows the results derived from 100 random pairings of the eight X-linked regions, one curve with no children assigned, the other with one child. Clearly, the accuracy of phase relations drops off much more rapidly when offspring information is excluded.
Figure 1.

Pairwise accuracy of phase relation calls versus intervening length in families with and without children. One hundred simulations of the eight X-linked loci were performed, and the binary accuracies of all pairwise phase relations were tabulated along with their distances. A logistic regression was performed in which accuracy was regressed on dichotomous variables corresponding to the placement of lengths in bins of 5 kb. Predicted values with corresponding 95% confidence intervals are plotted for phase relations whose intervening lengths were <100 kb.
Given that parents and children were simulated from the same empirically derived, X-linked sequences with missing data, the resultant correlations of dropped calls between parents and children could possibly confound the benefit observed upon including children's diplotypes. Moreover, only 40 haplotypes were available for each locus. Clearly, real-life studies requiring phase reconstruction will include many more chromosomes. As such, we turned to simulated haplotypes.
Each of the 50 groups of 40, 640, and 1280 haplotypes were randomly paired twice to form 10, 160, and 320 couples, respectively (see Methods). This data was input into our program with all couples having no children, one child, and so on up to three children. Ten percent of genotypes were randomly dropped to simulate missing data. From phase reconstructions of parents only, we constructed the pairwise accuracy versus distance curves shown in Figure 2, plots a through c. It is apparent that inclusion of diplotypes of more than one child does not greatly improve phase reconstruction. Also, when no children are used, the decay of accuracy over distance is less steep when 320 chromosomes are phased as opposed to 40, although no further improvement is observed for 1280. In general, these trends hold as well when only common sites (minor allele frequency >0.10) are considered (Fig. 2, plots e,f). When no children are used, the decay of accuracy over distance is less steep for sequences with only common sites compared with sequences with all sites.
Figure 2.

Pairwise accuracy of phase relation calls versus intervening length in families with both parents using simulated haplotypes. Results derived from 50 simulations of 40 haplotypes randomly paired twice to form 10 families (a, d); 640 haplotypes to form 160 families (b, e); and 1280 haplotypes to form 320 families (c, f). (a–c) All sites; (d–f) sites with minor allele frequency >0.1. The points for all graphs were calculated by the same procedure as in Figure 1 but without 95% confidence intervals.
Because Figure 2 was generated from input with 10% dropped calls and the simulated sequences had no missing data originally, we were able to calculate the accuracy of missing data calls as well (Table 2). Theoretically (and in data not shown) if single individuals are phased randomly, the baseline switch accuracy should be 0.5. An analogous baseline for missing data calls is not so obvious given the different frequencies of alleles at each site. Thus, the baselines for missing data calls were computed by filling dropped positions at random in accordance to the site's allelic frequencies. We see in Table 2 that accuracies of missing data inferences increase the more chromosomes are phased and when more children diplotypes are included but decrease when only common sites are considered.
Table 2.
Missing Call Accuracy for Two Parents
|
10 Families
|
160 Families
|
320 Families
|
||||
|---|---|---|---|---|---|---|
| Children | MCAa | BMCAb | MCA | BMCA | MCA | BMCA |
| All sitesc | ||||||
| 0 | 0.871 (0.003) | 0.773 (0.004) | 0.934 (0.001) | 0.868 (0.002) | 0.971 (0.0009) | 0.874 (0.002) |
| 1 | 0.944 (0.002) | 0.794 (0.003) | 0.969 (0.0006) | 0.907 (0.002) | 0.962 (0.0006) | 0.911 (0.001) |
| 2 | 0.969 (0.001) | 0.893 (0.003) | 0.978 (0.0004) | 0.939 (0.001) | 0.978 (0.0003) | 0.942 (0.0009) |
| 3 | 0.981 (0.001) | 0.934 (0.003) | 0.987 (0.0002) | 0.963 (0.0007) | 0.987 (0.0002) | 0.965 (0.0006) |
| q > 0.10c | ||||||
| 0 | 0.842 (0.004) | 0.649 (0.004) | 0.917 (0.002) | 0.644 (0.003) | 0.912 (0.002) | 0.642 (0.003) |
| 1 | 0.946 (0.002) | 0.746 (0.004) | 0.953 (0.001) | 0.747 (0.002) | 0.952 (0.001) | 0.746 (0.002) |
| 2 | 0.972 (0.001) | 0.841 (0.004) | 0.973 (0.0008) | 0.839 (0.002) | 0.972 (0.0006) | 0.837 (0.001) |
| 3 | 0.982 (0.001) | 0.903 (0.003) | 0.984 (0.0004) | 0.902 (0.0009) | 0.984 (0.0004) | 0.900 (0.001) |
Missing call accuracy (SEM).
Baseline missing call accuracy (SEM).
Minor allele frequency.
To measure the performance of our program in the face of large amounts of missing information, we input data in which all the fathers were completely composed of dropped calls. The rate of dropped calls for all other input sequences was maintained at 10%. We constructed pairwise accuracy versus distance curves (Fig. 3) and measured missing call accuracy (Table 3) as before for the mothers only. From Figure 3, it can be seen that phase accuracy plateaus when two children are included as opposed to one child. Otherwise, the various trends observed previously for input in which fathers' diplotypes are included hold just as well, although both phase and missing call accuracies are in general higher for the previous data set.
Figure 3.

Pairwise accuracy of phase relation calls versus intervening length in families in which fathers' genotypes are missing data. Results derived from 50 simulations of 40 haplotypes randomly paired twice to form 10 families (a, d); 640 haplotypes to from 160 families (b, e); and 1280 haplotypes to form 320 families (c, f). (a–c) All sites; (d–f), sites with minor allele frequency >0.1. The points for all graphs were calculated by the same procedure as in Figure 1 but without 95% confidence intervals.
Table 3.
Missing Call Accuracy in Genotyped Parent With Other Parent Missing
|
10 Families
|
160 Families
|
320 Families
|
||||
|---|---|---|---|---|---|---|
| Children | MCAa | BMCAb | MCA | BMCA | MCA | BMCA |
| All sitesc | ||||||
| 0 | 0.855 (0.003) | 0.771 (0.004) | 0.929 (0.001) | 0.869 (0.002) | 0.930 (0.001) | 0.874 (0.002) |
| 1 | 0.916 (0.002) | 0.812 (0.003) | 0.950 (0.0007) | 0.894 (0.002) | 0.948 (0.0007) | 0.899 (0.002) |
| 2 | 0.944 (0.002) | 0.844 (0.004) | 0.964 (0.0006) | 0.914 (0.002) | 0.964 (0.0005) | 0.917 (0.001) |
| 3 | 0.964 (0.002) | 0.893 (0.004) | 0.977 (0.0004) | 0.940 (0.001) | 0.976 (0.0003) | 0.942 (0.001) |
| q < 0.10c | ||||||
| 0 | 0.812 (0.005) | 0.646 (0.005) | 0.889 (0.002) | 0.647 (0.003) | 0.881 (0.002) | 0.642 (0.003) |
| 1 | 0.923 (0.003) | 0.707 (0.005) | 0.938 (0.002) | 0.717 (0.002) | 0.932 (0.001) | 0.711 (0.002) |
| 2 | 0.951 (0.002) | 0.763 (0.005) | 0.960 (0.001) | 0.770 (0.002) | 0.959 (0.0009) | 0.765 (0.002) |
| 3 | 0.970 (0.002) | 0.832 (0.005) | 0.975 (0.0007) | 0.840 (0.002) | 0.974 (0.0005) | 0.837 (0.002) |
Missing call accuracy (SEM).
Baseline missing call accuracy (SEM).
Allele frequency.
To demonstrate the input capacity of our new program, we input 20 sets of data simulating 1 Mb of diplotypes from 250 couples, some having as many as nine children. Moreover, we varied the amount of missed data and simulated recombination events in the input data. The results of these runs are shown in Table 4. Running these files using the same parameters as before on Pentium 4 3.06-Ghz processors took less than a half hour each.
Table 4.
Test of Input Capacity: Phasing 1000 Chromosomes of Length 1 Mb
| Fraction missing data | Switch accuracy | Switch accuracy baseline | Missing call accuracy | Baseline missing call accuracy |
|---|---|---|---|---|
| 0 | 0.9989 (0.00004)a | 0.889 (0.002) | — | — |
| 0.05 | 0.9949 (0.00009) | 0.865 (0.002) | 0.974 (0.0004) | 0.936 (0.001) |
| 0.10 | 0.9902 (0.0002) | 0.842 (0.002) | 0.972 (0.0004) | 0.932 (0.001) |
| 0.15 | 0.9848 (0.0003) | 0.819 (0.002) | 0.969 (0.0005) | 0.927 (0.001) |
(SEM)
DISCUSSION
To improve haplotype reconstruction, other groups have accounted for LD in their algorithms. Li and Stephens (2003) integrated a model of recombination into PHASE. Excoffier et al. (2003) took a more heuristic approach by phasing within dynamically changing windows of high LD.
To improve phasing, we instead took the route of including familial information because individuals are, for many study designs, genotyped within familial contexts. It has been demonstrated both theoretically and by simulation that inclusion of family information significantly increases the accuracy of haplotype reconstruction (Rohde and Fuerst 2001; Schaid 2002). Our finding that including the genotype of one child is sufficient to effect marked improvement verifies the theoretical predictions of others (Becker and Knapp 2002).
Our work represents an implementation that increases the accuracy of phase reconstructions for random population samples and greatly expands the input capacity over the current alternative methodology. Another distinction of our computer program is its ability to handle most instances of recombination. The two cases it cannot handle, that is, families with more than one child in which every child inherits at least one recombined haplotype or in which a child inherits two recombined haploypes, occurs with probability one in 10,000 or less when regions of 1 Mb are considered (a rate of 30 recombination events/3 × 109 bp is assumed).
In our experience applying the program to real data sets, the recombination feature is most commonly invoked due to unexpected and most likely nonbiological scenarios. For instance, in a data set comprised of 21 segregating sites covering 120 kb from 33 families, the haplotypes of one family with five children were reconstructed such that one child inherited a haplotype with two recombination events, another with six. Recombination events were not inferred in the other children. Clearly, such a result cannot be taken at face value, biologically speaking. Much more probable explanations are genotyping error, data mishandling, or nonpaternity. Thus, our method can uncover such anomalies, which warrants further inspection.
In the analysis of our program's capability to fill in missing genotype calls, we find that our program performs with a high degree of accuracy. However, in contrast to phase accuracy, missing call accuracy decreases when the input is comprised of only common sites. Both these phenomena are more easily understood when rare sites are considered. In terms of phasing, such sites are difficult to reconstruct because haplotypes with the minor allele appear rarely in the sample. In other words, there is little information in the sample with which to correctly reconstruct such haplotypes. On the other hand, dropped calls with corresponding sites that have low minor allele frequency are correctly filled in at a higher rate merely by dint of chance. Indeed, this explains the pronounced increase in baseline missing call accuracy when the input data consist of 640 and 1280 chromosomes as opposed to 40, all sites considered. Samples with greater numbers of chromosomes are sure to turn up more segregating sites, the vast majority of which are rare. By excluding sites in which these two influences are operating, the observed results follow.
For the tested data sets in which all the fathers' genotypes were assigned missing data, the program did output haplotype reconstructions for these individuals. However, a cautionary word should be given to those who may be tempted to use such sequences for downstream analysis. In examinations of such output, it was observed that the number of inferred heterozygous sites was markedly undercalled (data not shown). The extent of this phenomenon gradually diminished as couples were assigned more children, until it all but disappeared at five children. The same was observed in examinations of haplotype reconstructions for parents in simulated input of sibship data. Clearly, haplotyping parents for whom diplotype data are unavailable is difficult. Apparently, the probability that all four parental haplotypes will be represented in children is quite high when families have five children, thus fully determining at least parental diplotypes if not haplotypes.
With regard to phasing sequences without children, Stephens and Donnelly (2003) recently reported modifications of their original haplotyping program to allow handling of larger data sets. Their results show their new computer program phases sequences more accurately than our own. With regard to our own methodology, in light of their recent work, our program performed most poorly in the case in which haplotypes were derived from a bottleneck population with sequence sites that were in complete linkage equilibrium, a feature Stephens and Donnelly (2003) remarked as unusual. (Of course, this situation would be completely solved by using offspring data, if available.) Indeed, our reformulation of the infinite-alleles model, believed by some to be biologically implausible, increases switch accuracy to a degree approaching theirs reported for the X-linked loci—a difference of 2.6% is calculated when switch accuracies are averaged. In assessing overall performance, the issue at hand is what significance a slight decrease in accuracy should be given in the face of far greater input capacity. Our new program still completes input from the largest X-linked locus, TRPC5, under 10 sec compared with the 2 min required for Stephens and Donnelly's (2003) new program. It should be kept in mind that the goal of haplotype reconstruction is rarely an end in and of itself, but will invariably be performed proximal to some downstream analysis, which may itself be computationally intensive. If it is to be believed that disease haplotypes are indeed 10s to 100s of kilobytes long (Pritchard 2001), then testing for association may involve “sliding window analyses” on the order of such lengths across megabases of reconstructed haplotypes. In the end, the trade-off between accuracy and computational speed/input capacity may be altogether moot. Low-frequency sites will rarely be used in mapping studies, and in analyzing data sets of high-frequency alleles, according to Stephens and Donnelly (2003), differences in accuracy between the programs implementing infinite sites and infinite-alleles models are largely diminished.
The development of a haplotyping program that is capable of handling data on the order of megabases and incorporates family information is timely. Every month, a plethora of positive findings from genome-wide linkage analyses are entered into the literature. Unless promising candidates are immediately implicated within these linkage loci, follow-up fine-mapping of regions spanning megabases will be required. Tests of association with SNPs are the mainstay of such endeavors, and procuring phase information may potentially increase the power for detection. Simply using test statistics that expend degrees of freedom to test all different haplotypes depresses power (Chapman et al. 2003). Clearly then, to realize the potential of haplotypes, further development of analytical methodologies is required.
Of late, several groups have published methods in which EM algorithm-based procedures are used to infer haplotypes to be used for transmission disequilibrium tests (Zhao et al. 2000; Cheng et al. 2003). A natural extension of our work will be to couple our haplotype reconstruction program to statistical procedures for testing disease association. Given our algorithm's input capacity of >1000 chromosomes of an megabyte length, it has the potential to serve as a key component in fine-mapping as well as genome-wide tests for association.
METHODS
Data Analyzed
Five sets of haplotypes were used in the various simulations. The first was comprised of eight X-linked genomic regions derived from 40 male subjects from the National Institutes of Health Polymorphism Discovery Resource at the Coriell Institute for Medical Research (Collins et al. 1998). The Polymorphism Discovery Resource consists of anonymous DNA samples from individuals who, in aggregate, have equally represented ancestry from Africa, America, Asia, and Europe. The loci were sequenced with Affymetrix Resequencing arrays, and genotyping calls were made with ABACUS (Cutler et al. 2001) at a stringent quality threshold that assigned 10% of sites as missing data. The eight loci were 87 to 327 kb in length with segregating sites ranging from 45 to 165 (Table 1).
The second, third, and fourth sets of haplotypes were generated from a computer program similar to Hudson's (2002), which conforms to standard coalescent approximations to the Wright-Fisher model (Ewens 1979). Fifty groups of 40, 640, and 1280 haplotypes were produced with the standard population genetics parameters θ (nucleotide diversity) and 4Nr (N is the effective population size; r, the recombination rate per individual per generation) set at 80 and 40, respectively. θ per site was chosen to be 8 × 10–4 in accordance with empirically reported estimates (Halushka et al. 1999). N was set to the traditional 10,000; r, to 30 exchanges/3 × 109 bp/meiosis. These sequences were generated to simulate variation patterns in 100 kb of genomic sequence. The groups of 40, 640, and 1280 haplotypes had ∼300 to 450, 400 to 650, and 500 to 700 segregating sites, respectively. All figures were constructed as if the sites were evenly spaced across 100 kb.
The final set of haplotypes was similarly generated from the aforementioned program. Twenty groups of 1000 haplotypes were produced with θ and 4Nr set at 800 and 400, respectively, to simulate 1 Mb of data. These sequences had ∼5600 to 6400 segregating sites.
Infinite-Alleles Algorithm
The infinite-alleles coalescent algorithm (algorithm 2 in Stephens et al. 2001), implemented in our previous program, was recast to use explicitly the probabilities of Hoppe's urn model (Hoppe 1987). We also incorporated a variant of Niu et al.'s (2002) partition ligation method for piecemeal reconstruction of haplotypes. We set the boundaries of the atomistic units to coincide with those of high LD blocks. These regions were defined to be contiguous sequences in which all pairwise |D′| (Lewontin 1988) among segregating sites were >0.8. The two-locus haplotype frequencies needed for the calculation of these values were estimated by a Weir-Cockerham EM algorithm (Weir 1989). In our program, LD blocks longer than six sites were split to make atomistic units computationally manageable. Also, orphaned segregating sites that were not linked with any high LD blocks were arbitrarily absorbed into adjacent blocks.
With nuclear family data, our program reconstructs the haplotypes of parents with children's genotypes used to constrain the former's haplotype space. The iterative stochastic sampling process underlying both SSD and our previous program is retained. In all, the program still captures the strategy set forth in Stephens et al. (2001) of dynamically updating individuals' haplotypes to resemble other haplotypes in the sample at each iteration in a Markov chain Monte Carlo (MCMC) series. On a more technical note, the algorithm still relies on a Dirichlet prior, for whenever an individual's haplotypes cannot be reconstructed to be equivalent to other haplotypes found in the population sample, a parent-independent mutation model is assumed. However, owing to the size of the atomistic units used in the algorithm, this situation is rarely encountered.
The algorithm implemented in our program is as follows. First, the constraints placed on couples with children are taken into account. For a given family i, let element a represent the four parental haplotypes and am1, am2, af1, and af2 be the two haplotypes of the mother and father, respectively. At the start, certain sites of a will be ambiguous due to heterozygosity and missing data. The children are queried in the following manner to fill in these ambiguities. am1 and af1 are arbitrarily chosen to be the inherited haplotypes of the first child considered, whereupon certain sites in a may be definitively filled in. However, other arrangements only allow a constraint to be placed. For example, if the mother, father, and child are all heterozygous at a particular site, then whatever am1 is designated to be for that site, af1 must have the other allele. A list of all constraints is stored.
If family i has more than one child, the same process is repeated. However, instead of am1 and af1 being necessarily chosen, in certain cases enough information has already been filled in such that the inherited haplotypes of the second child are apparent. If more than one out of the four possible pairings of the parental haplotypes are consistent with the second child, each one of these possibilities, designated ab, where b = 1...c, c ≤ 4, are considered (c = 1 refers to the case in which the inherited haplotypes are apparent). In filling in the various ab by examining the genotype of the second child, some constraints from the first child will resolve ambiguities, whereupon they will be unnecessary and be discarded. It is possible that none of the four possible pairings of parental haplotypes are consistent with the second child. At this point, a recombination is posited.
Within the framework of the above process, inconsistencies from recombination events appear as if distinct regions of a child's genotype appear to be from two different pairings of parental haplotypes. In this situation, the child's genotype is replaced by two genotypes. The original genotype is split into two sequences at the site where a recombination event is posited. The resultant partial sequences are then padded with missing data. The process of filling in ambiguous sites and enumerating constraints occurs as before, except for the replacement. For example, in a particular family, after having extracted information from the first child's genotype, suppose the first half of the second child's genotype is consistent with the pairing of the first halves of haplotypes am1 and af1 and those the second half with of am1 and af2. Say the second child's sequence is GGGG. This child's genotype is then replaced with the GGNN and NNGG.
If family i has more than two children, the process of filling in ambiguous sites and enumerating constraints described above is repeated with the third (and subsequent) child(ren) for each of the ab. Some ab may spawn yet more possibilities whereupon c will be incremented. On the other hand, all four pairings of the parental haplotypes of certain ab may be inconsistent with the third child. These ab are discarded, and c is decremented.
If at any time in the process c reaches zero, a recombination event is posited for that child. The process for handling recombination is order dependent in that different numbers of recombinations will be inferred in different places depending on the order in which the children are processed. As such, the order of the children are permuted, and the arrangement (possibly multiple ones) with the least number of recombinations is retained as ab. It should be noted that this program can handle multiple recombinations along the transmitted haplotype from one parent. However, because in our scheme a recombinant child's genotype is split in two (and not more), families with multiple children in which every child inherits a recombined chromosome or in which a child inherits two recombined chromosomes cannot be analyzed. In these exceedingly rare cases, the program exits.
At the end of this process, family i will have a corresponding set {ab}i. Each element in set{ab}i has a different list of constraints and residual ambiguous sites. For single individuals or equivalently, couples without children, each individual will have an analogous set {ab} with necessarily one element consisting of two haplotypes and a list of ambiguous sites.
Second, phase reconstruction is first carried forth in blocks of high LD. Restricting all operations to the portions of {ab} relevant to the block at hand for family i, all four haplotype elements consistent with any element in {ab}i are enumerated and collected into a set, say H. The MCMC chain is then started. Let hi denote the four parental haplotypes of family i (or two haplotypes of single individual i) at a given point in the MCMC chain and h = (h1,..., hn), the collection of such families and individuals. h is initially constructed by randomly choosing elements from the corresponding sets H before the first iteration. The algorithm then proceeds as follows.
Randomly pick i from 1 to n. If i corresponds to a family, remove the family's four current haplotypes from h, and with the remainder, create a list of unique haplotypes y = (y1,..., yk) with corresponding counts r = (r1,..., rk). Let N – 4 = Σjrj. The four parental haplotypes of family are reconstructed in steps 2 and 3.
- First, choose how many of the updated haplotypes will have already been seen in y. Calculate pT for T = 0,...,4 where pT is proportional to the probability that T alleles match the k distinct types in the sample upon having chosen four new alleles from the population. Let S be the set of (ordered) four-tuples of T ones and 4-T zeros. Let s be an element of S, and sj represent the jth tuple. Then,
where θ is the infinite-alleles scaled mutation rate and depends on the numbers of both unique and total alleles (Ewens 1972). This parameter is calculated with respect to h without family i. Choose T = t with probability pT/ΣT′pT′.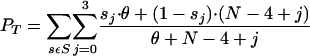
Let W be the set of all elements in Hi for which t of the haplotypes are found in y. For t > 0, there are t haplotypes in the elements of W which have corresponding values in the count set r. Choose an element from W uniformly if t = 0, with probability proportional to its corresponding count in r if t = 1, or with probability proportional to the product of the counts if t > 1. (In the case in which W is null, its corresponding pT would have been set to zero in step 2.)
Add this chosen element of W back into h as the reconstructed parental haplotypes for family i.
An analogous procedure is followed if i corresponds to a single individuals without children. More specifically, instead of four haplotypes being considered in the various steps, two are.
As the chain is run, realizations h are stored periodically in accordance with parameters specified by the user. For each saved realization, the haplotypes of hi are consistent with some elements of {ab}i. A running tabulation of these occurrences is kept for each element of {ab}i. After the chain is stopped, the element in {ab}i with the highest tabulation is saved and the rest are discarded. The haplotype space for each family is trimmed in this way.
Third, the whole process in the prior step is repeated with each set {ab}i containing only one element. At the termination of the chain, the stored realizations are used to make final reconstructions of haplotypes within the blocks. For families, the most frequently saved haplotype reconstruction is chosen. For individuals, sites with missing data are filled in with the most common nucleotide found at corresponding sites among the stored haplotypes. The first heterozygous position is called arbitrarily. For the second heterozygous position, the most common nucleotide is chosen from those haplotypes with the specific nucleotide of the first call. Likewise, for all subsequent positions, the most common nucleotide is chosen conditional upon the specific call made at the immediate prior heterozygous site.
Fourth, phasing is next performed between blocks again by iterating an MCMC chain.
Randomly pick family i. Scan the genotypes of the mother and father block by block, from left to right, and by referencing the corresponding abi; stop at a block in which a segregating site in one of the parents needs to be phased. Find the closest block that contains a segregating site that has already been phased, say from children's information. Restricting attention to the sequences of these two blocks (and anything possibly in between) for all h, all possible block arrangements consistent with abi are enumerated; call the collection of these elements set H′i. Follow step 2, 1 through 4, except with H′i replacing Hi. Repeat the process for all blocks of family i that require phasing. A similar process is performed if i is a single individual except that the first block with a segregating site is skipped, thereby acting as the anchor in reference to which the next block with a segregating is phased.
Realizations of h are stored periodically. Final haplotypes are reconstructed in a fashion analogous to step 3.
Measures of Accuracy
Diplotypes were prepared by randomly pairing haplotypes in both the empirically derived and computer-generated data sets. Of note, such sampling conforms the frequency of genotypes observed to Hardy-Weinberg expectations. Offspring diplotypes, when assigned, were assumed to follow Mendelian (independent) assortment of haplotypes.
In all simulations in this article, the following input parameters were specified for the execution of our program. For phasing within regions of high LD and between blocks, our computer program was run at 10,000 iterations at each stage, with the first 5000 discarded as “burn-in” and the remainder thinned by storing every 20th iteration.
To measure the accuracy of phase relations, the outputs of the parental and single individual haplotypes from the various programs tested in this study were compared with the original haploid sequences, and for genotypes with more than one heterozygous site, the accuracy was scored by switch accuracies (Lin et al. 2002). The latter is applied to each individual reconstruction and is defined as (n – 1 – sw)/(n – 1), where n denotes the number of heterozygous sites; sw, the number of switches between neighboring heterozygous sites needed in the computer-phased haplotype to recover the original haploid sequence. Statistical analyses were carried out with STATA version 7.0 (Stata Corp.). Of note, global measures of whole haplotype accuracy, such as individual error rate and mean square error of haplotype frequency estimates, were avoided, because these measures are negatively associated with sizes of haplotypes. Intuitively, for arbitrarily long haplotypes, if each phase relationship has a finite probability of error, the probability that there is at least one error somewhere in the haplotype approaches one for all haplotypes and is thereby not meaningful. Furthermore, for long haplotypes and finite sample sizes n, the frequency of every haplotype in the sample approaches 1/n and is therefore not informative either.
To compare between our previous and current algorithm, 100 simulations were performed for each X-linked region. For the latter program, the process was repeated except the diplotypes were randomly paired to form couples, and zero to three children were randomly assigned to each family (thus, fathers were assigned two X-chromosome haplotypes). Of note, a family without children was treated as two single individuals in the algorithm. The Wilcoxon rank-sum test was used to test for statistical significance (Rosner 2000)
In comparisons of our current program to that of Rohde and Fuerst (2001), 100 sites were randomly chosen within the eight X-linked regions. At each point, 40 haplotypes of 12 contiguous segregating sites were randomly paired twice to form 20 couples. Couples were assigned zero to three children each. The length reduction of the sequences was necessary to accommodate the recommended input capacity of the Rohde-Fuerst program. Haplotype reconstruction was carried out at the group's Web site. Statistical significance was calculated with the Wilcoxon signed-rank test.
To test the input capacity of our new program, each of the 20 groups of 1000 haplotypes were randomly paired twice to form 250 couples. These couples were assigned numbers of children based on the Poisson distribution with the average set to one. Given that the sequences were supposed to simulate 1 Mb, recombinations were chosen to occur at a rate of 0.01 per meiosis (30 recombination events/3 × 109 bp/meisosis was assumed).
To simulate input with missing data, the individual sites of the diplotypes of parents and children, if assigned, were dropped at rates of 0, 0.05, 0.10, or 0.15. To determine the baseline rates of accuracy for missing data, dropped calls were filled in randomly in proportion to the binomial frequencies of corresponding sites and scored for accuracy. (In the same spirit, baseline switch accuracy is switch accuracy if phase is determined at random, i.e., 0.5.)
To measure the accuracy of missing data handling in parents and single individuals, the closest heterozygous sites were located to haplotype sites for which the corresponding diplotype calls were dropped. This step allowed the two reconstructed haplotypes of each diplotype to be matched with the two corresponding true haplotypes in a local manner.
Acknowledgments
We would like to thank Y. Lin for his technical assistance. This research was supported by National Institutes of Health grants HG02757 and HL54466 to A.C. and D.J.C.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be herebymarked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2204604. Article published online ahead of print in July 2004.
References
- Akey, J., Jin, L., and Xiong, M. 2001. Haplotypes vs. single marker linkage disequilibrium tests: What do we gain? Eur. J. Hum. Genet. 9: 291–300. [DOI] [PubMed] [Google Scholar]
- Becker, T. and Knapp, M. 2002. Efficiency of haplotype frequency estimation when nuclear family information is included. Hum. Hered. 54: 45–53. [DOI] [PubMed] [Google Scholar]
- Chapman, J.M., Cooper, J.D., Todd, J.A., and Clayton, D.G. 2003. Detecting disease associations due to linkage disequilibrium using haplotype tags: A class of tests and the determinants of statistical power. Hum. Hered. 56: 18–31. [DOI] [PubMed] [Google Scholar]
- Cheng, R., Ma, J.Z., Wright, F.A., Lin, S., Gao, X., Wang, D., Elston, R.C., and Li, M.D. 2003. Nonparametric disequilibrium mapping of functional sites using haplotypes of multiple tightly linked single-nucleotide polymorphism markers. Genetics 164: 1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A.G. 1990. Inference of haplotypes from PCR-amplified samples of diploid populations. Mol. Biol. Evol. 7: 111–122. [DOI] [PubMed] [Google Scholar]
- Collins, F.S., Guyer, M.S., and Charkravarti, A. 1997. Variations on a theme: Cataloging human DNA sequence variation. Science 278: 1580–1581. [DOI] [PubMed] [Google Scholar]
- Collins, F.S., Brooks, L.D., and Chakravarti, A. 1998. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 8: 1229–1231. [DOI] [PubMed] [Google Scholar]
- Cutler, D.J., Zwick, M.E., Carrasquillo, M.M., Yohn, C.T., Tobin, K.P., Kashuk, C., Mathews, D.J., Shah, NA., Eichler, E.E., Warrington, J.A., et al. 2001. High-throughput variation detection and genotyping using microarrays. Genome Res. 11: 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, E., Abecasis, G.R., Bumpstead, S., Chen, Y., Hunt, S., Beare, D.M., Pabial, J., Dibling, T., Tinsley, E., Kirby, S., et al. 2002. A first-generation linkage disequilibrium map of human chromosome 22. Nature 418: 544–548. [DOI] [PubMed] [Google Scholar]
- Douglas, J.A., Boehnke, M., Gillanders, E., Trent, J.M., and Gruber, S.B. 2001. Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies. Nat. Genet. 28: 361–364. [DOI] [PubMed] [Google Scholar]
- Ewens, W.J. 1972. The sampling theory of selectively neutral alleles. Theor. Popul. Biol. 3: 87–112. [DOI] [PubMed] [Google Scholar]
- Ewens, W. 1979. Mathematical population genetics. Springer-Verlag, New York.
- Excoffier, L. and Slatkin, M. 1995. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol. Biol. Evol. 12: 921–927. [DOI] [PubMed] [Google Scholar]
- Excoffier, L., Laval, G., and Baldin, D. 2003. Gametic phase estimation over large genomic regions using an adaptive window approach. Hum. Genom. 1: 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halushka, M.K., Fan, J.B., Bentley, K., Hsie, L., Shen, N., Weder, A., Cooper, R., Lipshutz, R., and Chakravarti, A. 1999. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat. Genet. 22: 239–247. [DOI] [PubMed] [Google Scholar]
- Hoppe, F.M. 1987. The sampling theory of neutral alleles and an urn model in population genetics. J. Math. Biol. 25: 123–159. [DOI] [PubMed] [Google Scholar]
- Hudson, R.R. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R.R. and Kaplan, N.L. 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111: 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak, L., Daly, M.J., Reeve-Daly, M.P., and Lander, E.S. 1996. Parametric and nonparametric linkage analysis: A unified multipoint approach. Am. J. Hum. Genet. 58: 1347–1363. [PMC free article] [PubMed] [Google Scholar]
- Lander, E.S. 1996. The new genomics: Global views of biology. Science 274: 536–539. [DOI] [PubMed] [Google Scholar]
- Lewontin, R.C. 1988. On measures of gametic disequilibrium. Genetics 120: 849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N. and Stephens, M. 2003. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 165: 2213–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S., Cutler, D.J., Zwick, M.E., and Chakravarti, A. 2002. Haplotype inference in random population samples. Am. J. Hum. Genet. 71: 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, R.W. and Kaplan, N.L. 2002. On the advantage of haplotype analysis in the presence of multiple disease susceptibility alleles. Genet. Epidemiol. 23: 221–233. [DOI] [PubMed] [Google Scholar]
- Muller-Myhsok, B. and Abel, L. 1997. Genetic analysis of complex diseases. Science 275: 1328–1329. [PubMed] [Google Scholar]
- Niu, T., Qin, Z.S., Xu, X., and Liu, J.S. 2002. Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms. Am. J. Hum. Genet. 70: 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos, N., Leach, F.S., Kinzler, K.W., and Vogelstein, B. 1995. Monoallelic mutation analysis (MAMA) for identifying germline mutations. Nat. Genet. 11: 99–102. [DOI] [PubMed] [Google Scholar]
- Pritchard, J.K. 2001. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 69: 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch, N. and Merikangas, K. 1996. The future of genetic studies of complex human diseases. Science 273: 1516–1517. [DOI] [PubMed] [Google Scholar]
- Rohde, K. and Fuerst, R. 2001. Haplotyping and estimation of haplotype frequencies for closely linked biallelic multilocus genetic phenotypes including nuclear family information. Hum. Mutat. 17: 289–295. [DOI] [PubMed] [Google Scholar]
- Rosner, B. 2000. Fundamentals of biostatistics. Duxbury Press, Pacific Grove, CA.
- Schaid, D.J. 2002. Relative efficiency of ambiguous vs. directly measured haplotype frequencies. Genet. Epidemiol. 23: 426–443. [DOI] [PubMed] [Google Scholar]
- Schaid, D.J., McDonnell, S.K., Wang, L., Cunningham, J.M., and Thibodeau, S.N. 2002. Caution on pedigree haplotype inference with software that assumes linkage equilibrium. Am. J. Hum. Genet. 71: 992–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager, S.L., Huang, J., and Vieland, V.J. 2000. Effect of allelic heterogeneity on the power of the transmission disequilibrium test. Genet. Epidemiol. 18: 143–156. [DOI] [PubMed] [Google Scholar]
- Sobel, E. and Lange, K. 1996. Descent graphs in pedigree analysis: Applications to haplotyping, location scores, and marker-sharing statistics. Am. J. Hum. Genet. 58: 1323–1337. [PMC free article] [PubMed] [Google Scholar]
- Sobel, E., Lange, K., O'Connel, J.R., and Weeks, D.E. 1996. Haplotyping algorithms. In Genetic mapping and DNA sequencing (eds. T. Speed and M.S. Waterman), pp. 89–110. Springer-Verlag, New York.
- Spielman, R.S., McGinnis, R.E., and Ewens, W.J. 1993. Transmission test for linkage disequilibrium: The insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet. 52: 506–516. [PMC free article] [PubMed] [Google Scholar]
- Stephens, M. and Donnelly, P. 2003. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 73: 1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., Smith, N.J., and Donnelly, P. 2001. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68: 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J.D. and Pritchard, J.K. 2003. Haplotype blocks and linkage disequilibrium in the human genome. Nat. Rev. Genet. 4: 587–597. [DOI] [PubMed] [Google Scholar]
- Weir, B.S. and Cockerham, C.C. 1989. Complete characterization of disequilibrium at two loci. In Mathematical evolutionary theory (ed. M.W. Feldman), pp. 86–110. Princeton University Press, Princeton, NJ.
- Zhao, H., Zhang, S., Merikangas, K.R., Trixler, M., Wildenauer, D.B., Sun, F., and Kidd, K.K. 2000. Transmission/disequilibrium tests using multiple tightly linked markers. Am. J. Hum. Genet. 67: 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick, M.E., Cutler, D.J., and Chakravarti, A. 2001. A genetic variation analysis of neuropsychiatric traits. In Methods in genomic neuroscience, pp. 289–302. CRC Press, Boca Raton, FL.
WEB SITE REFERENCES
- http://www.bioinf.mdc-berlin.de/∼rob/; The Rohde-Fuerst haplotyping program.
- http://archimedes.well.ox.ac.uk/pise; PHamily.