

Abstract
It is widely hoped that the study of sequence variation in the human genome will provide a means of elucidating the genetic component of complex diseases and variable drug responses. A major stumbling block to the successful design and execution of genome-wide disease association studies using single-nucleotide polymorphisms (SNPs) and linkage disequilibrium is the enormous number of SNPs in the human genome. This results in unacceptably high costs for exhaustive genotyping and presents a challenging problem of statistical inference. Here, we present a new method for optimally selecting minimum informative subsets of SNPs, also known as “tagging” SNPs, that is efficient for genome-wide selection. We contrast this method to published methods including haplotype block tagging, that is, grouping SNPs into segments of low haplotype diversity and typing a subset of the SNPs that can discriminate all common haplotypes within the blocks. Because our method does not rely on a predefined haplotype block structure and makes use of the weaker correlations that occur across neighboring blocks, it can be effectively applied across chromosomal regions with both high and low local linkage disequilibrium. We show that the number of tagging SNPs selected is substantially smaller than previously reported using block-based approaches and that selecting tagging SNPs optimally can result in a two- to threefold savings over selecting random SNPs.
In anticipation of cost-effective SNP genotyping technologies and the availability of databases of a large number of candidate SNPs, many investigators are seriously considering genome-wide SNP scans with the hope of performing hypothesis-free disease association studies as opposed to hypothesis-driven candidate gene or region studies. Although the cost of SNP genotyping may be rapidly decreasing, it is still infeasible to genotype all available SNPs across the human genome. In this paper we examine the challenging problem of choosing an optimal or “minimal informative subset” of SNPs to be used in such a study. The end objective is to be able to identify DNA sequence variation within human populations that is associated with elevated risk of disease or adverse drug reaction caused by linkage disequilibrium (LD) with a causative variant. Challenges for this approach are the many unknowns regarding the nature of common or complex disease. We do not know how many genes influence the susceptibility to a given disease and their type of interaction or what the frequency of the causative alleles is; nor do we know the magnitude of the increased risk associated with those alleles in the study population. In the absence of this information, the best we can do is try to identify subsets of SNPs that at least allow us to reconstruct the haplotypes inferred by genotyping all the other previously known SNPs. In this sense, the problem is like a data compression problem: We start with the full pattern of all available SNPs for a relatively small sample, and want to devise an algorithm to select SNPs that will allow us to reconstruct the full data set. We hope that even though only a subset of all SNPs would be genotyped, that the statistical power for identifying phenotype–genotype associations would be minimally compromised.
In this paper, we give an algorithmic framework for selecting a minimum informative set of SNPs avoiding any reference to haplotype blocks. We argue that the selection of tagging SNPs can be partitioned into the three following steps:
Determining neighborhoods of linkage disequilibrium: Determine which sets of SNPs can be meaningfully used to infer each other.
Tagging quality assessment: Define a quality measure that describes how well a set of tagging SNPs captures the variance observed.
Optimization: Minimize the number of tagging SNPs.
Identifying those pairs of SNPs for which each member of the pair can be used to infer the other meaningfully is essential when dealing with large regions, because occasional spurious, long-range correlations can be observed but are not biologically relevant. The very large number of SNPs in the genome implies that in even a fairly large study population, a pair of SNPs will be found to be correlated by random chance when in a larger cohort these observed correlations may prove to be unsubstantiated. Selecting tagging SNPs based on these correlations is thus likely to identify only the study population (training set) and not be extensible to the overall population. In this paper, we define LD neighborhoods and argue that they are a reasonable way to restrict our attention to extensible trends.
Several papers have recently been published on selecting tagging SNPs (Avi-Itzhak et al. 2003; Hampe et al. 2003; Ke and Cardon 2003; Meng et al. 2003; Sebastiani et al. 2003; Stram et al. 2003; Thompson et al. 2003; Wang and Todd 2003; Weale et al. 2003). The main concern of these papers is the problem of defining a quality measure, that is, how well a set of tagging SNPs captures the variance observed (step 2). The investigators generally assume that the region dealt with is not very large, and thus the number of candidate SNPs is not very large. The investigators can then assume that most observed correlations are true correlations and LD neighborhoods need not be defined. The main contribution of this paper is to present an optimal algorithm for minimizing the number of tagging SNPs (step 3) that is applicable to large data sets. This paper does not attempt to determine which quality measure is best suited for a particular purpose, and the generic algorithm presented can be extended to incorporate the quality measures presented elsewhere.
We evaluate the algorithm on a data set of 4102 SNPs covering most of the genomic span of Chromosome 22 genotyped on 45 DNA samples of individuals of European ancestry. These evaluations show that selecting tagging SNPs optimally can result in a two- to threefold cost savings over selecting tagging SNPs randomly. Secondly, our work is also evaluated on publicly available data sets of SNP haplotypes across Chromosome 21 (Patil et al. 2001) and the human lipoprotein lipase (LPL) gene (Clark et al. 1998; Nickerson et al. 2000). These evaluations show that the number of tagging SNPs selected is substantially smaller than using haplotype-block-based approaches.
METHODS
The primary concern of the present work is defining subsets of “haplotype tagging” SNPs that can characterize the overall genetic diversity of a region. More specifically, we seek to identify a subset of SNPs that allow reconstruction, with some margin of error, of the full set of haplotypes determined by the entire SNP set. This section is organized into three subsections corresponding to the three steps described above. We start by showing how neighborhoods of predictive SNPs can be determined and contrast this with the more rigid notion of haplotype blocks. Subsequently, we present a measure of “informativeness” of an SNP that is related to measures of haplotype diversity (Johnson et al. 2001), but provides a direct measure of how an SNP, or a set of SNPs, can be used to characterize another SNP, or a set of SNPs. This is an easily computable function that correlates well with alternative multiloci linkage disequilibrium measures such as haplotype r2 (Weale et al. 2003). As we showed in previous work (Bafna et al. 2003), the computation of sets of SNPs with maximal informativeness is NP-hard in the general case, but is tractable in most cases of practical interest when the number of SNPs predicting each SNP is small. Lastly, we present an algorithm for obtaining a minimal set of tagging SNPs.
Finding Neighborhoods of Potentially Predictive SNPs
Although significant LD can occur between SNPs physically distant on the genome, such distant relationships are likely to reflect common ancestry only in the case of very recent admixture. In practice, if high LD is observed at greater distances than a reasonable threshold, such as 200 kb, it is commonly ignored (Gabriel et al. 2002) and is considered to be an artifact of a small sample size. Our primary concern in locating SNPs in LD with one another is finding those sets of SNPs that are predictive not only of other SNPs in our sample, but also of those SNPs that were not typed in our sample population. We therefore wish primarily to identify those SNPs that characterize regions of common recent ancestry (conserved haplotypes) rather than those distant SNPs that might be associated because of selection, admixture, or random chance. SNPs associated by recent common ancestry are likely to be those that are sufficiently close that recombination has not occurred frequently between them. We therefore restrict our search for predictive SNPs to those that are in relatively close proximity to the targets for which they might be predictive.
In practice, it is neither efficient nor desirable to have a fixed neighborhood in which to look for SNPs. Recombination rates (Kong et al. 2002) and historical LD (Gabriel et al. 2002) vary across the genome. Because of variability in SNP density and recombination rate, we note that for most SNPs, the neighborhood of SNPs that are in LD with it, or are otherwise informative for it, is highly variable. We exploit this by making a neighborhood graph with SNP sites as vertices. Two vertices are connected by an edge only if one can be used for predicting another. For each SNP s we thus define a set of neighbors, N(s).
In determining whether one SNP can be used for predicting another, one must determine whether the allelic states of two SNPs are significantly correlated. A variety of statistics have been devised for quantifying this correlation, or linkage disequilibrium. But the special case of selecting informative SNPs requires a bit more than simply quantifying correlation. In the haplotype-block-tagging approach (Johnson et al. 2001; Patil et al. 2001; Zhang et al. 2002, 2003a,b), two SNPs are considered to have a useful level of correlation if they occur in the same haplotype block, that is, if they both occur in the same region with little evidence of recombination. We note that this is not entirely the same as saying that one SNP can be used to predict another if there is little evidence of recombination between the two SNPs. Hence, if a given method is effective in detecting regions of low recombination rate and that low recombination rate between SNPs allows one to use one SNP to predict another, then the set of SNPs that can be used to predict an SNP s can be found by taking the union of all putative haplotype blocks that contain SNP s. Figure 1 shows how the union of possible haplotype blocks over a region can differ from a partition of the region into haplotype blocks. The latter situation is common with the rule-based algorithms to find haplotype blocks, because many overlapping possible block decompositions that meet the rules are often possible (cf. Schwartz et al. 2003). Figure 1 illustrates the potential value of using the union of blocks by showing optimal SNP sets selected based on two distinct minimal block decompositions using the method of Zhang et al. (2003a) and those selected from the union of the two decompositions by our block-free method. The figure shows how the broader correlations allow one to characterize the information content of the region with fewer SNPs, each of which has predictive power across the boundaries of any one block decomposition.
Figure 1.

The three panels display a region of Chromosome 22. The lower triangle of each panel shows the LD between pairs of SNPs, where red denotes high LD and blue low LD. The upper triangles show the pairs of SNPs where one is used to predict the other. (A,B) The haplotype blocks as determined by two different runs of the block detection method of Gabriel et al. (2002). (C) The neighborhoods determined from taking the union of all possible Gabriel et al. blocks in the interval. The diamonds on the diagonal display the tagging SNPs selected for the three sets, using the algorithm of Zhang et al. (2003a) for the first two figures and the algorithm presented in this paper for the third figure.
Another way to determine a meaningful neighborhood of predictive SNPs is to use the metric LD maps described by Maniatis et al. (2002); only those SNPs that are within a distance of <1 LD unit are considered to be significantly correlated to each other. Beyond this distance, the correlation (LD) between SNPs falls to levels unlikely to be useful for mapping (Morton et al. 2001).
Using either one of these methods, neighborhoods are generally small in the data sets studied here (about five SNPs for the genotype data, using the Gabriel et al. [2002] definition of blocks to define neighborhoods).
Defining Informativeness
In this section, we define a measure of how well a set of SNPs can predict a single target SNP. Informativeness measures how well one can reconstruct a target SNP, t, from a set of its neighbors, N(t); given the haplotype pattern of a set of its neighbors, look at the pairs of haplotypes that have a different allele at t, count how many of these also do not have the same set of alleles on all of the SNPs in N(t), and divide by the total number of pairs. This is one of many such measures one could use; we show below that this is highly correlated with alternate measures such as haplotype r2 (Weale et al. 2003).
A set S of n SNPs specifying a collection of m haplotypes can be denoted by an n × m matrix M. The columns of M correspond to SNPs in the population, and rows correspond to haplotypes. For an SNP s, denote si = M[i, s]. We assume for simplicity that all SNPs are biallelic (taking on only two values); consequently M[i, s] ∈ {0, 1} ∀ i, s. Let a “target” SNP t be associated with a trait of interest. If t is not typed, its state is predicted using proximal SNPs. We define a measure of”informativeness” of an SNP s with respect to t to quantify the accuracy with which we can make this prediction.
For an SNP s and haplotypes i, j, let Dsi,j be the event that M[i, s] ≠ M[j, s]. We define the “informativeness” of SNP s with respect to an SNP t as
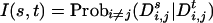 |
(1) |
where i, j are two haplotypes drawn uniformly at random from the set of all distinct haplotype pairs. Observe that I(s, t) = 1 implies complete predictability, and I(s, t) = 0 when t is monomorphic in the population. I(s, t) is estimated easily from a sample as follows: Consider the complete graph GH on m nodes labeled 1, 2,..., m. Each SNP s defines a subgraph that is a bipartite clique with m nodes. The edge set E(s) is defined by the rule (i, j) ∈ E(s) if and only if si ≠ sj. Then
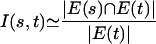 |
is the informativeness of s with respect to t. The definition is easily extended to a subset of SNPs. For S′ ⊆ S, let 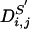 be the event that M[i, s] ≠ M[j, s] for some s ∈ S′. Likewise, let E(S′) = ∪s∈S′E(s). Then,
be the event that M[i, s] ≠ M[j, s] for some s ∈ S′. Likewise, let E(S′) = ∪s∈S′E(s). Then,
 |
(2) |
When predicting a set of SNPs from a set of tagging SNPs, we need to be aware that each SNP can only be predicted from its set of neighbors, and we define for all S′, T ⊆ S,
 |
In Supplemental material 2, we make connections between Informativeness with measures of LD and Diversity.
Dealing With Genotype Data
The definition of informativeness above assumes the availability of haplotype phases. In practice, genotype data are much more easily determined experimentally than haplotypes. We overcome this problem by computationally inferring haplotypes over each neighborhood using a maximum likelihood/expectation maximization approach (Abecasis et al. 2001). For the purpose of our discussion, we assume that this method can infer accurately all common haplotypes of a neighborhood. Further work into the subject of how genotype data can be used for selecting tagging SNPs has been done by Zhang et al. (2004).
SNP Selection Algorithm
We now present our algorithm for optimizing a measure I of informativeness, such as the metrics presented in the previous section, or one of those presented in Stram et al. (2003), Weale et al. (2003), and Meng et al. (2003).
We start by defining the k-most informative SNPs problem.
k-MIS: k Most Informative SNPs
Input: A set of n SNPs S, 0 < k ≤ n.
Output: Find the subset S′ ⊆ S such that I(S′, S) = maxR⊂S, |R|≤kI(R, S).
Consider again the informativeness of a subset S′ with respect to an SNP t. For ease of exposition, we define the distance between SNPs s and t simply by the number of SNPs in between s and t. We now show how we can solve the minimum informative SNPs problem if we assume that the neighborhoods are not overly large, that is, their size is bounded by some constant (say, 13 or 21). Informally, we would like to find a most informative subset of SNPs given that only SNPs that are a distance w apart can be used in the prediction.
In Figure 2 we give pseudocode for an algorithm that solves the k-MIS problem. In Supplemental material 1, we show that this algorithm can be used to solve the k-MIS efficiently, when the size of each neighborhood is bounded by a constant w.
Figure 2.

An O(nk2w) algorithm for the k-MIS problem, assuming a maximum size w on all neighborhoods.
Test Data Sets
For evaluation, we rely on three data sets. The first is a chromosome-wide data set from human Chromosome 21 described by Patil et al. (2001), which consists of 24,047 SNPs typed on 20 haploid copies of the chromosome. This data set contains a large contiguous set of closely spaced SNPs, but the small number of sampled chromosomes, the cosmopolitan origin of the population sample, and high rate of missing data (21.7%) make this data set less suitable for our purposes. To limit the amount of computation, those experiments that leave out one test were done on only the first 1000 SNPs of this data set. This subset was found to be highly representative of the overall data set.
We also use a data set derived from 71 individuals typed at 88 polymorphic sites in the human lipoprotein lipase (LPL) gene (Clark et al. 1998; Nickerson et al. 2000), from which we ignored one multiallelic site to simplify our analysis. The greater sample size allows us to draw more confident predictions. The haplotype phase is known in this data set, allowing for comparisons with the method of Zhang et al. (2003a) and cross-validation studies.
The third data set consists of 4102 SNPs distributed along most of the genomic span of Chromosome 22 with a median spacing of 4 kb, genotyped by the 5′ nuclease assay (TaqMan Assays-on-Demand SNP Genotyping Products, Applied Biosystems; de la Vega et al. 2002) on 45 DNA samples of Caucasian individuals obtained from the NIGMS Human Variation Panel (Coriell Institute for Medical Research, Camden, NJ). The SNP density and sample size of this data set are close to that used by the International HapMap Project (International HapMap Consortium 2003), and thus is particularly interesting to analyze.
Cross-Validation Procedure
To assess the quality of the solution given by the tagging SNP selection algorithm, we performed leave-one-out cross-validation. For each haplotype in our data set, we trained our algorithm on the rest of the data set to determine a minimum informative SNP set. The performance of the SNP selection for the haplotype left out was evaluated by counting the number of alleles in that haplotype that were correctly imputed from the SNPs that were “typed.” The accumulated accuracy over all haplotypes gives a global measure of the accuracy for the given data set. Implicitly, all SNPs that were typed in the test haplotype were considered to be correctly imputed. SNPs that were not typed were imputed by looking at the typed SNPs in their neighborhood; if there are training haplotypes that had the same allele call on all the typed SNPs in their neighborhoods, then the allele was determined by a majority vote of those haplotypes (cf. Fig. 3). If no such haplotype existed, then the majority vote was taken over all training haplotypes that have the same allele call on all but one of the typed SNPs in the neighborhood. Furthermore, if no haplotype existed having the same allele call on all but k SNPs typed in the neighborhood, then the allele call was determined over all training haplotypes with the same allele call on all but k + 1 SNPs typed. If the majority vote was ambiguous, then we counted the SNP as being one-half predicted.
Figure 3.

Method of imputing the second SNP present in the test haplotype from the four training haplotypes. The “haplotype” column shows the original haplotypes. The “typed” column shows only the SNPs that are typed, the first and the fourth SNP. The “imputed” column shows the haplotypes used in the imputation of the second SNP; only those haplotypes that are identical to the test haplotype on the first and fourth SNP are used in the imputation. A is imputed as of those haplotypes that agreed with the test haplotype on the first and fourth SNP, two haplotypes have an A, and one has a C.
Comparison With Other Tagging Methods
We compared our method with the tagging SNP selection method presented by Zhang et al. (2003a), which like our method can deal with large SNP data sets. The method presented there partitions a chromosome into haplotype blocks and within each block selects a set of tagging SNPs. To select a set of tagging SNPs with limited resources, a cost is imposed with not tagging a given SNP. In our experiments, we fixed the cost of an untagged SNP to be 1, the same as the cost of typing a SNP. A block was considered to be tagged if 90% of the haplotype diversity (Johnson et al. 2001) was captured by the tagging SNPS. To judicially compare our approach and the approach presented in Zhang et al. (2003a), we eliminated the effect of neighborhood or block definition by assuming that an SNP i is in the neighborhood of an SNP j if i and j both occur in the same putative block.
To look at the effect of different quality measures, we also performed tagging SNP selection replacing the informativeness quality measure described here with the haplotype r2 metric of Weale et al. (2003).
RESULTS
We first consider how well our formal definition of informativeness correlates with a key practical benchmark of the value of an SNP subset: its utility in predicting missing values. In Figure 4, we plot informativeness and the number of SNPs correctly imputed in a leave-one-out cross-validation test as a function of the number of SNPs typed. Informativeness and fraction of SNPs imputed in cross-validation studies closely track one another until >80% of their maximal value is achieved by both measures. The informativeness measure thus appears to be minimally affected by overfitting on sparse data up to this accuracy level. For higher numbers of SNPs, cross-validated imputation fraction slightly lags informativeness. This latter observation is consistent with the idea that a small fraction of the SNPs capture the common haplotype variants that account for most population variation and that are easily inferred from even very small population samples, but that a minority of the variation is explained by rarer haplotype patterns that are more difficult to infer accurately from small population samples. Nonetheless, if we type only 20% of the SNPs, we can correctly impute 90% of them using our algorithm. We would expect informativeness to track imputation accuracy even more closely when larger population samples are used for inference.
Figure 4.

The x-axis shows the number of SNPs typed, and the y-axis shows the fraction of total informativeness or total number of SNPs correctly imputed in a leave-one-out experiment. The solid (upper) curve represents informativeness and the dashed (lower) curve shows the fraction of SNPs that are correctly imputed in a leave-one-out experiment. The data set used is the first 1000 SNPs of the Chromosome 21 data set of Patil et al. (2001) using the no-four-gamete violation definition of blocks and neighborhoods. Neighborhoods were restricted to have sizes no larger than 13.
We can further ask how this block-free method compares with block-based informative SNP selection through comparison to the Zhang et al. (2003a) algorithm. In Figure 5, we examine the fraction of SNPs correctly imputed with leave-one-out cross-validation as a function of the number of SNPs typed. We perform this comparison under two different conditions—the long-range, small chromosome sample of the Patil et al. (2001) data and the short-range, larger individual sampling of the LPL data— allowing us to judge not just overall accuracy of the methods but also their relative sensitivities to sample size. For almost all values in both data sets, the number of correctly imputed SNPs is higher for this method than for the one presented by Zhang et al. (2003a). For the Chromosome 21 data, both methods show the sort of rapid initial increase followed by a more gradual approach to 100% informativeness that we observed in Figure 4. The block-based method, however, almost immediately shows a slower rate of growth in informativeness. This effect presumably reflects the cost imposed by artificially restricting the range of influence of the few SNPs chosen based on block boundaries. Both methods level off and approach similar slow rates of growth upon reaching ∼80% accuracy, suggesting that both methods may encounter similar problems of inferring rare haplotype patterns from the smaller sample size. The LPL data show a slower initial growth, perhaps reflecting the relatively high apparent historical recombination rate of that genetic region or the presence of comparatively rare variants in that data set. For both methods, the slope of the curve falls off more slowly with increasing SNP count for LPL, consistent with the hypothesis that the larger population sample for LPL versus Chromosome 21 reduces the difficulty of inferring rare haplotypes.
Figure 5.

The x-axis shows the number of SNPs typed, and the y-axis shows the fraction of SNPs correctly imputed in a leave-one-out experiment. The solid (upper) curve represents the fraction of SNPs correctly imputed by the block-free method presented in this paper, and the dashed (lower) curve represents the fraction of SNPs correctly imputed by the method of Zhang et al. (2003a). The no-four-gamete violation definition was used for blocks and neighborhoods. (A) Results from the first 1000 SNPs of Chromosome 21 data set. (B) Results from the LPL data set.
We next investigated whether informative SNP selection provides a significant advantage over random SNP selection using the genotype data from Chromosome 22. In Figure 6, we compare the results of our algorithm with the random selection of SNPs across the chromosome. The plot shows a substantial advantage to informative SNP selection over random selection in preserving informativeness. That advantage persists across the range of SNP set sizes. For example, with randomly chosen SNPs, we must genotype half of the data set (2051 SNPs) to reach 78.8% accuracy, whereas an optimally chosen set can exceed that accuracy with only 636 SNPs (15.5%). Approximately the same accuracy can thus be achieved with a more than threefold reduction in cost by choosing optimal tagging SNPs as opposed to random SNPs. To compare the methods another way, the 2051 SNPs we require to capture 78.8% of informativeness when the SNPs are randomly chosen are enough to yield 99.6% of the informativeness when the SNPs are optimally selected. Thus, whether our goal is to choose a specific subset size of maximum utility or to achieve a fixed level of utility with minimum cost, informative SNP selection appears to have considerable value.
Figure 6.

The x-axis shows the number of SNPs genotyped and the y-axis the fraction of the informativeness captured by those SNPs. The upper (solid) line represents the optimal solution and the lower (dashed) line a random solution. Computations are done for the Chromosome 22 Caucasian genotype data set.
One final issue we explore is the relationship between the informativeness measure used in this paper and other multiloci metrics of linkage disequilibrium. For this purpose, we compare our informativeness measure with the haplotype r2 described by Weale et al. (2003), a generalization of the pairwise r2 measure to haplotypes. We compare with this measure because it provides a simple analog into the space of haplotypes of a well-understood and widely used traditional measure of pairwise LD. In Figure 7, we examine the informativeness captured by SNPs when we select them to optimize average haplotype r2 compared with that derived when we select SNPs so as to optimize directly for informativeness. For comparison, the figure also shows the informativeness captured by randomly chosen SNPs. The figure shows that the results of optimizing for informativeness and for haplotype r2 are extremely close and are both very different from results achieved with randomly chosen SNPs. In Figure 8, we reverse this test and explore the fraction of total haplotype r2 correlation captured when we optimize separately for informativeness and for haplotype r2, with randomly chosen SNPs again included for comparison. The figure again shows that optimizing for either measure optimizes very well for the other. The results thus suggest that informativeness and haplotype r2, although distinct measures, are closely related in practice.
Figure 7.

The effects of changing the quality function being optimized on the Chromosome 22 Caucasian genotype data set. The x-axis represents the number of SNPs genotyped, and the y-axis shows the fraction of haplotype r2 captured. The solid line shows the haplotype r2 if tagging SNPs are chosen optimally, the dashed line shows the haplotype r2 if the tagging SNPs are selected by maximizing haplotype informativeness, and the dotted line shows the informativeness from choosing random sets of tagging SNPs.
Figure 8.

The effects of changing the quality function being optimized on the Chromosome 22 Caucasian genotype data set. The x-axis represents the number of SNPs genotyped, and the y-axis shows the fraction of informativeness captured. The solid line shows the informativeness if tagging SNPs are chosen optimally, the dashed line shows the informativeness if the tagging SNPs are selected by maximizing average haplotype r2 correlation, and the dotted line shows the informativeness from choosing random sets of tagging SNPs.
Table 1 shows a summary of the number of tagging SNPs obtained by optimally selecting tagging SNPs maintaining reasonable informativeness values ranging from 85% to 100% on the Chromosome 22 data set. As a comparison, we also show the results using the haplotype r2 metric instead of informativeness, and for randomly selected SNPs. As shown in Table 1, when maintaining 100% of informativeness or haplotype r2, a reduction in genotyping of 34% is observed. Furthermore, if one is willing to accept lower levels of informativeness, savings in genotyping up to 80% are achievable at 85% of informativeness. Similarly savings of up to 70% are achievable at 85% of haplotype r2.
Table 1.
Number of Tagging SNPs Needed at Various Thresholds, Using Either the Informativeness or Haplotype r2 Measures and Either an Optimal or Random Choice of Tagging SNPs
|
Threshold
|
|||||||
|---|---|---|---|---|---|---|---|
| Measure | Algorithm | # SNPs | 85% | 90% | 95% | 99% | 100% |
| Informativeness | Optimal | 4102 | 817 | 1001 | 1274 | 1798 | 2715 |
| Informativeness | Random | 4102 | 2633 | 3160 | 3618 | 3930 | 4100 |
| Haplotype r2 | Optimal | 4102 | 1243 | 1461 | 1757 | 2234 | 2724 |
| Haplotype r2 | Random | 4102 | 2497 | 2907 | 3248 | 3887 | 4102 |
DISCUSSION
We present a new measure for the identification of subsets of SNPs that are predictive of other SNPs identified in population samples. As we argue above (and in Supplemental material 2), this measure avoids some of the difficulties traditional linkage disequilibrium measures have experienced when applied to tagging SNP selection, particularly when dealing with small population samples. Furthermore, the concept of pairwise LD does not reliably capture the higher-order dependencies implied by observed haplotype structures, whereas the extension to second-order and higher LD would be tremendously complex analytically. The latter problem often leads to very inflated estimates of the number of tagging SNPs required for genome-wide association studies by requiring that all pairwise values of a LD metric like r2 surpasses a minimum arbitrary threshold (e.g., r2 > 0.85; Wang and Todd 2003; Carlson et al. 2004). These estimates are flawed because they ignore the fact that multiple tagging SNPs can predict the state of another SNP, as has been pointed out by Goldstein et al. (2003). In addition, the sampling properties of statistics for higher-order LD are quite poor, so that much larger sample sizes are needed. Our notion of informativeness provides a practical framework for formalizing the problem of tagging SNP selection suitable for generalized structures of higher-order local dependency. On the other hand, a consequence of using metrics that leverage higher-order interactions, is that the data analysis of association studies that used the tagging SNPs might need a deconvolution function to map back the SNPs that were tagged but not genotyped, in particular if a marker-by-marker analysis is used. However, this may not be necessary if a haplotype-based analysis is performed. One possible drawback of our new metric is that it may not seem intuitive in the context of population genetics theory and thus its relationship to other study design parameters like power and sample size may not be obvious. Our empirical studies confirm the practical value of this measure and its relevance to prior work in the field. We have shown that the informativeness measure closely follows both the r2 measure when extended to haplotypes (Stram et al. 2003; Weale et al. 2003) and the intuitive notion of imputation accuracy of missing data. It has been suggested that the haplotype r2 measure also correlates well to sample size requirements (Stram et al. 2003; Weale et al. 2003), and owing to the high correlation between the latter and informativeness, we expect that setting thresholds on our new metric can also inform on the power loss and sample size trade-offs. We have presented algorithms for efficiently solving the problem of optimally finding minimum informative SNP subsets in most practical cases. The bounded-width method provides a means of solving the problem without resorting to a haplotype block decomposition, which may be an important advantage given the uncertainty about the definition and utility of haplotype blocks (Wall and Pritchard 2003).
By not relying on haplotype block structures, we overcome the limitations of tagging methods that are restricted to those blocks and cannot tag SNPs outside these (Avi-Itzhak et al. 2003; Sebastiani et al. 2003; Zhang et al. 2003a), and that ignore block-to-block LD. We have shown that there is considerable value to our particular method for block-independent selection of informative SNP subsets, compared with both random selection and a leading block-based selection method. We further show that our method is not significantly impaired by overfitting even when inferring from small population samples. These results are established across several data sets representing a range of numbers of SNPs and depths of coverage.
As one exemplar method for defining sensible neighborhoods of SNPs that can predict the state of another, we took advantage of the uncertainty in the heuristics for finding haplotype blocks, and used as neighborhood the union of alternative block decompositions that are possible for a given region. However, a better alternative would be to use map distance thresholds not in the physical map, but in the metric LD map that has additive distances proportional to the strength of LD (Maniatis et al. 2002). This would provide a truly block-free method that would factor in map location in a meaningful way, using a neighborhood description that is resilient to the issues of pairwise LD metrics described above, and robust to SNP density (Ke et al. 2004). We also emphasize that this algorithm can be used with our informativeness measure or with other quality metrics more appropriate for a particular study design. For example, when there is more knowledge on the disease mode of inheritance or on the range of disease allele frequency, alternate objective functions may be considered such as maximizing the power for detecting association.
There are several avenues for future work in this area. We note that although we have presented an optimal algorithm for selecting tagging SNPs in the three-step framework presented here, methods that are demonstrably optimal for all cases have not been presented for the first two steps; selecting sets of predictive SNPs and defining the best quality measure. Another question to explore is the necessary sample size to select a tagging SNP set that can be extrapolated to larger sample sizes. Thompson et al. (2003) suggested that samples as small as 25 individuals may be sufficient for the screening of a panel of SNPs from which to select a useful tagging SNP set. This sample size is smaller than the one used in our Chromosome 22 data set and in the HapMap project (International HapMap Consortium 2003). However, further exploration of this parameter in our framework is warranted. Finally, the issue of which starting density of sampled SNPs is appropriate to select a tagging SNP set with suitable coverage of all genomic regions needs to be studied. Clearly, the data sets being generated by the HapMap project and others (Ke et al. 2004) will be helpful to perform this assessment.
Acknowledgments
We thank Michell Cargill, Fiona Hyland, Francis Kalush, and Kit Lau for many valuable discussions and technical suggestions about SNP and haplotype data analysis challenges as well as Charles Scafe for providing access to the Chromosome 22 data.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2570004.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Abecasis, G., Martin, R., and Lewitzky, S. 2001. Estimation of haplotype frequencies from diploid data. Am. J. Hum. Genet. 69: 114. [Google Scholar]
- Avi-Itzhak, H.I., Su, X., and de la Vega, F.M. 2003. Selection of minimum subsets of single nucleotide polymorphisms to capture haplotype block diversity. In Proceedings of Pacific Symposium on Biocomputing, Vol. 8, pp. 466–477. [DOI] [PubMed] [Google Scholar]
- Bafna, V., Halldórsson, B.V., Schwartz, R.S., Clark, A.G., and Istrail, S. 2003. Haplotypes and informative SNP selection algorithms: Don't block out information. In Proceedings of the Seventh Annual International Conference on Computational Molecular Biology (RECOMB), pp. 19–27.
- Carlson, C., Eberle, M., Rieder, M., Yi, Q., Kruglyak, L., and Nickerson, D. 2004. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 74: 106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A., Weiss, K., Nickerson, D., Taylor, S., Buchanan, A., Stengard, J., Salomaa, V., Vartiainen, E., Perola, M., Boerwinkle, E., et al. 1998. Haplotype structure and population genetic inferences from nucleotide-sequence variation in human lipoprotein lipase. Am. J. Hum. Genet. 63: 595–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Vega, F., Dailey, D., Ziegle, J., Williams, J., Madden, D., and Gilbert, D. 2002. New generation pharmacogenomic tools: A SNP linkage disequilibrium map, validated SNP assay resource, and high-throughput instrumentation system for large-scale genetic studies. Biotechniques Suppl 48–50: 54. [PubMed] [Google Scholar]
- Gabriel, S.B., Schaffner, S.F., Nguyen, H., Moore, J.M., Roy, J., Blumenstiel, B., Higgins, J., DeFelice, M., Lochner, A., Faggart, M., et al. 2002. The structure of haplotype blocks in the human genome. Science 296: 2225–2229. [DOI] [PubMed] [Google Scholar]
- Goldstein, D., Ahmadi, K., Weale, M., and Wood, N. 2003. Genome scans and candidate gene approaches in the study of common diseases and variable drug responses. Trends Genet. 19: 615–622. [DOI] [PubMed] [Google Scholar]
- Hampe, J., Schreiber, S., and Krawczak, M. 2003. Entropy-based SNP selection for genetic association studies. Hum. Genet. 114: 36–43. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium. 2003. The international HapMap project. Nature 426: 789–796. [DOI] [PubMed] [Google Scholar]
- Johnson, G.C.L., Esposito, L., Barratt, B.J., Smith, A., Heward, J., Di Genova, G., Ueda, H., Cordell, H., Eaves, I., Dudbridge, F., et al. 2001. Haplotype tagging for the identification of common disease genes. Nat. Genet. 29: 233–237. [DOI] [PubMed] [Google Scholar]
- Ke, X. and Cardon, L. 2003. Efficient selective screening of haplotype tag SNPs. Bioinformatics 19: 287–288. [DOI] [PubMed] [Google Scholar]
- Ke, X., Hunt, S., Tapper, W., Lawrence, R., Stavrides, G., Ghori, J., Whittaker, P., Collins, A., Morris, A., Bentley, D., et al. 2004. The impact of SNP density on fine-scale patterns of linkage disequilibrium. Hum. Mol. Genet. 13: 577–588. [DOI] [PubMed] [Google Scholar]
- Kong, A., Gudbjartsson, D.F., Sainz, J., Jonsdottir, G.M., Gudjonsson, S.A., Richardsson, B., Sigurdardottir, S., Barnard, J., Hallbeck, B., Masson, G., et al. 2002. A high-resolution recombination map of the human genome. Nat. Genet. 31: 241–247. [DOI] [PubMed] [Google Scholar]
- Maniatis, N., Collins, A., Xu, C.F., McCarthy, L.C., Hewett, D.R., Tapper, W., Ennis, S., Ke, X., and Morton, N.E. 2002. The first linkage disequilibrium (LD) maps: Delineation of hot and cold blocks by diplotype analysis. Proc. Natl. Acad. Sci. 99: 2228–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, Z., Zaykin, D., Xu, C., Wagner, M., and Ehm, M. 2003. Selection of genetic markers for association analyses, using linkage disequilibrium and haplotypes. Am. J. Hum. Genet. 73: 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, N., Zhang, W., Taillon-Miller, P., Ennis, S., Kwok, P., and Collins, A. 2001. The optimal measure of allelic association. Proc. Natl. Acad. Sci. 98: 5217–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson, D.A., Taylor, S.L., Fullerton, S.M., Weiss, K.M., Clark, A.G., Stengaard, J.H., Salomaa, V., Boerwinkle, E., and Sing, C.F. 2000. Sequence diversity and large-scale typing of SNPs in the human apolipoprotein E gene. Genome Res. 10: 1532–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil, N., Berno, A.J., Hinds, D.A., Barrett, W.A., Doshi, J.M., Hacker, C.R., Kautzer, C.R., Lee, D.H., Marjoribanks, C., McDonough, D.P., et al. 2001. Blocks of limited haplotype diversity revealed by high resolution scanning of human Chromosome 21. Science 294: 1719–1723. [DOI] [PubMed] [Google Scholar]
- Schwartz, R.S., Halldórsson, B.V., Bafna, V., Clark, A.G., and Istrail, S. 2003. Robustness of inference of haplotype block structure. J. Comput. Biol. 10: 13–20. [DOI] [PubMed] [Google Scholar]
- Sebastiani, P., Lazarus, R., Weiss, S., Kunkel, L., Kohane, I., and Ramoni, M. 2003. Minimal haplotype tagging. Proc. Natl. Acad. Sci. 100: 9900–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stram, D., Leigh Pearce, C., Bretsky, P., Freedman, M., Hirschhorn, J., Altshuler, D., Kolonel, L., Henderson, B., and Thomas, D. 2003. Modeling and E-M estimation of haplotype-specific relative risks from genotype data for a case-control study of unrelated individuals. Hum. Heredity 55: 179–190. [DOI] [PubMed] [Google Scholar]
- Thompson, D., Stram, D., Goldgar, D., and Witte, J.S. 2003. Haplotype tagging single nucleotide polymorphisms and association studies. Hum. Heredity 56: 48–55. [DOI] [PubMed] [Google Scholar]
- Wall, J. and Pritchard, J. 2003. Assessing the performance of the haplotype block model of linkage disequilibrium. Am. J. Hum. Genet. 73: 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. and Todd, J. 2003. The usefulness of different density SNP maps for disease association studies of common variants. Hum. Mol. Genet. 12: 3145–3149. [DOI] [PubMed] [Google Scholar]
- Weale, M., Depondt, C., Macdonald, S., Smith, A., Lai, P., Shorvon, S., Wood, N., and Goldstein, D. 2003. Selection and evaluation of tagging SNPs in the neuronal-sodium-channel gene SCN1A: Implications for linkage-disequilibrium gene mapping. Am. J. Hum. Genet. 73: 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K., Deng, M., Chen, T., Waterman, M.S., and Sun, F. 2002. A dynamic programming algorithm for haplotype block partitioning. Proc. Natl. Acad. Sci. 99: 7335–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K., Sun, F., Waterman, M.S., and Chen, T. 2003a. Haplotype block partition with limited resources and applications to human chromosome 21 haplotype data. Am. J. Hum. Genet. 73: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K., Sun, F., Waterman, M.S., and Chen, T. 2003b. Dynamic programming algorithms for haplotype block partitioning: Applications to human chromosome 21 haplotype data. In Proceedings of the Seventh Annual International Conference on Computational Molecular Biology (RECOMB).
- Zhang, K., Qin, Z., Liu, J., Chen, T., Waterman, M., and Sun, F. 2004. Haplotype block partitioning and tag SNP selection using genotype data and their applications to association studies. Genome Res. 14: 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]