

Myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) are usually sporadic diseases, however, rare familial cases have helped to identify human disease genes and provide crucial insight into hematopoiesis. Multiple predisposition genes have been identified, including RUNX1, CEBPA, GATA2, ETV6 and DDX41 (1). These syndromes are characterized by autosomal dominant inheritance and heterozygous germline mutations. Latency periods preceding the onset of MDS or other hematologic malignancies are widely variable, and appearance of the malignant phenotype is thought to require additional somatic mutations. In addition to improving diagnosis and treatment of hematologic malignancies, identification of disease-predisposing mutations has broadened our understanding of the role of these genes in normal hematopoiesis. Nevertheless, a large portion of familial MDS/AML cases remain unexplained. One potentially distinct form is MDS/AML with erythroid hyperplasia (OMIM 133180). Patients typically present in the erythroleukemic phase with multilineage dysplasia and complex cytogenetics; the prognosis is poor, with an overall survival of 3–9 months (2). There are at least five families reported in the literature, all with an autosomal dominant mode of inheritance and an unexplained genetic basis. We investigated one of these families, first reported in 1982 and again in 1987 (3,4).
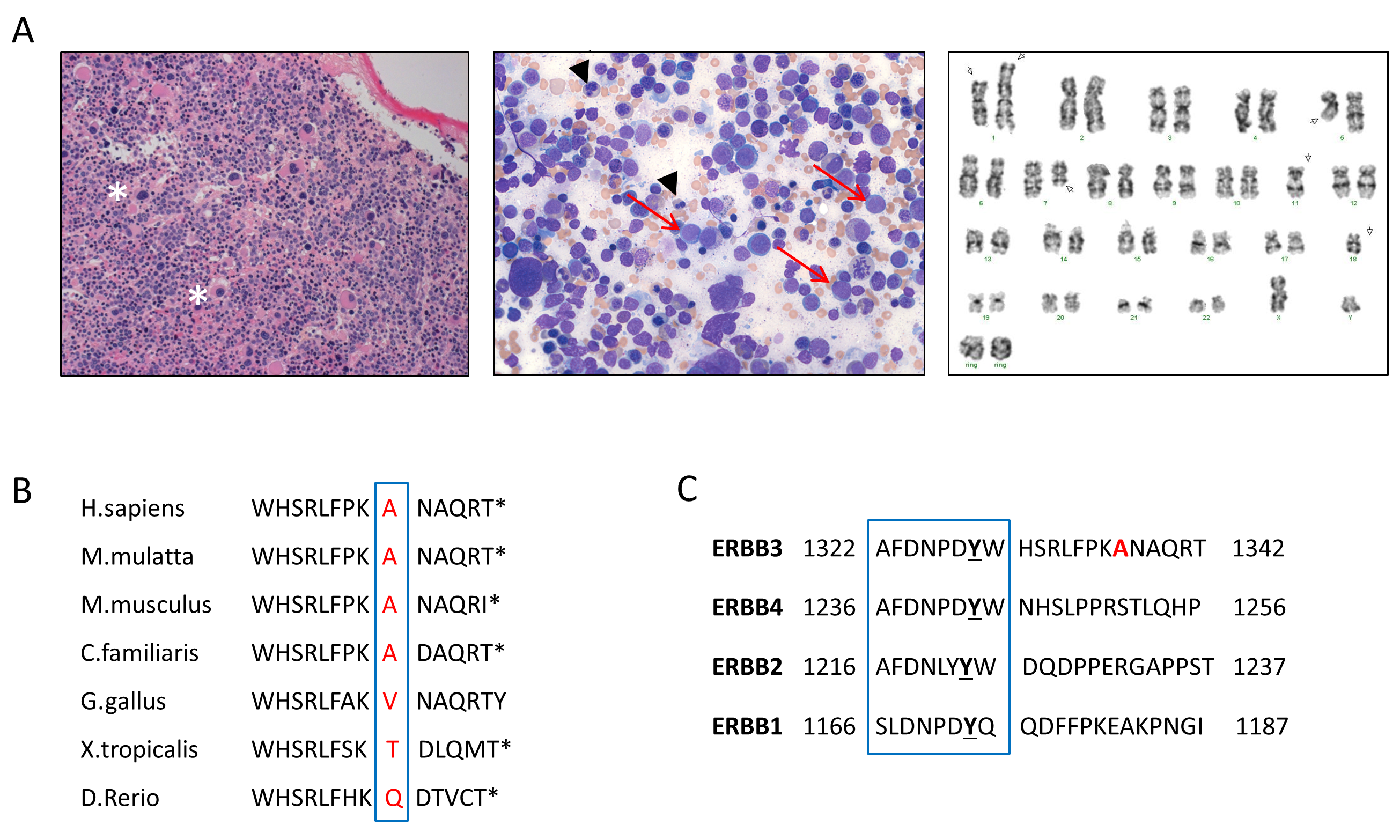
The proband was diagnosed with evolving erythroleukemia following a bone marrow biopsy that revealed a hypercellular marrow with multilineage dysplasia, marked erythroid hyperplasia, excess blasts and complex cytogenetics (Figure S1A). An updated pedigree was established (Figure 1A), exhibiting an inheritance pattern consistent with autosomal dominance with a variable latency period (range 27–73 years, median 54). Family members and published reports confirmed that all affected individuals in this family were diagnosed with erythroleukemia or erythroid MDS. Further, multiple affected family members displayed normal hematopoiesis during evaluations prior to disease onset, indicating that overt hematologic abnormalities were absent during the latency period (Table S1). An initial screen of 12 known predisposition genes failed to detect a pathogenic variant in the proband (5). This, along with the consistent erythroid phenotype suggested that the germline variant predisposing to disease in this family was distinct from those currently known.
Figure 1. Identification of the germline ERBB3 variant.

(A) Family pedigree: filled circles/boxes denote affected family members, slashes denote deceased individuals. A single asterisk (*) marks genotyped individuals with homozygous wildtype ERBB3; while (**) marks genotyped individuals heterozygous for ERBB3 p.A1337T. Representative chromatograms for those with the variant are shown below. Diamonds denote multiple individuals of both sexes, with the number of offspring given. Current age or age of death is noted for all affected as well as unaffected individuals involved in this study. A black arrow denotes the proband. Whole exome sequencing was performed on individuals III-01, III-02, III-03, III-07, III-08, III-09, III-15. (B) Schematic diagram of the ERBB3 protein demonstrating the four extracellular domains (I-IV), the transmembrane domain (TM), and the tyrosine kinase domain. The C-terminus is responsible for transactivation and effector protein binding, leading to signaling. The A1337T variant (red) is located at the far C-terminal end of the protein. Other frequent somatic mutations are noted in black.
Whole exome sequencing of the proband identified a potential pathologic c.4009G>A;p.A1337T variant in the erb-b2 receptor tyrosine kinase 3 (ERBB3) gene. This variant is not found in the Exome Variant Server (NHLBI Exome Sequencing Project [EVS] (6)) or the Catalogue Of Somatic Mutations In Cancer (COSMIC) (7). The Exome Aggregation Consortium (ExAC) identifies the p.A1337T variant in 7/119,654 alleles, indicating it is rare (8). Further analysis revealed the presence of the variant another affected individual (II-03) and in a female who died at the age of 47 of metastatic breast cancer with bone marrow infiltration (IV-03). She inherited the variant from affected individual III-12. One currently unaffected family member, age 62, is heterozygous for the p.A1337T variant (III-14). In total, our analysis confirmed the presence of the ERBB3 p.A1337T variant in two affected individuals and identified two affected individuals as obligate carriers, as well as one asymptomatic carrier. In addition, it confirmed the absence of the variant in eight unaffected individuals (Figure 1A, Table S1). This indicates that the ERBB3 variant co-segregates with disease in this large multigenerational pedigree family, with one individual who is non-penetrant at this time.
The ERBB3 gene is a member of the epidermal growth factor receptor (EGFR) tyrosine kinase family that also includes ERBB2/HER2, and is frequently somatically mutated in several types of solid tumor malignancies (9). Ligand binding produces a conformational change that results in homo- or heterodimerization with other members of the ERBB family, most commonly ERBB2 (10). The C-terminal tyrosine-rich tail is responsible for recruitment and activation of downstream binding partners (11). The ERBB3 p.A1337T variant is located at the far C-terminal end of the protein and is conserved across most vertebrate species (Figure 1B, S1B). The closest known phosphorylation site is at Tyr1328, and the sequence surrounding this tyrosine is conserved (Figure S1C). Because multiple ERBB3 mutations were found to cause increased proliferation and activation of ERBB3 in the mouse pro-B cell line BaF3 (9), we hypothesized that the p.A1337T variant may cause a similar phenotype.
BaF3 stable transfectants overexpressing ERBB3 p.A1337T (Mut) and wild-type (WT) ERBB3 were generated (Figure S2A), together with tranfectants co-expressing WT ERBB2 (Figure S2B). When cells lacking ERBB2 expression were cultured in the presence of interleukin (IL)-3 and Neuregulin (NRG)-1β ligand, cell proliferation was similar (Figure S2C). However, both ERBB2+WT ERBB3 and ERBB2+Mut ERBB3 cells displayed IL-3 independent proliferation when cultured with NRG-1β alone. Interestingly, a small but consistent 20% increase in proliferation was observed in Mut ERBB3 cells compared to WT ERBB3 cells when cultured with IL-3 (Figure 2A).
Figure 2. Functional analysis of ERBB3 p.A1337T.

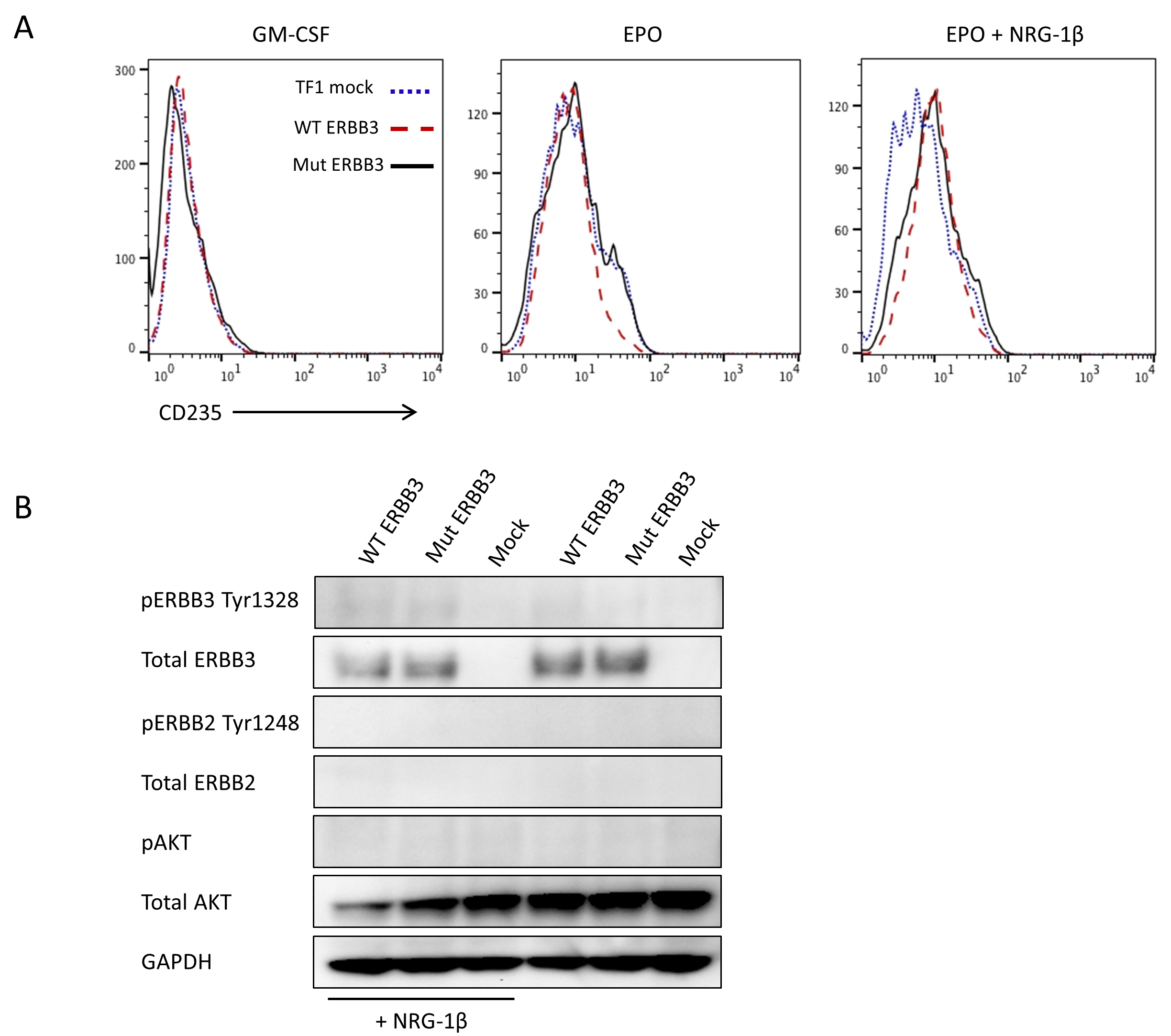
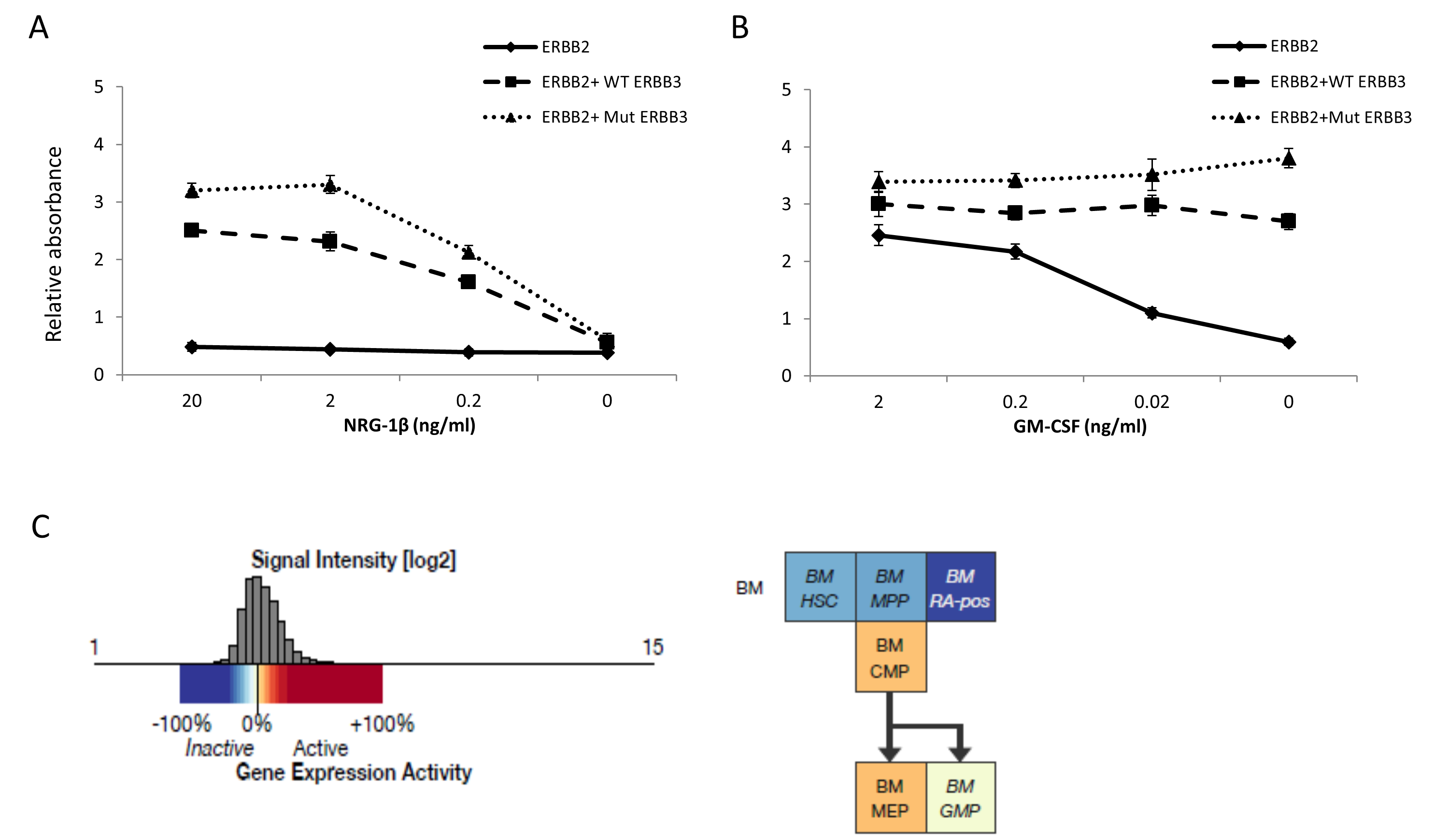
(A) Cell proliferation measured by WST-1 assay of stable BaF3 transfected cells expressing ERBB2 alone or co-expressing WT or Mut ERBB3. In the absence of growth factor (control), all three cell types fail to proliferate, while in the presence of IL-3, cells expressing Mut ERBB3 display a slight growth increase over cells expressing WT ERBB3. (B) Analogous experiment to (A) using TF-1 cells. In the absence of growth factor, or in the presence of GM-CSF or EPO, proliferation was not significantly different. The addition of NRG-1β, either with or without GM-CSF, led to a small but consistent proliferation advantage in ERBB2+Mut ERBB3 cells compared to ERBB2+WT ERBB3 cells. This effect was significantly pronounced in the presence of EPO and NRG-1β. (C). Expression of CD235 is upregulated in the presence of EPO (center) compared to GM-CSF (left) CD235 expression in all cell types. The addition of NRG-1β (right) results in a reduction of CD235 expression in ERBB2+Mut ERBB3 TF-1 cells. (D) When ERBB2 is co-expressed in the absence of NRG-1β, low levels of pERBB3 are detected, however no downstream activation is observed. In the presence of ERBB2 and NRG-1β, significant expression of pERBB2, pERBB3 and pAKT is observed, with an increase in cells expressing Mut ERBB3 compared to WT. GM-CSF, granulocyte macrophage colony stimulating factor; EPO, erythropoietin; IL-3, interleukin-3; NRG-1β, neuregulin-1beta, * p≤0.01, ** p≤0.001
We then sought to investigate the ERBB3 p.A1337T variant in cells more relevant to the disease phenotype by using the human hematopoietic progenitor cell line TF-1 (derived from an individual with erythroleukemia). Cell lines analogous to those described in BaF3 cells were generated and cultured in the presence of GM-CSF or EPO, as well as NRG-1β (Figure S3A, B). Again, TF-1 cells lacking ERBB2 expression behaved similar in all culture conditions (Figure S3C). Further, in the absence of NRG-1β, cells expressing ERBB2 alone or in combination with ERBB3 displayed similar proliferation (Figure 2B). However, when cultured with NRG-1β, ERBB2+WT ERBB3 and ERBB2+Mut ERBB3 cells displayed GM-CSF independent growth, and ERBB2+Mut ERBB3 cells produced a non-statistically significant growth advantage over ERBB2+WT ERBB3 cells (Figure 2B, S4B). This growth advantage was also observed in the presence of GM-CSF and was dependent on the dose of NRG-1β (Figure S4A). When cultured in EPO and NRG-1β, ERBB2+Mut ERBB3 cells displayed a two-fold growth advantage over ERBB2+WT ERBB3 cells (Figure 2B). These data indicate that the ERBB3 p.A1337T variant confers a proliferation advantage over WT ERBB3 in the presence of EPO that appears dependent on ligand binding and heterodimerization.
Cell cycle analysis revealed that in the absence of NRG-1β, all three TF-1 cell types (ERBB2 alone, ERBB2+WT ERBB3, ERBB2+Mut ERBB3) displayed similar fractions of cells in G1, S and G2/M phases (Figure S5A). However, when stimulated by NRG-1β, ERBB2+Mut ERBB3 cells displayed a decrease in the G1 phase and an increase the in S and G2/M phases compared to ERBB2+WT ERBB3 cells (Figure S5B). This suggests that the ERBB3 p.A1337T variant overcomes a G1 phase cell cycle block that results in increased proliferation.
Because of the consistent erythroid phenotype in this family, we hypothesized that Mut ERBB3 TF-1 cells may also possess an erythroid differentiation defect. After culture in EPO for 48-hours, TF-1 cells lacking ERBB2 expression displayed increased expression of the red cell marker Glycophorin A (CD235), while the addition of NRG-1β had minimal effect (Figure S6A). Similarly, ERBB2 and ERBB2+WT or Mut ERBB3 also displayed increased CD235 expression when cultured in EPO (Figure 2C). The addition of NRG-1β had no effect on ERBB2 expressing cells, while ERBB2+WT ERBB3 cells displayed partial loss of CD235 expression. However, ERBB2+Mut ERBB3 cells showed near complete loss of CD235 expression, close to the level seen when cultured in GM-CSF, indicating a block in erythroid differentiation (Figure 2C). This defect was associated with augmented ERBB3 signaling, evidenced by increased expression of phosphorylated ERBB3 (pERBB3), pERBB2, and pAKT in ERBB2+Mut ERBB3 compared to ERBB2+WT ERBB3 cells in the presence of EPO (Figure 2D).
The known genes that predispose to hematologic malignancies are frequently somatically mutated in acquired disease. Somatic mutations in the coding region of ERBB3 have been reported rare cases of acute lymphoblastic leukemia, mixed Langerhans cell histiocytosis, multiple myeloma, and diffuse large B cell lymphoma (7). Currently, there is one case of AML reported to have a variant of unknown origin in the C-terminus of ERBB3 (Table S4). Further, hematopoietic expression of ERBB3 appears to be highest in bone marrow common myeloid and megakaryocyte-erythrocyte progenitors (Figure S4B) (12), and analysis of The Cancer Genome Atlas data reveals increased expression of ERBB3 in 4% of AML samples (13).
In summary, we have identified ERBB3 as a candidate gene for predisposition to erythroid MDS/AML. Due to the limited number of available samples in this study, a definitive causal relationship will require identification of additional families. It is of interest to note that the avian erythroblastosis virus (which contained ERBB1) was unable to transform non-erythroid hematopoietic cells, and the C-terminal tail was found to be critical for its erythroblast transforming property (14,15). Consistent with this, the mutation found in this study is located in the far C-terminal end of ERBB3, while the most frequent somatic mutations found in non-hematologic malignancies lie in the N-terminus (Figure 1B). It is possible that N-terminal mutations produce ligand independent activation with more oncogenic potential, while C-terminal mutations only produce a phenotype under the certain conditions (similar to our TF-1 cell model). In addition, while multiple members of this family were diagnosed with other malignancies prior to the onset of MDS or AML (Table S1), it was not possible to draw any conclusions regarding the influence of this germline ERBB3 variant on solid tumor malignancy risk. However, the prevalence of the p.A1337T variant in the ExAC database could explained by an increased risk of non-hematologic malignancies, as well as incomplete penetrance.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank all family members for their participation in this research study. We thank Zhaohui Ye for helpful discussions. This work was supported by NHLBI K12HL087169 (EMB) and by NHGRI 1U54HG006542 to the Baylor-Hopkins Center for Mendelian Genomics. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Sanger sequencing was conducted at the Genetic Resources Core Facility, Johns Hopkins Institute of Genetic Medicine, Baltimore, MD.
Footnotes
Supplementary information accompanies this paper on the Leukemia website.
The authors have no conflicts of interest to disclose.
Authorship Contributions
E.M.B., R.A.B and L.C. conceived the project and designed the experiments. E.M.B. and R.L. performed the experiments. K.D., B.M. and K.H. performed exome sequencing and sequence quality control. E.M.B., R.L., N.S., D.V., L.C. and R.A.B. analyzed the data. E.M.B., N.S., D.V and C.G. identified study subjects, performed clinical phenotyping and contributed biological samples. E.M.B. wrote the manuscript. R.A.B and L.C. supervised the research. All authors approved the final manuscript.
References
- 1.Babushok DV, Bessler M. Genetic predisposition syndromes: when should they be considered in the work-up of MDS? Best Pract Res Clin Haematol. 2015 Mar;28(1):55–68. doi: 10.1016/j.beha.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santos FP, Bueso-Ramos CE, Ravandi F. Acute erythroleukemia: diagnosis and management. Expert review of hematology. 2010 Dec;3(6):705–718. doi: 10.1586/ehm.10.62. [DOI] [PubMed] [Google Scholar]
- 3.Nissenblatt MJ, Bias W, Borgaonkar D, Dixon S, Cody RP. Familial erythroleukemia: four cases of the Diguglielmo syndrome in close relatives. The Johns Hopkins medical journal. 1982 Jan;150(1):1–9. [PubMed] [Google Scholar]
- 4.Lee EJ, Schiffer CA, Misawa S, Testa JR. Clinical and cytogenetic features of familial erythroleukaemia. British journal of haematology. 1987 Mar;65(3):313–320. doi: 10.1111/j.1365-2141.1987.tb06859.x. [DOI] [PubMed] [Google Scholar]
- 5.Churpek JE, Pyrtel K, Kanchi KL, Shao J, Koboldt D, Miller CA, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood. 2015 Nov 26;126(22):2484–2490. doi: 10.1182/blood-2015-04-641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Exome Variant Server. Seattle, WA: NHLBI GO Exome Sequencing Project (ESP); 2015. [Accessed November 2015]. Available at: http://evs.gs.washington.edu/EVS/ [Google Scholar]
- 7.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015 Jan;43(Database issue):D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, et al. Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv 2015 Cold Spring Harbor Labs Journals. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013 May 13;23(5):603–617. doi: 10.1016/j.ccr.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 10.Steinkamp MP, Low-Nam ST, Yang S, Lidke KA, Lidke DS, Wilson BS. erbB3 is an active tyrosine kinase capable of homo- and heterointeractions. Mol Cell Biol. 2014 Mar;34(6):965–977. doi: 10.1128/MCB.01605-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sithanandam G, Anderson LM. The ERBB3 receptor in cancer and cancer gene therapy. Cancer Gene Ther. 2008 Jul;15(7):413–448. doi: 10.1038/cgt.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seita J, Sahoo D, Rossi DJ, Bhattacharya D, Serwold T, Inlay MA, et al. Gene Expression Commons: an open platform for absolute gene expression profiling. PLoS One. 2012;7(7):e40321. doi: 10.1371/journal.pone.0040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012 May;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graf T, Beug H. Role of the v-erbA and v-erbB oncogenes of avian erythroblastosis virus in erythroid cell transformation. Cell. 1983 Aug;34(1):7–9. doi: 10.1016/0092-8674(83)90130-7. [DOI] [PubMed] [Google Scholar]
- 15.Pelley RJ, Maihle NJ, Boerkoel C, Shu HK, Carter TH, Moscovici C, et al. Disease tropism of c-erbB: effects of carboxyl-terminal tyrosine and internal mutations on tissue-specific transformation. Proc Natl Acad Sci U S A. 1989 Sep;86(18):7164–7168. doi: 10.1073/pnas.86.18.7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.