

 This work is licensed under a
This work is licensed under a Summary
Co-secretion of growth hormone (GH) and prolactin (PRL) from a single pituitary adenoma is common. In fact, up to 25% of patients with acromegaly may have PRL co-secretion. The prevalence of acromegaly among patients with a newly diagnosed prolactinoma is unknown. Given the possibility of mixed GH and PRL co-secretion, the current recommendation is to obtain an insulin-like growth factor-1 (IGF-1) in patients with prolactinoma at the initial diagnosis. Long-term follow-up of IGF-1 is not routinely done. Here, we report two cases of well-controlled prolactinoma on dopamine agonists with the development of acromegaly 10–20 years after the initial diagnoses. In both patients, a mixed PRL/GH-cosecreting adenoma was confirmed on the pathology examination after transsphenoidal surgery (TSS). Therefore, periodic routine measurements of IGF-1 should be considered regardless of the duration and biochemical control of prolactinoma.
Learning points:
Acromegaly can develop in patients with well-controlled prolactinoma on dopamine agonists.
The interval between prolactinoma and acromegaly diagnoses can be several decades.
Periodic screening of patients with prolactinoma for growth hormone excess should be considered and can lead to an early diagnosis of acromegaly before the development of complications.
Background
Mammotrophs and somatotrophs originate from common progenitor cells in the anterior pituitary gland (1). Based on studies, up to 25% growth hormone (GH)-producing adenomas cosecrete prolactin (PRL) (2). By contrast, the prevalence of GH secretion among prolactinomas is unknown but thought to be rare. The current literature emphasizes screening for GH excess by checking insulin-like growth factor 1 (IGF-1) in patients with prolactinoma at the time of the initial diagnosis, however, follow-up screening is not generally recommended (3).
Here, we present two patients with well-controlled prolactinoma on dopamine agonists, who each developed clinical and biochemical evidence of acromegaly many years after the initial diagnosis of prolactinoma. Both patients underwent transsphenoidal surgery (TSS), and the pathology was consistent with mixed PRL/GH co-secreting adenoma.
Thus, our cases indicate that in patients with prolactinoma, routine screening for GH excess should be considered even if patients are well controlled with medical treatment and regardless of the duration of the disease.
Case #1
Case presentation
A white female diagnosed in 2001, at the age of 30, with hyperprolactinemia associated with galactorrhea and oligomenorrhea. Her initial serum prolactin level was not available. Head MRI demonstrated a 9 mm pituitary microadenoma. She was started on bromocriptine but did not tolerate it due to nausea and dizziness. She was then referred to an endocrinologist in 2006. Her prolactin level was elevated at 111 ng/mL (2.8–29.2 ng/mL), and IGF-1 was normal for age and gender at 120 ng/mL (58–318 ng/mL). She was treated with cabergoline with resolution of symptoms. Over the following years, she developed hirsutism, weight gain, widening of teeth, heat intolerance, acne and multiple skin tags. Thus, an IGF-1 level was obtained in 2011, which was mildly elevated at 352 ng/mL (58–318 ng/mL) and, consequently, this led to her referral to the pituitary clinic at the University of Rochester. On her initial visit, she confirmed taking cabergoline 0.5 mg twice weekly since 2006. In addition to the previously mentioned symptoms, she also complained of intermittent headaches, vision problems, arthralgia and polyuria, although she did not notice any change in her ring or shoe size. On examination, her blood pressure was 130/70 mm/Hg, pulse 80/min, weight 214 pounds and body mass index 34.0. She had hirsutism and wide spaced teeth without other clinical features of acromegaly.
Investigation
Laboratory investigation revealed that her IGF-1 level was 396 ng/mL (58–318 ng/mL), prolactin level 6.7 ng/mL (2.8–29.2 ng/mL), TSH 1.77 μIU/mL (0.35–5.50 μIU/mL) and free T4 of 1.2 ng/dL (0.9–1.8 ng/dL). She underwent a 75 g glucose tolerance test, which showed inadequate suppression with GH level of 2.35 ng/mL at 2 h, thus confirming the diagnosis of acromegaly. GH was measured by immunoenzymatic assay. Head MRI demonstrated a 6 mm pituitary mass which has decreased in size compared with previous imaging (Fig. 1).
Figure 1.
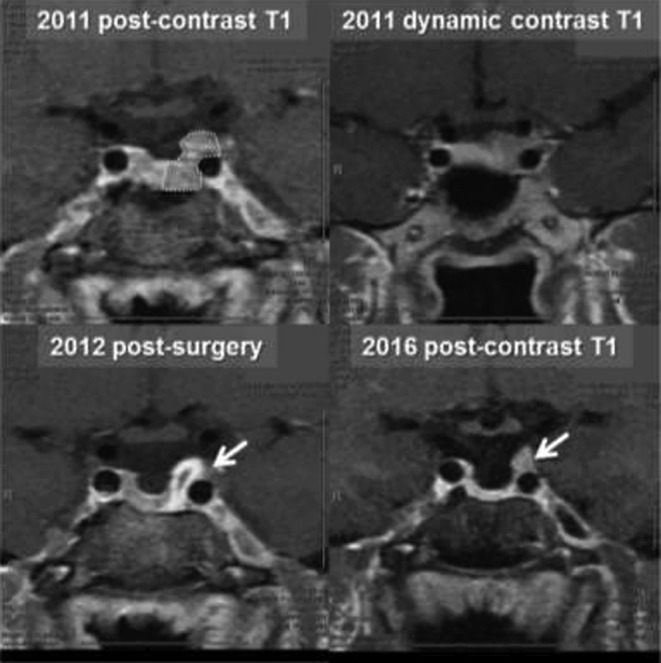
Case #1. An MRI scan before and after the surgery. Dotted area is an original adenoma. Arrows point to a possible area of residual tumor in the left cavernous sinus.
Treatment
She underwent TSS in January 2012. Microscopic examination of the resected tissue was consistent with pituitary adenoma. Immunohistochemical stains for pituitary hormone markers confirmed prolactin and focal GH expression (Fig. 2). Immunohistochemistry for ACTH, FSH, LH and TSH showed no significant immunoreactivity.
Figure 2.
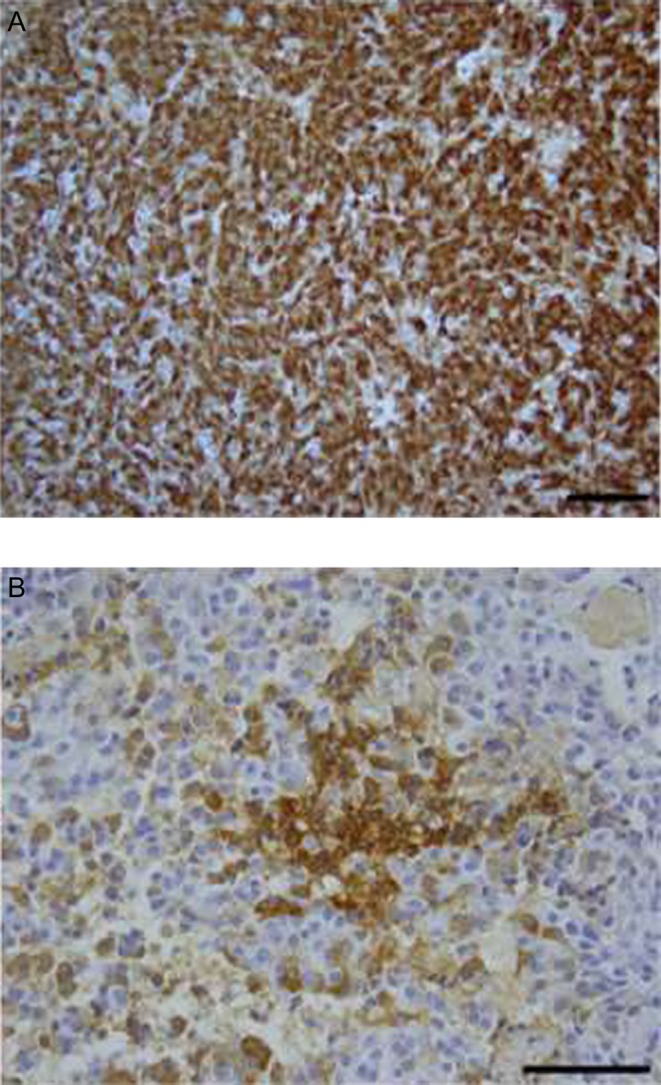
Case #1. Immuno-histochemical stains for hormone markers showed strong prolactin expression (Panel A, magnification 200×), but focal reactivity for growth hormone (Panel B, magnification 400×). Scale bar is 100 µm.
Outcome and follow-up
Post-operative MRI showed a possible residual adenoma and a newly developed partial empty sella syndrome (Fig. 1). Both PRL and IGF-1 levels initially normalized without cabergoline use (Fig. 3A and B). During follow-up, her IGF-1 stayed in a normal range with the most recent values of 208 ng/mL in 2016 (118–298 ng/mL). However, her prolactin level, which had initially normalized, began to slowly increase to the most recent level of 103 ng/mL (4.8–23.3 ng/mL) in 2016 (Fig. 3B). At the most recent visit in 2016, she complained of irregular menses and expressive galactorrhea. We speculate that hyperprolactinemia is due to an enlarging residual lesion and she may need to restart dopamine agonists.
Figure 3.

Case #1. (A) IGF-1 levels over time. The latest normal range is 118–298 ng/mL. (B) Prolactin levels over time. Latest normal level is 4.8–23.3 ng/mL. Gray dotted lines represent normal range for the used assay and age of the patient. TSS, transsphenoidal surgery.
Case #2
Case presentation
An African American female presented to an endocrinologist in 1989 at the age of 30 with galactorrhea and a serum prolactin level of 47.4 ng/mL (2.8–29.2 ng/mL). Medication-induced hyperprolactinemia and hypothyroidism were excluded. Initial head CT demonstrated a 2 mm pituitary microadenoma and a partial empty sella. Elevated prolactin was thought to be due to either an empty sella with pituitary stalk disruption or due to microprolactinoma. Given disturbing galactorrhea, she was started on bromocriptine 5 mg daily. This led to normalization of her prolactin level and resolution of her symptoms. She was not screened for acromegaly at the time of diagnosis. Her initial IGF-1 level in 1998 was within normal range, and she had neither signs nor symptoms of acromegaly. Head MRI in 1994 confirmed a stable pituitary microadenoma. Over time, she developed impaired glucose tolerance, hypertension, arthritis and carpal tunnel syndrome. In 2006, her IGF-1 level was in the upper end of normal range at 283 ng/mL (54–307 ng/mL). Over the next 3 years, her IGF-1 slowly rose to above the normal range (Fig. 4A) and in 2009, she was referred to the pituitary clinic at the University of Rochester. At that time, she confirmed taking bromocriptine for the past 20 years. She denied any headaches, vision problems or galactorrhea. The patient had regular menses. On examination, her blood pressure was 138/72 mm/Hg with pulse 80/min, weight 260 pounds and BMI 42.0. She had broadening of her nose, coarsening of facial features and widened incisor spaces but no prognathism, macroglossia, or changes in extremities. There were no skin tags.
Figure 4.

Case #2. (A) IGF-1 levels over time. The latest normal range is 53–287 ng/mL. (B) Prolactin levels over time. Latest normal level is 4.8–23.3 ng/mL. Gray dotted lines represent normal range for the used assay and age of the patient. TSS, transsphenoidal surgery.
Investigation
Laboratory investigation revealed IGF-1 level of 471 ng/mL (94–252 ng/mL), prolactin level 23.3 ng/mL (2.8–29.2 ng/mL), TSH 0.77 μIU/mL (0.35–5.50 μIU/mL) and free T4 of 1.0 ng/dL (0.9–1.8 ng/dL). Her 75 g glucose challenge test showed inadequate suppression with GH level of 2.5 ng/mL at 2 h, thus confirming the diagnosis of acromegaly. GH was measured by electrochemiluminescence assay. Head MRI demonstrated a 5 mm pituitary mass, which has increased in size compared with prior imaging and an empty sella (Fig. 5).
Figure 5.

Case #2. An MRI scan before and after the surgery. Arrows point to an adenoma and the optic nerves.
Treatment
She underwent TSS in 2009 and had normalization of her IGF-1 level. Microscopic examination of the resected tissue was consistent with pituitary adenoma. Immunohistochemical stains for the pituitary hormone markers showed diffuse reactivity for growth hormone and strong, but patchy, prolactin expression (Fig. 6). Immunohistochemistry for ACTH, FSH, LH and TSH showed no significant immunoreactivity.
Figure 6.
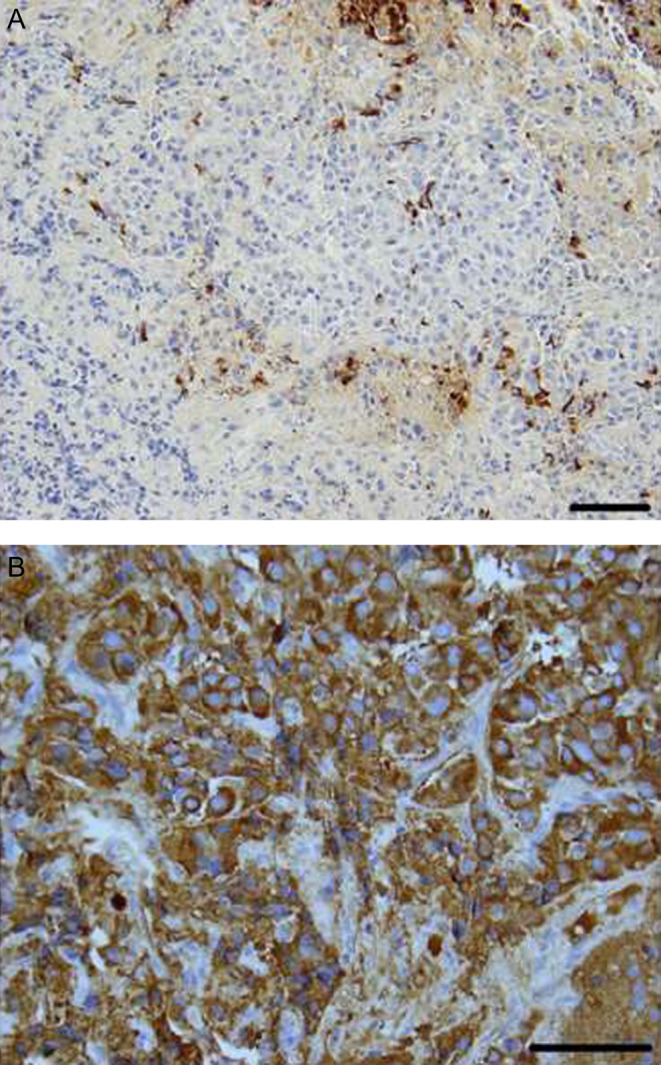
Case #2. Immunohistochemical stains for the pituitary hormone markers showed patchy prolactin expression (Panel A, magnification 200×) and diffuse reactivity for growth hormone (Panel B, magnification 400×). Scale bar is 100 µm.
Outcome and follow-up
After surgery, bromocriptine was discontinued and her latest prolactin and IGF-1 levels remained normal (Fig. 4A and B).
Discussion
Here, we report late development of acromegaly in two patients with well-controlled prolactin-secreting adenomas for 10–20 years.
The first case describes a woman with a well-established diagnosis of prolactinoma based on the presence of pituitary microadenoma, prolactin level >100 ng/mL, and an adequate response to cabergoline treatment. She was diagnosed with acromegaly 9 years later. Pathology confirmed the diagnosis of a mixed GH/PRL lesion. She had biochemical normalization of prolactin and IGF-1 levels after surgery.
A second case describes a woman with mild prolactin elevation in the settings of the pituitary microadenoma and an empty sella syndrome. Twenty years later, she was diagnosed with acromegaly. Her pathology also confirmed a mixed GH/PRL-secreting pituitary lesion. After surgery both IGF-1 and prolactin levels normalized.
The pituitary gland develops from the oral epithelia. Somatotrophes and lactotrophes originate from common progenitor cells under regulation of Pit1 transcription factor (1). Up to 25% of growth hormone adenomas cosecrete prolactin (2, 4). In the majority of cases, this is due to mixed adenomas originating from somatotrophs and lactotrophes. These adenomas are bimorphous and have two separate cell types on the immunohistochemistry staining (5). However, in contrast to our cases, patients are typically diagnosed with acromegaly first and may not even have symptoms of elevated prolactin at the time of diagnosis.
A second type of dual GH/PRL-secreting adenomas is an aggregation of monomorphous mammosomatotroph cells with both GH and PRL hormones secreted from a single cell. It is found in approximately 8–10% of GH-secreting tumors. These patients come to medical attention due to the development of acromegaly and typically do not have symptoms of hyperprolactinemia at presentation (5).
A third type of GH/PRL co-secreting adenomas is primitive acidophilic stem cell adenomas with dual hormone secretion from a single cell. This subtype is very rare and described only in case reports. In contrast to other types, patients often present with symptoms of prolactin excess, and shortly after, develop features of acromegaly. Acidophilic stem cell adenomas usually have a very aggressive clinical course and require multiple surgical interventions or adjuvant therapy within a short period of time (6). Lastly, there is a case report of aggressive prolactinoma that transformed into growth hormone-secreting adenoma presumably due to GNAS gene mutation (7).
As discussed, the percentage of GH/PRL-secreting lesions among prolactinomas is unknown. In a series of 48 patients with galactorrhea, hyperprolactinemia and the presence of pituitary adenoma, eight patients (17%) had an increased GH level and clinical acromegaly (8). Given the possibility of GH/PRL co-secretion, the current guidelines recommend screening all patients with hyperprolactinemia for GH excess at the time of diagnosis (3). However, there are no recommendations for routine IGF-1 measurements in remote follow-up.
Two studies describe the development of acromegaly in patients with well-controlled prolactinoma and initial normal screening for GH excess (9, 10). Anderson et al. (10) routinely obtained IGF-1 levels on 78 patients with prolactinoma. It is not specified if the patients were diagnosed with prolactinoma upon entering the study or previously. Three out of 78 patients developed acromegaly during the 5-year study period. In addition, 5 out of 78 patients had isolated IGF-1 elevation with a normal glucose suppression test. It is unclear if the isolated IGF-1 elevation is an early stage of acromegaly or if it is a normal variant.
Rosario et al. (9) performed a cross-sectional study of 121 patients with diagnosis of prolactinoma. All patients were lacking symptoms of acromegaly. Patients received dopamine agonists with appropriate response for up to 5 years before the study. Five patients were diagnosed with acromegaly. Two of them underwent pituitary surgery and had mixed GH/PRL adenomas. The interval between prolactinoma diagnosis and biochemical evidence of GH excess was not reported in this study. Taken together, both studies (9, 10) suggest that acromegaly can develop in about 4% of the prolactinoma patients during long-term follow-up. The interval from the diagnosis of prolactinoma to the development of acromegaly can be up to 20 years (our “case #2” section).
Acromegaly is a rare disease with an incidence rate of 3–4 cases per one million, and a mortality rate 2–3 times higher, compared with the general population (4). Despite improved diagnostic tests, acromegaly remains to be under-diagnosed. Most of the diagnoses occur when patients present with prominent facial features and co-morbidities of acromegaly, some of which are irreversible. As a result, it is important to screen patients with prolactinoma for GH excess periodically.
Conclusion
Despite diagnostic advances and more sensitive GH assays, acromegaly remains under-diagnosed. There are no recommendations for screening prolactinoma patients with IGF-1 in remote follow-up. Based on published studies, up to 4% of well-controlled prolactinoma patients can develop acromegaly. The interval between the two diagnoses can be up to 20 years. Therefore, based on the presented cases and literature review, periodic measurement of IGF-1 should be considered. More studies are needed to determine the optimal interval and duration of screening.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
The authors confirm that written informed consent was obtained from the patients for publication of the submitted article and accompanying images.
Author contribution statement
E Manuylova performed the review of cases and wrote the first draft. I Shafiq reviewed the draft and made corrections. Drs. Calvi, Vates, Cave and Catherine Hastings participated in the patients’ care. Dr Johnson prepared pathological data.
Acknowledgements
The authors would like to thank Barbara Morabito for her help in preparation of the manuscript.
References
- 1.Dasen JS, Rosenfeld MG. 2001. Signaling and transcriptional mechanisms in pituitary development. Annual Review of Neuroscience 24 327–355. () [DOI] [PubMed] [Google Scholar]
- 2.Wang M, Mou C, Jiang M, Han L, Fan S, Huan C, Qu X, Han T, Qu Y, Xu G. 2012. The characteristics of acromegalic patients with hyperprolactinemia and the differences in patients with merely GHsecreting adenomas: clinical analysis of 279 cases. European Journal of Endocrinology 166 797–802. () [DOI] [PubMed] [Google Scholar]
- 3.Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, Wass JA, Endocrine Society 2011. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism 96 273–288. ( 10.1210/jc.2010-1692) [DOI] [PubMed] [Google Scholar]
- 4.Melmed S. 2006. Medical progress: acromegaly. New England Journal of Medicine 355 2558–2573. ( 10.1056/NEJMra062453) [DOI] [PubMed] [Google Scholar]
- 5.Lopes MB. 2010. Growth hormone-secreting adenomas: pathology and cell biology. Neurosurgical Focus 29 E2 () [DOI] [PubMed] [Google Scholar]
- 6.Horvath E, Kovacs K, Singer W, Smyth HS, Killinger DW, Erzin C, Weiss MH. 1981. Acidophil stem cell adenoma of the human pituitary: clinicopathologic analysis of 15 cases. Cancer 47 761–771. () [DOI] [PubMed] [Google Scholar]
- 7.Lania AG, Ferrero S, Pivonello R, Mantovani G, Peverelli E, Di Sarno A, Beck-Peccoz P, Spada A, Colao A. 2010. Evolution of an aggressive prolactinoma into a growth hormone secreting pituitary tumor coincident with GNAS gene mutation. Journal of Clinical Endocrinology and Metabolism 95 13–17. ( 10.1210/jc.2009-1360) [DOI] [PubMed] [Google Scholar]
- 8.Kleinberg DL, Noel GL, Frantz AG. 1977. Galactorrhea: a study of 235 cases, including 48 with pituitary tumors. New England Journal of Medicine 296 589–600. ( 10.1056/NEJM197703172961103) [DOI] [PubMed] [Google Scholar]
- 9.Rosario PW, Purisch S. 2010. Biochemical acromegaly in patients with prolactinoma during treatment with dopaminergic agonists. Arquivos Brasileiros de Endocrinologia and Metabologia 54 546–549. ( 10.1590/S0004-27302010000600006) [DOI] [PubMed] [Google Scholar]
- 10.Andersen M, Hagen C, Frystyk J, Schroeder HD, Hagen C. 2003. Development of acromegaly in patients with prolactinomas. European Journal of Endocrinology 149 17–22. ( 10.1530/eje.0.1490017) [DOI] [PubMed] [Google Scholar]