

Abstract
Background
As cancer progresses, methylation patterns change to promote the tumorigenic phenotype. However, stability of methylation markers over time and the extent that biopsy samples are representative of larger tumor specimens are unknown. This information is critical for clinical use of such biomarkers.
Methods
Ninety-eight patients with tumor specimens from two time points were measured for DNA methylation in the promoter regions across four genes.
Results
There were no significant differences in overall methylation of CCNA1, DCC, CD1A or NDN within paired specimens (p-values= 0.56, 0.17, 0.66 and 0.58, respectively). All genes showed strong correlations between paired specimens across time. Methylation was most consistent for CCNA1 and NDN over time.
Conclusions
This report provides the first evidence that methylation markers measured in biopsy samples are representative of gene methylation in later specimens and suggests that biopsy markers could be representative biomarkers for use in defining personalized treatment utilizing epigenetic changes.
Keywords: DNA methylation, head and neck cancer, stability, time, tumor
Introduction
There is a growing body of literature showing associations between molecular markers and head and neck cancer. These markers are being developed as potential clinical tools to direct treatment, to identify low-risk patients that may benefit from less harsh treatments and to predict prognosis. The use of epigenetic markers is a promising tool in this regard. These markers do not change the sequence of DNA, may be reversible and are indicative of tumor biology(1). Specifically, variation in DNA methylation is one of the hallmark processes of cancer and potentially, these markers might be used as therapeutic targets alone, or to select patients for more effective therapy. For example, gene promoter hypermethylation of the DNA repair gene MGMT is a prognostic marker for glioma patients and is currently being evaluated as marker for patient selection for with carmustine and temozolomide in clinical trials(1, 2). Methylation of the mismatch repair gene, hMLH1, was found to significantly increase upon relapse of epithelial ovarian cancer patients and was associated with poor survival(3). Hypermethylation of a DNA helicase gene involved in DNA replication, recombination and DNA repair, WRN, increases sensitivity of colorectal tumors to topoisomerase inhibitors. Combined therapy with DNA damaging agents showed significantly better prognosis in patients with hypermethylated WRN than in patients with unmethylated WRN(4). Such markers offer high translatability into the clinical setting and can allow for personalized therapy with high efficacy depending upon the methylation profile of a patient’s tumor.
An inherent limitation of incorporating methylation markers clinically is that the persistence of methylation in a tumor is unknown. As cancer progresses, methylation patterns can change to promote the tumor phenotype(5, 6). Further, methylation of specific genes could differ significantly depending on timing and site of tumor sampling. However, methylation markers that are known to persist over time may potentially be used to direct treatment. Whether biopsy specimens would be representative of samples obtained at surgical resection is particularly important in head and neck cancer where non-surgical primary treatment is becoming more common. This report addresses this important limitation and provides evidence that tumor biopsy specimens can be used to promote the development of epigenetically based treatments for cancer in a clinical setting. Here, we measure the methylation of four genes across time: CCNA1 (cyclin A1), NDN (necdin), DCC (deleted in colorectal carcinoma) and CD1a (cluster of differentiation 1a). A discovery-based study previously published by our group, was designed to identify novel prognostic epigenetic biomarkers for patients with HNSCC(7, 8). CCNA1 (cyclin A1) was found to be differentially methylated by HPV status(7). NDN (necdin) and CD1a (cluster of differentiation 1a) were also differentially methylated in this discovery analysis, however they were not significant, potentially due to small sample size. NDN is an imprinted gene previously implicated in epithelial ovarian, bladder, breast, colorectal, and urothelial cancers, as well as premalignant lesions such as vulval intraepithelial neoplasia and Barrett’s oesophagus, although has not been studied in the context of HNSCC(7–14). CD1A was the first immune gene found to be differentially methylated in the discovery analysis. CD1A methylation has not been previously studied in HNSCC, however significant hypermethylation of CD1B, CD1C, CD1D and CD1E has been found in HPV (+) HNSCC tumors compared to HPV(−) tumors (15). DCC (deleted in colorectal carcinoma), GADD45 (growth arrest and DNA damage 45) and p16 (cyclin-dependent kinase inhibitor) were previously found to be hypermethylated in HNSCC and were chosen for their role as tumor suppressors and potential involvement with HPV(16–20). Previous literature on the importance of these genes in HNSCC highlights their potential clinical relevance. However, validation of their methylation stability across time is critical in determining the clinical utility of these epigenetic biomarkers.
Materials and Methods
Study Population
This study takes advantage of an established cohort of head and neck cancer patients from the University of Michigan’s Head and Neck Cancer Specialized Program of Research Excellence (UM HN SPORE). Details on the cohort can be found in a separate study(21). Eligible subjects were biopsied pretreatment and diagnosed with head and neck squamous cell carcinoma at an outside hospital (OSH) before referral to the University of Michigan (UM) for treatment. Upon presentation at UM, patients may be rebiopsied and staged during treatment planning. Ninety-eight subjects that signed a written, informed consent, had both a formalin-fixed paraffin-embedded (FFPE) biopsy specimen from an OSH and a surgical resection (n=70) or biopsy (n=28) specimen from UM at a second time point available for microdissection and methylation analysis. Histology was confirmed on all samples by a qualified pathologist (JM). Areas of >70% tumor cellularity were specified for use in microdissection. Subjects completed an epidemiological questionnaire of behavioral and pathophysiological information. This study was approved as being within the ethical standards of the Institutional Review Board of the University of Michigan.
Microdissection/DNA Extraction/Bisulfite Conversion/HPV testing
Designated areas of FFPE tissue were microdissected from unstained slides and DNA was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. DNA concentration was measured with a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA, USA). Sodium bisulfite treatment was performed on 250ng of DNA using the Epitect Bisulfite Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s recommended protocol. HPV status was determined by an ultrasensitive method using real-time competitive polymerase chain reaction (PCR) and matrix-assisted laser desorption/ionization mass spectroscopy as described and validated previously, due to its low DNA input requirement and rapid identification of HPV types, with high sensitivity and specificity(18, 22–25).
Methylation Analysis
Methylation assays for promoter regions of DCC, CD1A, and NDN, were designed using PyroMark Assay Design 2.0 software and conducted via pyrosequencing across 5 , 2, and 3 CpG sites, respectively (Qiagen, Valencia, CA, USA). The promoter region of CCNA1 was sequenced across 4 CpG sites using the Sequenom EpiTyper, a MALDI-TOF mass spectrometry based platform, due to its CpG-dense promoter region and subsequent difficulty in using pyrosequencing methodology. These assays were designed to cover CpG sites at or near the CpG sites found in our previous study to be prognostic indicators of head and neck squamous cell carcinoma. All primer sets and PCR conditions are listed in Table 1S. Bisulfite singleplex PCR amplification was performed using FastStart Taq Polymerase (Roche Diagnostics, Indianapolis, IN, USA) for CCNA1, and HotStar Taq® Master Mix Kit (Qiagen Valencia, CA, USA) for all other genes, with a forward and reverse primer concentration of 0.2 mM and 30ng of bisulfite-converted DNA. Fifteen microliters of each PCR product was combined with the respective sequencing primer and methylation analysis by pyrosequencing was conducted using the Pyromark™ MD System (Biotage, Charlotte, NC, USA) according to manufacturer’s protocol, including single strand binding protein (PyroGold reagents). Measurement of all samples for every methylation marker selected was not possible if there was insufficient quantity of total extracted DNA.
Statistical Analysis
Methylation values were calculated as means across all CpG sites of each gene. Locations of each CpG site and distance to transcription start site are listed in Table 2S. Site-specific and mean methylation from matched tissue specimens across time for CCNA1, DCC, and CD1A were compared using a non-parametric Wilcoxon-signed rank test due to skewed distributions. Methylation values for NDN were compared using a paired t-test due to its Gaussian distribution. Pearson (NDN) and Spearman (CCNA1, DCC, and CD1A) correlation coefficients and 95% confidence intervals (CIs) were calculated for methylation across both time points. The difference in methylation between time points was calculated for each gene and the differences and their absolute values were tested for correlation with the number of days between specimens. Correlation coefficients were also calculated subsetting by HPV status, smoking status, days between time points and specimen type of second sample. Differences in the amount of change in methylation values across subsets were tested using Wald tests from linear regression models and a correction for false discovery was applied to the p-values to adjust for multiple comparisons of the various subgroup tests using q-values described by Storey et al.(26) Multivariable analyses was conducted separately for each gene using a linear model to measure the association of days between sample collection and methylation differences, adjusting for HPV status, age, site, stage and comorbidity status. Comorbidity data were abstracted from the medical record and graded by severity (none, mild, moderate, severe) using the Adult Comorbidity Evaluation of 27 conditions organized by 12 systems (ACE-27).
Results
The study population consisted of 98 paired samples with the median time between first and second tumor tissue specimens at 44 days (range: 8–156 days). Approximately 74% of the population was male. Tumor sites were primarily distributed across larynx, oral cavity and oropharynx (16%, 53%, and 29%, respectively) with 2% in the hypopharynx. Most patients were HPV-negative (69%). Only 16% were nonsmokers, while 46% were current smokers, or having quit within the past 12 months, and 38% were former smokers (quit more than one year ago). Mean age was 60 years (SD=13 years). All genes showed a wide range of methylation levels across samples, as expected of epigenetic markers. There were no significant differences in overall methylation within paired specimens of CCNA1, DCC, and CD1A or NDN (p-values = 0.56, 0.17, 0.66 and 0.58, respectively; Table 1). The lack of significant differences in methylation across time persisted even when considering site-specific methylation within each gene (Figure 1). Patterns of methylation across CpG sites within each gene were similar for both OSH and MI samples, justifying the use of mean methylation across CpG sites as an appropriate measure to compare methylation across time.
Table 1.
Percent Methylation Distribution for Paired Samples
| Gene | Number of patients | Initial Biopsy | Re-Biopsy/Surgery | Differenceb | p-valuec |
|---|---|---|---|---|---|
| CCNA1a | 86 | 23.5 (4.5, 78) | 24.8 (6.3, 67.3) | 0.9 (−31.3, 53.3) | 0.56 |
| CD1Aa | 94 | 69.2 (21.8,95.9) | 69.1 (21.4, 91.1) | −0.2 (−28.2, 36.3) | 0.66 |
| DCCa | 96 | 33.1 (5.7,91.2) | 32.2 (3.9, 85.8) | −0.7 (−36.9, 43.1) | 0.17 |
| NDNa | 94 | 42.2 (34.9, 51.2) | 43.0 (36.3, 52.3) | 0.4 (−5.2, 7.7) | 0.58d |
Median(range)
Re-Biopsy or Surgery-Initial Biopsy
p-value for paired test
parametric test
Figure 1.

Site-specific comparison of methylation at both time points. There are no significant differences in methylation at each CpG site for each gene. Methylation of each gene was measured in promoter regions at four sites for CCNA1 (a), 3 sites for NDN (b), five sites for DCC (c) and two sites for CD1A (d). Locations of each site, distance to transcription start sites and assay specifications are available in supplementary material.
All genes showed strong correlations between paired specimens across time. CD1A and DCC had identical correlation coefficients (rho(95% CI) = 0.70(0.58, 0.79) and 0.70(0.58, 0.79), respectively) (Figure 2c, Figure 2d), while CCNA1 and NDN had slightly lower correlations (rho(95% CI) = 0.65 (0.50, 0.75) and 0.65 (0.51, 0.75), respectively) (Figure 2a, Figure 2b). There were no correlations between the differences in methylation between the two time points and the number of days between specimens for any gene (rho(95%CI): CCNA1: −0.04 (−0.25,0.17); NDN: −0.07 (−0.26, 0.13); CD1A: 0.06 (−0.14, 0.26); DCC: −0.08 (−0.28, 0.12)). Additionally, there were no correlations between the absolute values of these differences and the number of days between specimens for any gene ((rho(95%CI): CCNA1: 0.11 (−0.10,0.31); NDN: −0.16 (−0.35, 0.04); CD1A:−0.008 (−0.21, 0.19); DCC: −0.04 (−0.24, 0.16)). Multivariable models run to assess the association of days between samples and methylation difference across time, adjusting for HPV status, age, site, stage and comorbidity status, also showed no significant association between methylation differences and collection times (data not shown). These results demonstrate that methylation at both time points was strongly correlated and did not differ by the number of days between specimens.
Figure 2.
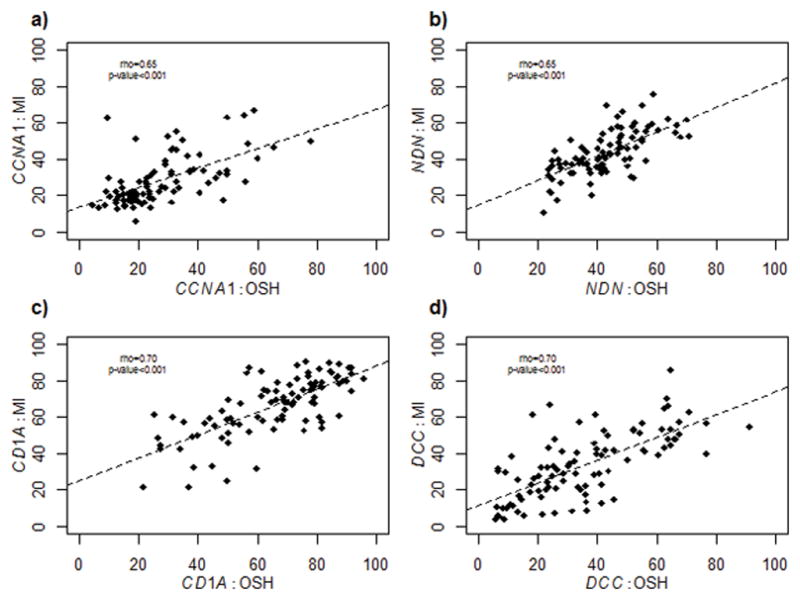
Correlations of each marker between paired specimens across time. CCNA1 (a) and NDN (b) have similar correlation coefficients while CD1A (c) and DCC (d) have similar correlation coefficients.
As temporal changes in methylation levels may be associated with patient and tumor characteristics, correlations were also calculated separately by HPV status, smoking parameters, and whether the second specimen was from a biopsy or surgery resection; correlations were also calculated by the length of time between specimen sampling (Table 2). CD1A was most stable across time in HPV- patients (rho = 0.77, 95% CI = (0.65, 0.85)). Patients who had a biopsy at their second time point showed the most stable methylation at NDN (rho = 0.77, 95% CI = (0.53, 0.89)) whereas patients with a surgery resection specimen at the second time point showed the most stable methylation at CD1A and DCC (rho(95% CI) = 0.74 (0.61, 0.83) and 0.75 (0.62, 0.84), respectively). Patients with shorter times between their tumor samples (0–44 days) showed the most stable methylation at CCNA1 and CD1A (rho (95% CI) = 0.71 (0.52, 0.83) and 0.74 (0.58, 0.85), respectively). Patients who had their second tissue sample beyond 44 days showed the most stable methylation at DCC (rho = 0.72, 95% CI = (0.55, 0.83)). Strong correlations across time were found for CD1A and DCC in former smokers (rho (95% CI) =0.81 (0.65, 0.90) and 0.79 (0.62, 0.88), respectively), CCNA1 in current smokers (rho (95% CI) = 0.74 (0.55, 0.85), and DCC and NDN in never smokers (rho (95% CI) = 0.84 (0.59, 0.94) and 0.74 (0.39, 0.90), respectively). To determine correlations accounting for intensity and duration of smoking, pack-years were also considered, using 20 pack-years as a cutoff(27). Patients with less than 20 pack-years and with 20 pack-years or greater showed the most stable methylation at CD1A (rho (95% CI) =0.78 (0.57, 0.89) and 0.73 (0.57, 0.84) respectively). None of the subset differences we observed proved statistically significant after p-values were corrected for multiple comparisons.
Table 2.
Correlations within subsets of population
| Parameter |
CCNA1
|
CD1A
|
DCC
|
NDN
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nc | rd | 95% CI | Nc | rd | 95% CI | Nc | rd | 95% CI | Nc | rd | 95% CI | |
| Second Specimen Type | ||||||||||||
| Biopsy | 25 | 0.64 | (0.33, 0.83) | 27 | 0.63 | (0.33, 0.81) | 27 | 0.57 | (0.24, 0.78) | 26 | 0.77 | (0.54, 0.89) |
| Surgery | 61 | 0.64 | (0.46, 0.77) | 67 | 0.74 | (0.61, 0.83) | 69 | 0.75 | (0.62, 0.84) | 68 | 0.56 | (0.37, 0.70) |
| HPV Status | ||||||||||||
| HPV+ | 27 | 0.52 | (0.17, 0.75) | 29 | 0.45 | (0.10, 0.70) | 29 | 0.49 | (0.15, 0.73) | 28 | 0.57 | (0.25, 0.78) |
| HPV− | 59 | 0.52 | (0.30, 0.68) | 65 | 0.77 | (0.65, 0.85) | 67 | 0.60 | (0.42, 0.73) | 66 | 0.58 | (0.39, 0.72) |
| Days between specimensa | ||||||||||||
| 0–44 days | 43 | 0.71 | (0.52, 0.83) | 49 | 0.74 | (0.58, 0.85) | 49 | 0.64 | (0.44, 0.78) | 48 | 0.67 | (0.48, 0.80) |
| >44 days | 43 | 0.52 | (0.26, 0.71) | 45 | 0.68 | (0.48, 0.81) | 47 | 0.72 | (0.55, 0.83) | 46 | 0.65 | (0.44, 0.79) |
| Smoking Status | ||||||||||||
| Current | 39 | 0.74 | (0.55, 0.86) | 44 | 0.67 | (0.46, 0.81) | 44 | 0.48 | (0.21, 0.68) | 44 | 0.59 | (0.36, 0.75) |
| Former | 33 | 0.58 | (0.30, 0.77) | 34 | 0.81 | (0.65, 0.90) | 36 | 0.79 | (0.62, 0.88) | 34 | 0.68 | (0.44, 0.83) |
| Never | 14 | 0.64 | (0.17, 0.87) | 16 | 0.49 | (−0.01, 0.79) | 16 | 0.84 | (0.59, 0.94) | 16 | 0.74 | (0.39, 0.90) |
| Pack-yearsb | ||||||||||||
| <20 pack-years | 26 | 0.65 | (0.35, 0.83) | 27 | 0.78 | (0.57, 0.89) | 28 | 0.67 | (0.40, 0.83) | 28 | 0.59 | (0.28, 0.79) |
| ≥20 pack-years | 45 | 0.60 | (0.37, 0.76) | 50 | 0.73 | (0.57, 0.84) | 51 | 0.66 | (0.47, 0.79) | 50 | 0.67 | (0.48, 0.80) |
cutoff based on median.
cutoff based on Gillison, et al. paper(17)
Number of patients
Correlation Coefficient
Probability of stable methylation across time
It is difficult to define methylation cutoffs that are biologically relevant. To compare consistency across time, we determined the proportion of specimens that fell within 10% and 20% of methylation at the first time point. Methylation was most consistent across time for CCNA1 and NDN. Approximately 91% and 96% of patients, respectively, had methylation levels of these markers at the second time point within 20% of methylation at the first time point. CD1A and DCC methylation at the second time point was within 20% of methylation at the first time point for 85% and 79% of the patient population, respectively. This consistency persisted when restricting methylation change to 10%. Approximately 66% and 68% of patients had methylation of CCNA1 and NDN at the second time point within 10% of methylation at the first time point, respectively. The probability of CD1A and DCC methylation at the second time point staying within 10% of the first time point was 60% and 53%, respectively.
Discussion
These findings in head and neck cancer patients demonstrate the stability of DNA methylation changes in tumor specimens from the time of biopsy to time of surgical treatment or second biopsy ranging from 8 to 156 elapsed days. To date, this is the first study to examine changes in methylation of specific genes across time and from different tumor samples within the same patients.
Correlations across time and by patient characteristic were positive and statistically significant, although the strength of correlations differed slightly based on patient characteristics, potentially due to underlying biological mechanisms associated with these genes. For example, we found that methylation of our genes was more strongly correlated across time in HPV- tumors, likely due to that fact that HPV+ tumors tend to have more DNA methylation events in genic regions(8). The strength of correlations was higher in specific genes when considering patient characteristics, indicating that a gene chosen for diagnostic purposes may depend on a patient’s clinical profile.
A limitation of this study is the variability of methylation within each gene. Since the differences observed between paired specimens were uncorrelated with length of time separating the specimens, they are instead likely due to heterogeneity within the tumors, measurement variability in the assay itself, measurements made across mixed cell populations, averages taken across several CpG sites in promoter regions or intra-individual variability in methylation across time. It is important to note that although our biopsies came from a separate institution, the management of the biopsy material is fairly standardized across hospitals. The sample is placed in formalin immediately upon excision and eventually embedded in paraffin. There are many factors that may potentially affect methylation, the most significant being sampling error due to samples being taken from differing locations in the tumor (i.e. periphery for the biopsy and perhaps more central location for the resection). However, because minimal differences were noted in methylation between these two time points and locations, it is unlikely that differing institutions would be a significant variable. In addition, our findings showed no significant differences in paired distributions, relatively strong correlation coefficients as high as 0.84 and high probabilities of stable methylation within patients across time. These findings support the conclusion that when targeting epigenetic changes, alterations in gene methylation after initial biopsy likely reflects biologic changes rather than sampling errors. Additionally, these are issues that are likely to impact any clinical measurement, and thus these results represent a realistic assessment of the persistence of methylation levels.
The amount of methylation change needed to instigate a biological effect is currently unknown. Therefore, it is important that methylation levels remain relatively consistent across time when considered in a clinical setting. Here, we show high probabilities of CCNA1 and NDN methylation to be within 10% and 20% of the first measurement. However, CD1A and DCC methylation had lower probabilities indicating that in the tumor microenvironment, some genes are stably methylated while others are not, presumably to promote the tumorigenic phenotype.
Although stability of methylation of other specific genes could differ, our current findings are significant since these genes have been shown to be important in HNSCC(7, 8). We report CCNA1 and DCC methylation levels similar to previous studies(28–30). CD1A and NDN methylation has not been previously reported. The results of this study provide evidence for the stability over time of specific gene methylation measured in biopsy samples and supports the use of biopsy results as representative of the entire tumor, and as a potential prognostic indicator that could aid in defining personalized treatment.
References
- 1.Olson RA, Brastianos PK, Palma DA. Prognostic and predictive value of epigenetic silencing of MGMT in patients with high grade gliomas: a systematic review and meta-analysis. Journal of neuro-oncology. 2011;105(2):325–35. doi: 10.1007/s11060-011-0594-5. [DOI] [PubMed] [Google Scholar]
- 2.Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. The New England journal of medicine. 2000;343(19):1350–4. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 3.Gifford G, Paul J, Vasey PA, Kaye SB, Brown R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10(13):4420–6. doi: 10.1158/1078-0432.CCR-03-0732. [DOI] [PubMed] [Google Scholar]
- 4.Agrelo R, Cheng WH, Setien F, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(23):8822–7. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith IM, Mydlarz WK, Mithani SK, Califano JA. DNA global hypomethylation in squamous cell head and neck cancer associated with smoking, alcohol consumption and stage. International journal of cancer Journal international du cancer. 2007;121(8):1724–8. doi: 10.1002/ijc.22889. [DOI] [PubMed] [Google Scholar]
- 6.Steinmann K, Sandner A, Schagdarsurengin U, Dammann RH. Frequent promoter hypermethylation of tumor-related genes in head and neck squamous cell carcinoma. Oncology reports. 2009;22(6):1519–26. doi: 10.3892/or_00000596. [DOI] [PubMed] [Google Scholar]
- 7.Colacino JA, Dolinoy DC, Duffy SA, et al. Comprehensive analysis of DNA methylation in head and neck squamous cell carcinoma indicates differences by survival and clinicopathologic characteristics. PloS one. 2013;8(1):e54742. doi: 10.1371/journal.pone.0054742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sartor MA, Dolinoy DC, Jones TR, et al. Genome-wide methylation and expression differences in HPV(+) and HPV(−) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics : official journal of the DNA Methylation Society. 2011;6(6):777–87. doi: 10.4161/epi.6.6.16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3. 0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9(2):166–80. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asai T, Liu Y, Nimer SD. Necdin, a p53 target gene, in normal and cancer stem cells. Oncotarget. 2013 doi: 10.18632/oncotarget.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Faveri LE, Hurst CD, Platt FM, et al. Putative tumour suppressor gene necdin is hypermethylated and mutated in human cancer. British journal of cancer. 2013;108(6):1368–77. doi: 10.1038/bjc.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haviland R, Eschrich S, Bloom G, et al. Necdin, a negative growth regulator, is a novel STAT3 target gene down-regulated in human cancer. PloS one. 2011;6(10):e24923. doi: 10.1371/journal.pone.0024923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(5):778–89. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- 14.Tan AC, Jimeno A, Lin SH, et al. Characterizing DNA methylation patterns in pancreatic cancer genome. Molecular oncology. 2009;3(5–6):425–38. doi: 10.1016/j.molonc.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–82. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butz K, Whitaker N, Denk C, Ullmann A, Geisen C, Hoppe-Seyler F. Induction of the p53-target gene GADD45 in HPV-positive cancer cells. Oncogene. 1999;18(14):2381–6. doi: 10.1038/sj.onc.1202557. [DOI] [PubMed] [Google Scholar]
- 17.Gillison ML. Human papillomavirus and prognosis of oropharyngeal squamous cell carcinoma: implications for clinical research in head and neck cancers. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(36):5623–5. doi: 10.1200/JCO.2006.07.1829. [DOI] [PubMed] [Google Scholar]
- 18.Kumar B, Cordell KG, Lee JS, et al. EGFR, p16, HPV Titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(19):3128–37. doi: 10.1200/JCO.2007.12.7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ying J, Srivastava G, Hsieh WS, et al. The stress-responsive gene GADD45G is a functional tumor suppressor, with its response to environmental stresses frequently disrupted epigenetically in multiple tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(18):6442–9. doi: 10.1158/1078-0432.CCR-05-0267. [DOI] [PubMed] [Google Scholar]
- 20.Langevin SM, Butler RA, Eliot M, et al. Novel DNA methylation targets in oral rinse samples predict survival of patients with oral squamous cell carcinoma. Oral oncology. 2014;50(11):1072–80. doi: 10.1016/j.oraloncology.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arthur AE, Peterson KE, Shen J, et al. Diet and proinflammatory cytokine levels in head and neck squamous cell carcinoma. Cancer. 2014;120(17):2704–12. doi: 10.1002/cncr.28778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Worden FP, Kumar B, Lee JS, et al. Chemoselection as a strategy for organ preservation in advanced oropharynx cancer: response and survival positively associated with HPV16 copy number. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(19):3138–46. doi: 10.1200/JCO.2007.12.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maxwell JH, Kumar B, Feng FY, et al. HPV-positive/p16-positive/EBV-negative nasopharyngeal carcinoma in white North Americans. Head & neck. 2010;32(5):562–7. doi: 10.1002/hed.21216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maxwell JH, Kumar B, Feng FY, et al. Tobacco use in human papillomavirus-positive advanced oropharynx cancer patients related to increased risk of distant metastases and tumor recurrence. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(4):1226–35. doi: 10.1158/1078-0432.CCR-09-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walline HM, Komarck C, McHugh JB, et al. High-risk human papillomavirus detection in oropharyngeal, nasopharyngeal, and oral cavity cancers: comparison of multiple methods. JAMA otolaryngology-- head & neck surgery. 2013;139(12):1320–7. doi: 10.1001/jamaoto.2013.5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Storey JD, Taylor JE, Siegmund D. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. J Roy Stat Soc B. 2004;66:187–205. [Google Scholar]
- 27.Gillison ML, D’Souza G, Westra W, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. Journal of the National Cancer Institute. 2008;100(6):407–20. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- 28.Rettori MM, de Carvalho AC, Longo AL, et al. TIMP3 and CCNA1 hypermethylation in HNSCC is associated with an increased incidence of second primary tumors. Journal of translational medicine. 2013;11:316. doi: 10.1186/1479-5876-11-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiss D, Basel T, Sachse F, Braeuninger A, Rudack C. Promoter methylation of cyclin A1 is associated with human papillomavirus 16 induced head and neck squamous cell carcinoma independently of p53 mutation. Molecular carcinogenesis. 2011;50(9):680–8. doi: 10.1002/mc.20798. [DOI] [PubMed] [Google Scholar]
- 30.Carvalho AL, Chuang A, Jiang WW, et al. Deleted in colorectal cancer is a putative conditional tumor-suppressor gene inactivated by promoter hypermethylation in head and neck squamous cell carcinoma. Cancer research. 2006;66(19):9401–7. doi: 10.1158/0008-5472.CAN-06-1073. [DOI] [PubMed] [Google Scholar]