

Abstract
Inhibitors of poly(ADP-ribose) polymerase (PARP) are clinically used as single-agent therapy for tumors with BRCA1 or BRCA2 mutations. One approach to expanding the use of PARP inhibitors to a wider range of tumors is to combine them with cytotoxic chemotherapy or radiotherapy. Preclinical studies in experimental animals and tumor cells in culture indicate that PARP inhibition modestly sensitizes most tumor cells to ionizing radiation. Studies of cell behavior after these combined treatments show that radiosensitization is manifested predominantly in an increase in prolonged growth arrest and senescence, with little or no contribution from apoptosis. The secretory phenotype associated with senescence can target these tumor cells for immune surveillance, and therefore increased senescence can effectively contribute to tumor control. However, the possible recovery of senescent cells and re-entry into cell cycle after prolonged arrest also needs to be considered. Such recovery could lead to tumor recurrence, yet may not be reflected in short-term assays commonly used to assess radiosensitization.
PARP AS A TARGET FOR RADIOSENSITIZATION
In mammalian cells, DNA polymerase beta (Polβ) and poly(ADP-ribose) polymerase (PARP) have been implicated in base excision repair (BER) and single-strand break (SSB) repair. Polβ−/−PARP-1−/− double-mutant mice are viable, but appear to be highly sensitive to DNA-alkylating agents or gamma radiation (1), raising the possibility that BER, and PARP in particular, can be targeted for chemo/radiosensitization of cells. The PARP enzyme acts as a “molecular sensor” to identify DNA SSBs. If PARP activity is compromised, SSBs collapse replication forks, which results in the formation of double-strand breaks (DSBs) (2), the most dangerous cytotoxic lesions. In recent years, there has been extensive interest in the possibility of enhancing the response to cancer therapies through the use of PARP inhibitors, a class of agents that interfere with BER and therefore with DNA repair (2–5). The development of PARP inhibitors as agents to treat tumors with certain sensitizing genetic lesions is based on the idea that cells with defects in DSB repair, such as BRCA-deficient cells, are more dependent on PARP and BER to maintain genomic integrity (6). The concept of “synthetic lethality”, first articulated by Dobzhansky in 1946 as a situation in which mutations in two genes have little or no effect individually but the combination results in cell death, is thus well illustrated by PARP inhibition in BRCA-deficient cells (7). This synthetic lethal approach has been validated in studies that show striking single-agent activity of PARP inhibitors in preclinical models of BRCA1 and BRCA2 inactivation in vitro and in vivo (2, 3). In patients with hereditary BRCA mutations, PARP inhibitors would be highly selective for tumor tissues (expected to be completely BRCA deficient) compared with normal tissues (expected to be heterozygous at the BRCA locus). In addition, the observation that cells deficient in other critical homologous recombination (HR) proteins are sensitive to PARP inhibitors provides the rationale for testing PARP inhibitors in other cancers (8). For instance, with the recent finding that the tumor suppressor PTEN is important for expression of the repair protein RAD51, it was shown that PTEN-deficient cells are exquisitely sensitive to PARP inhibitors, providing a rationale for ongoing studies of PARP inhibitors in PTEN-deficient tumors (9).
Initially, the toxicity of PARP inhibitors to HR-deficient cells was proposed to reflect an accumulation of SSBs due to lack of PARP-dependent SSB repair, followed by their conversion during S phase into DSBs that were not rejoined due to lack of homologous recombination (2, 3). It is now apparent that the mechanism is more complex, as maximal toxicity requires both PARP and the non-homologous end joining (NHEJ) DSB repair pathway. A comparison of the potencies of various PARP inhibitors suggests that persistent trapping of PARP on damaged DNA is more important than mere inhibition of SSB repair (10–12). Further, inappropriate channeling of the replication-associated DSBs into NHEJ, rather than simply an increase in their persistence, appears to account for most of the toxicity (13–16). Figure 1 show a diagrammatic summary of the primary concepts presented in this review.
FIG. 1.
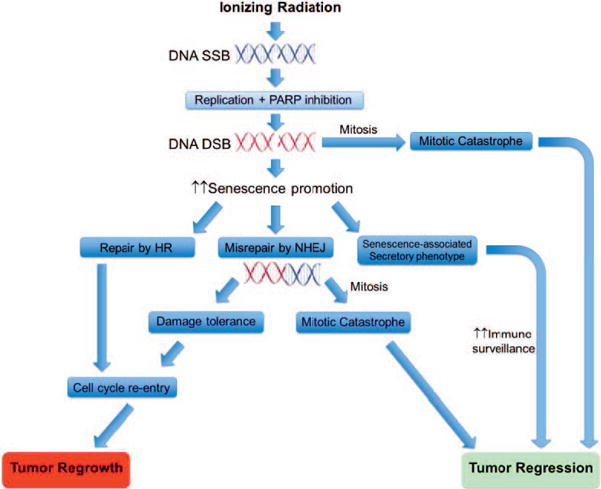
Divergent outcomes of radiation-induced senescence and its enhancement by PARP inhibitors. Senescence, at least initially, is maintained by signaling from unrepaired DSBs. PARP inhibitors block SSB repair, thus increasing the chances of collision with a replication fork to generate additional DSBs. PARP inhibition also promotes misjoining of these DSBs by NHEJ, which may be undetectable and inconsequential until the cell attempts to divide, at which point dicentric and other aberrant chromosomes could induce mitotic catastrophe. Alternatively, senescence, through the senescence-associated secretory phenotype, may promote cell death by triggering immune surveillance. However, some senescent cells may ultimately repair persistent damage or develop tolerance to it, and again begin to proliferate.
While there is evidence that PARP inhibitors alone might have utility against tumors that are intrinsically DNA DSB repair deficient, the focus of this review is on the literature describing the interaction of PARP inhibitors with radiation treatment. Specifically, we analyze the recent literature to address whether this treatment strategy results in growth arrest and/or cell death and the implications of those outcomes for clinical use of these agents.
INCREASE IN DNA DAMAGE
One fundamental underlying premise for the incorporation of PARP inhibitors into radiation therapy is that while tumor cells have the capacity to repair the bulk of radiation-induced DNA damage alone, inhibition of PARP allows SSBs to progress to DSBs. These DSBs are largely replication-dependent, as reflected in the reported finding that blocking replication largely eliminates PARP inhibitor-mediated radiosensitization of most cells (17, 18). The downstream consequences, including modes of cell death or growth arrest, could thus differ substantially from those of DSBs induced directly by radiation alone. For example, if, as in BRCA1-deficient cells, these replication-associated DSBs are misjoined by NHEJ, γ-H2AX foci would presumably disappear as free DSB ends are eliminated, yet cryptic potentially lethal damage would remain and its lethality would not be expressed until the cell attempts to enter mitosis. Moreover, production of these DSBs will only continue during the time PARP inhibitors are still present, which is typically limited to 24 h or less due to their toxicity.
Studies that have addressed the impact of PARP inhibitors with radiation treatment on DNA damage have, not surprisingly, provided clear evidence of increased damage; this result has generally been based on γ-H2AX foci formation and/or the comet assay (19–23). While most published studies have not attempted to actually distinguish between the induction of DNA SSBs and DSBs, the detection of γ-H2AX foci that are not resolved is generally considered to be a reliable indicator of unrepaired DSBs (24, 25). Moreover, neutral comet assays, which provide a more direct and specific measure of DSBs than γ-H2AX foci, also indicate that PARP inhibition increases radiation-induced damage (18). Nevertheless, it might be relevant to monitor the DNA damage induced by radiation with PARP inhibition for extended periods of time to reliably establish that the breaks are prolonged and essentially irreparable. This suggestion relates to the fact that, as indicated below, the incorporation of PARP inhibition with radiation has generally not demonstrated cell killing that might be expected with unrepaired DNA DSBs; instead, studies have shown that this experimental strategy tends to lead to inhibition of tumor cell proliferation and, in a number of studies, to the induction of a state of senescence (18, 26–28).
Our laboratories have confirmed that PARP inhibitors do not increase radiation-induced apoptosis in HCT-116 colon carcinoma cells, but instead markedly enhance growth arrest and senescence (18). In this context, a tumoristatic effect could be considered a less desirable outcome than a tumoricidal effect since we have further determined that the growth arrest induced by radiation with PARP inhibition is transient (as is also the case with that induced by radiation alone) (18) in at least a fraction of the cells. Although recovery of the culture as a whole could reflect outgrowth of a few undamaged or lightly damaged cells rather than proliferation of transiently arrested, senescent cells, we have succeeded in separating from the treated cultures β-galactoside-expressing cells that appear morphologically senescent, yet resume proliferation after a short delay (18).
INDUCTION OF APOPTOSIS
Because cell death is an expected effect of combined radiation and PARP inhibition treatment, given that unresolved DNA DSBs are being generated, it is logical to evaluate the promotion of apoptosis upon treatment. Some published studies of apoptosis induction by radiation alone as well as combined radiation and PARP inhibition treatment have relied on surrogate markers, particularly the cleavage of caspases, which does not necessarily represent a direct measurement of the actual extent of apoptosis in the irradiated tumor cell population. This would require assessment by such approaches as FACS analysis using propidium iodide/annexin or TUNEL assay. We have identified only one study where there was a significant increase in apoptosis from radiation treatment alone as well as from radiation treatment combined with a PARP inhibitor, but in only one of four cell lines examined (22). Otherwise, even in those studies demonstrating a statistically significant increase in apoptosis for radiation treatment with PARP inhibitors compared to radiation treatment alone, the extent of apoptosis has generally not been sufficient to account for the degree of radiosensitization observed (29–32). Furthermore, studies have rarely included complementary experiments where pharmacological or genetic suppression of apoptosis induced by radiation treatment with PARP inhibition was demonstrated to attenuate or reverse radiosensitization. Our own recent studies support the conclusion that neither radiation alone nor radiation with PARP inhibition results in a significant degree of apoptosis, which we have confirmed by demonstrating that radiosensitization is not altered by caspase inhibition (18).
GROWTH ARREST VERSUS CELL DEATH
In the reviewed literature where tumor cells were treated with radiation combined with PARP inhibitors, either in cell culture studies or in tumor-bearing animal experiments, the primary effect of the combination treatment almost uniformly reflected growth arrest rather than cell death (18, 19, 21, 22, 27–29, 33–35). Consequently, it does appear that modes of cell death such as apoptosis or necrosis may play, at most, only minor roles in the overall response to the combination treatment. In this context, when clonogenic assays are utilized to assess the effect of combined radiation and PARP inhibition, it should be recognized that growth arrest, if sufficiently prolonged, would be interpreted as reproductive death and would be indistinguishable from apoptosis or other modes of actual cell disintegration. Similarly, in assays where cell number is monitored in culture or tumor volume is measured in vivo, a prolonged delay in growth, followed by proliferation, is often observed (18, 19, 21, 22, 27–29, 33–35). However, it is generally unclear whether outgrowth resulted from a large fraction of cells that re-entered cell cycle after a long period of arrest, or from a minute fraction of cells that continued to proliferate with little or no arrest, and rarely has any attempt been made to distinguish between these two scenarios.
SENESCENCE
Given that the primary outcome of treatment with radiation and PARP inhibitor appears to be the promotion of a state of growth arrest as opposed to cell death, it is reasonable that, as reported in a number of studies, radiosensitization might be a consequence primarily of an increase in the extent of senescence (18, 27, 28). This outcome could also be a direct consequence of the fact that senescence has been shown to be the primary response to clinically relevant doses of radiation (36, 37). Again, in our own recent work combining radiation with PARP inhibitors, in HCT-116 colon carcinoma cells we observed a pronounced increase in the extent of senescence, as determined by β-galactosidase staining, release of IL-6 and morphological alterations (18).
As (chemotherapy and radiation induced) senescence has traditionally been considered an irreversible form of growth arrest (37), cells irradiated in the presence of a PARP inhibitor could prove to be reproductively dead, which would be a desirable outcome in cancer treatment even if the tumor cells do not formally die by e.g., apoptosis, necrosis or mitotic catastrophe. Furthermore, senescent cells have been reported to secrete factors that activate an immune response that is ultimately the basis for their recognition and elimination (38–40). Finally, it is possible that prolonged and functionally irreversible senescence may ultimately be succeeded by some other form of cell death, such as apoptosis, even if the latter is not a primary response to the combination treatment.
An alternative view that we recently reported on (41) is that senescence may actually prove to be reversible, even if only for a small subpopulation of tumor cells. In this scenario, senescent cells may actually not be functionally and reproductively dead but only in a prolonged state of (reversible) growth arrest, which is in some ways reminiscent of the stem cell population. At some subsequent time, and under the influence of an as yet undefined stimulus, the cells could recover proliferative function, allowing for resurgence of the tumor (and disease recurrence).
Sabisz and Skladanowski (42) reported in their studies that using A549 non-small cell lung cancer cells that regrowth from drug-induced premature senescence is associated with the presence of stem cells. In addition, Salmina et al. (43) present data in support of the premise that radiation induces senescence and a transient polyploid state, where polyploid tumor cells overcome senescence by upregulation of OCT4 and NANOG, characteristic of germ cell lines. Finally, studies by Chitikova et al. (44) using rat embryonic fibroblasts, also support the possibility that reversion of senescence is associated with polyploidy and expression of the stem cell marker OCT3/4.
If, in fact, senescent cells are actually in a state of pseudodormancy (either at the primary tumor site or at some distant site to which the cells have metastasized), then senescence could instead represent a pathway whereby tumor cells are able to escape the potential cytotoxic effect of radiation. Our recent work is consistent with the premise that senescence induced by either radiation alone or in combination with PARP inhibition is reversible, a finding that raises reservations as to the potential utility of this therapeutic strategy.
AUTOPHAGY
Studies from our laboratory as well as those by other research groups have demonstrated that when senescence occurs, it is often preceded by autophagy; that is, autophagy has been shown to accelerate the induction of senescence (by oncogenes and by chemotherapy), although it is also clear that senescence can occur even when autophagy has been abrogated (41). Whether this relationship also exists for radiation has not been fully resolved. Nevertheless, autophagy can actually be considered as a “first responder” to different forms of stress, such as that which occurs under conditions of nutrient deprivation, but also consistently in response to ionizing radiation. Given the evidence for the increased senescence induced by radiation with PARP inhibitors, it appeared likely that the radiosensitization observed under these experimental conditions would also be associated with the induction of autophagy. Since autophagy is generally considered to be a transient and reversible phenomenon, if autophagy is the mechanism of radiation sensitization by PARP inhibition, then the sensitization may prove to be transient as well, contributing to and/or facilitating proliferative recovery of the tumor cells from the accompanying state of senescence.
In our recently published work, we did, in fact, find that induction of autophagy correlated with the extent of senescence as well as the persistence of DNA damage based on γ-H2AX foci formation (18). Unexpectedly, however, inhibition of autophagy utilizing either pharmacologic or genetic approaches failed to alter the extent of radiosensitization by the PARP inhibitors, ostensibly via the promotion of senescence. These studies demonstrated dissociation between the induction of autophagy and senescence in response to radiation and unequivocally indicated that autophagy was not responsible for radiosensitization by PARP inhibition.
POTENTIAL CLINICAL RAMIFICATIONS
As indicated above, if the inclusion of PARP inhibitors with radiation in cancer therapy fails to result in tumor cell death, but instead leads to a state of growth arrest (associated with senescence), the critical question becomes whether the growth arrest is sustained and irreversible or transient and reversible. If the former, then having reproductively dead tumor cells is an appropriate and useful clinical outcome. If the latter, while this treatment approach might be useful in the short term by suppressing tumor growth initially, it might be lacking the decisive outcome that would also prevent (or at least delay or attenuate) reemergence of the tumor cell population that would otherwise lead to disease recurrence. Clinically, this could be detrimental to the long-term survival of the patient, since recovery of self-renewal capacity could be a precursor to active disease recurrence.
Acknowledgments
Dr. Povirk’s laboratory was supported by the National Cancer Institute, grant nos. CA40615 and CA166264. Dr. Gewirtz’s laboratory was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Breast Cancer Research Program under award no. W81XWH-14-1-0088 and by NIH-NCI Cancer Center support grant no. P30 CA016059 (Massey Cancer Center).
Footnotes
Meng Y, Efimova EV, Hamzeh KW, Darga TE, Mauceri HJ, Fu YX et al. Radiation-inducible immunotherapy for cancer: senescent tumor cells as a cancer vaccine. Mol Ther 2012; 20:1046–55.
References
- 1.Sugo N, Niimi N, Aratani Y, Masutani M, Suzuki H, Koyama H. Decreased PARP-1 levels accelerate embryonic lethality but attenuate neuronal apoptosis in DNA polymerase beta-deficient mice. Biochem Biophys Res Commun. 2007;354:656–61. doi: 10.1016/j.bbrc.2006.12.230. [DOI] [PubMed] [Google Scholar]
- 2.Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther. 2014;349:408–16. doi: 10.1124/jpet.113.210146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 4.Graziani G, Szabo C. Clinical perspectives of PARP inhibitors. Pharmacol Res. 2005;52:109–18. doi: 10.1016/j.phrs.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Powell C, Mikropoulos C, Kaye SB, Nutting CM, Bhide SA, Newbold K, et al. Pre-clinical and clinical evaluation of PARP inhibitors as tumour-specific radiosensitisers. Cancer Treat Rev. 2010;36:566–75. doi: 10.1016/j.ctrv.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol. 2005;25:7158–69. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobzhansky T. Genetics of natural populations. Xiii. Recombination and variability in populations of drosophila pseudoobscura. Genetics. 1946;31:269–90. doi: 10.1093/genetics/31.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 9.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–99. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–71. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13:433–43. doi: 10.1158/1535-7163.MCT-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011;108:3406–11. doi: 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Aroumougame A, Lobrich M, Li Y, Chen D, Chen J, et al. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014;28:2693–8. doi: 10.1101/gad.252478.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006;25:1305–14. doi: 10.1038/sj.emboj.7601015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther. 2010;9:1775–87. doi: 10.1158/1535-7163.MCT-09-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bridges KA, Toniatti C, Buser CA, Liu H, Buchholz TA, Meyn RE. Niraparib (MK-4827), a novel poly(ADP-ribose) polymerase inhibitor, radiosensitizes human lung and breast cancer cells. Oncotarget. 2014;5:5076–86. doi: 10.18632/oncotarget.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alotaibi M, Sharma K, Saleh T, Povirk LF, Hendrickson EA, Gewirtz DA. Radiosensitization by PARP inhibition in DNA repair proficient and deficient tumor cells: proliferative recovery in senescent cells. Radiat Res. 2016;185:229–45. doi: 10.1667/RR14202.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ali M, Kamjoo M, Thomas HD, Kyle S, Pavlovska I, Babur M, et al. The clinically active PARP inhibitor AG014699 ameliorates cardiotoxicity but does not enhance the efficacy of doxorubicin, despite improving tumor perfusion and radiation response in mice. Mol Cancer Ther. 2011;10:2320–9. doi: 10.1158/1535-7163.MCT-11-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang NY, Kim DH, Cho BJ, Choi EJ, Lee JS, Wu HG, et al. Radiosensitization with combined use of olaparib and PI-103 in triple-negative breast cancer. BMC Cancer. 2015;15:89. doi: 10.1186/s12885-015-1090-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shelton JW, Waxweiler TV, Landry J, Gao H, Xu Y, Wang L, et al. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. Int J Radiat Oncol Biol Phys. 2013;86:469–76. doi: 10.1016/j.ijrobp.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Chow JP, Man WY, Mao M, Chen H, Cheung F, Nicholls J, et al. PARP1 is overexpressed in nasopharyngeal carcinoma and its inhibition enhances radiotherapy. Mol Cancer Ther. 2013;12:2517–28. doi: 10.1158/1535-7163.MCT-13-0010. [DOI] [PubMed] [Google Scholar]
- 23.Nowsheen S, Bonner JA, Yang ES. The poly(ADP-ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol. 2011;99:331–8. doi: 10.1016/j.radonc.2011.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olive PL. Retention of gammaH2AX foci as an indication of lethal DNA damage. Radiother Oncol. 2011;101:18–23. doi: 10.1016/j.radonc.2011.05.055. [DOI] [PubMed] [Google Scholar]
- 25.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–94. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Azad A, Bukczynska P, Jackson S, Haupt Y, Cullinane C, McArthur GA, et al. Co-targeting deoxyribonucleic acid-dependent protein kinase and poly(adenosine diphosphate-ribose) polymerase-1 promotes accelerated senescence of irradiated cancer cells. Int J Radiat Oncol Biol Phys. 2014;88:385–94. doi: 10.1016/j.ijrobp.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 27.Barreto-Andrade JC, Efimova EV, Mauceri HJ, Beckett MA, Sutton HG, Darga TE, et al. Response of human prostate cancer cells and tumors to combining PARP inhibition with ionizing radiation. Mol Cancer Ther. 2011;10:1185–93. doi: 10.1158/1535-7163.MCT-11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Efimova EV, Mauceri HJ, Golden DW, Labay E, Bindokas VP, Darga TE, et al. Poly(ADP-ribose) polymerase inhibitor induces accelerated senescence in irradiated breast cancer cells and tumors. Cancer Res. 2010;70:6277–82. doi: 10.1158/0008-5472.CAN-09-4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu SK, Coackley C, Krause M, Jalali F, Chan N, Bristow RG. A novel poly(ADP-ribose) polymerase inhibitor, ABT-888, radiosensitizes malignant human cell lines under hypoxia. Radiother Oncol. 2008;88:258–68. doi: 10.1016/j.radonc.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Chen ZT, Zhao W, Qu S, Li L, Lu XD, Su F, et al. PARP-1 promotes autophagy via the AMPK/mTOR pathway in CNE-2 human nasopharyngeal carcinoma cells following ionizing radiation, while inhibition of autophagy contributes to the radiation sensitization of CNE-2 cells. Mol Med Rep. 2015;12:1868–76. doi: 10.3892/mmr.2015.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brock WA, Milas L, Bergh S, Lo R, Szabo C, Mason KA. Radiosensitization of human and rodent cell lines by INO-1001, a novel inhibitor of poly(ADP-ribose) polymerase. Cancer Lett. 2004;205:155–60. doi: 10.1016/j.canlet.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 32.Hocsak E, Cseh A, Szabo A, Bellyei S, Pozsgai E, Kalai T, et al. PARP inhibitor attenuated colony formation can be restored by MAP kinase inhibitors in different irradiated cancer cell lines. Int J Radiat Biol. 2014;90:1152–61. doi: 10.3109/09553002.2014.934927. [DOI] [PubMed] [Google Scholar]
- 33.Senra JM, Telfer BA, Cherry KE, McCrudden CM, Hirst DG, O’Connor MJ, et al. Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol Cancer Ther. 2011;10:1949–58. doi: 10.1158/1535-7163.MCT-11-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96:56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Mason KA, Ang KK, Buchholz T, Valdecanas D, Mathur A, et al. MK-4827, a PARP-1/-2 inhibitor, strongly enhances response of human lung and breast cancer xenografts to radiation. Invest New Drugs. 2012;30:2113–20. doi: 10.1007/s10637-011-9770-x. [DOI] [PubMed] [Google Scholar]
- 36.Gewirtz DA. Autophagy and senescence in cancer therapy. J Cell Physiol [Review] 2014;229:6–9. doi: 10.1002/jcp.24420. [DOI] [PubMed] [Google Scholar]
- 37.Gewirtz DA, Holt SE, Elmore LW. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochem Pharmacol. 2008;76:947–57. doi: 10.1016/j.bcp.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki M, Boothman DA. Stress-induced premature senescence (SIPS)–influence of SIPS on radiotherapy. J Radiat Res. 2008;49:105–12. doi: 10.1269/jrr.07081. [DOI] [PubMed] [Google Scholar]
- 39.Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, et al. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol. 2014;122:91–128. doi: 10.1016/B978-0-12-800267-4.00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9:75–89. doi: 10.1016/j.celrep.2014.08.044. [DOI] [PubMed] [Google Scholar]
- 41.Chakradeo S, Elmore LW, Gewirtz DA. Is senescence reversible? Curr Drug Targets. 2016;17:460–6. doi: 10.2174/1389450116666150825113500. [DOI] [PubMed] [Google Scholar]
- 42.Sabisz M, Skladanowski A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: implications for drug resistance and in vitro drug screening models. Cell Cycle. 2009;8:3208–17. doi: 10.4161/cc.8.19.9758. [DOI] [PubMed] [Google Scholar]
- 43.Salmina K, Jankevics E, Huna A, Perminov D, Radovica I, Klymenko T, et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp Cell Res. 2010;316:2099–112. doi: 10.1016/j.yexcr.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 44.Chitikova ZV, Gordeev SA, Bykova TV, Zubova SG, Pospelov VA, Pospelova TV. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces reversible senescence associated with mTOR downregulation and expression of stem cell markers. Cell Cycle. 2014;13:1424–39. doi: 10.4161/cc.28402. [DOI] [PMC free article] [PubMed] [Google Scholar]