

SUMMARY
Obesity is associated with adipose tissue dysfunction and multi-organ insulin resistance. However, the mechanisms of such obesity-associated systemic metabolic complications are not clear. Here, we characterized mice with adipocyte-specific deletion of nicotinamide phosphoribosyltransferase (NAMPT), a rate-limiting NAD+ biosynthetic enzyme known to decrease in adipose tissue of obese and aged rodents and people. We found that adipocyte-specific Nampt knockout mice had severe insulin resistance in adipose tissue, liver, and skeletal muscle, and adipose tissue dysfunction, manifested by increased plasma free fatty acids concentrations and decreased plasma concentrations of a major insulin-sensitizing adipokine, adiponectin. Loss of Nampt increased phosphorylation of CDK5 and PPARγ (serine-273) and decreased gene expression of obesity-linked phosphorylated PPARγ targets in adipose tissue. Remarkably, these deleterious alterations were normalized by administering rosiglitazone or a key NAD+ intermediate, nicotinamide mononucleotide (NMN). Collectively, our results provide important mechanistic and therapeutic insights into obesity-associated systemic metabolic derangements, particularly multi-organ insulin resistance.
Keywords: Adipocyte, NAMPT, NAD+, insulin resistance, obesity, PPARγ
INTRODUCTION
Obesity is associated with the development of systemic metabolic complications such as multi-organ insulin resistance, which is an important abnormality involved in the pathogenesis of type 2 diabetes, atherogenic dyslipidemia, non-alcoholic fatty liver disease (NAFLD), and cardiovascular disease (Reaven, 1988). Emerging evidence has suggested that adipose tissue has extraordinary capability of maintaining the functional integrity of whole-body metabolic health by modulating the production of insulin-sensitizing adipokines such as adiponectin, pro-inflammatory cytokines and chemokines, free fatty acids (FFA), and other metabolites. Thus dysfunction of adipose tissue contributes to obesity-associated metabolic disorder not only in adipose tissue, but also remote organs such as liver and skeletal muscle (Guilherme et al., 2008; Kadowaki et al., 2006; Perry et al., 2015; Scherer, 2006). However, the complex mechanisms responsible for the pathogenesis of such obesity-associated systemic metabolic complications are not still clear.
Nicotinamide adenine dinucleotide (NAD+) plays a pivotal role in energy metabolism in a wide variety of organisms (Belenky et al., 2007; Canto et al., 2015; Imai and Yoshino, 2013; Yoshino and Imai, 2013). In mammals, NAD+ is synthesized from four precursors, including tryptophan, nicotinic acid, nicotinamide, and nicotinamide riboside (NR). Nicotinamide phosphoribosyltransferase (NAMPT) functions as the rate-limiting NAD+ biosynthetic enzyme and converts nicotinamide into a key NAD+ intermediate, nicotinamide mononucleotide (NMN). NMN is then converted into NAD+ by the second enzymes, NMN adenylyltransferases (NMNATs). NAD+ exerts pleiotropic functions by modulating key NAD+-dependent metabolic regulators, such as sirtuins and poly(ADP-ribose) polymerases (Belenky et al., 2007; Canto et al., 2015; Imai and Yoshino, 2013). Recent studies have revealed that NAMPT-mediated NAD+ biosynthesis in adipose tissue is dynamically regulated by nutritional and environmental cues. For example, adipose tissue expression of Nampt mRNA and NAMPT protein displays a robust diurnal oscillation pattern (Ando et al., 2005; Ramsey et al., 2009). Caloric restriction markedly increases expression of Nampt mRNA and NAMPT protein and NAD+ content in adipose tissue (Chen et al., 2008; Song et al., 2014). In contrast, genetic or diet-induced obesity decreases adipose tissue NAMPT and NAD+ content (Caton et al., 2011; Mercader et al., 2008; Yoshino et al., 2011) and dampens diurnal oscillation of Nampt gene expression (Ando et al., 2005). Aging, which is another major risk factor of metabolic complications, also impairs NAMPT-mediated NAD+ biosynthesis in adipose tissue (Yoshino et al., 2011). Interestingly, data obtained from recent studies conducted in people demonstrate that the alterations in adipose NAMPT-mediated NAD+ biosynthesis are associated with obesity and its metabolic complications (Jukarainen et al., 2015; Kovacikova et al., 2008; Varma et al., 2007; Xu et al., 2012). For example, adipose tissue NAMPT protein content or NAMPT gene expression is decreased in subjects who are insulin-resistant, compared to body mass index matched insulin-sensitive counterparts (Varma et al., 2007; Xu et al., 2012). Contrarily, diet-induced weight loss increases adipose tissue gene expression of NAMPT in people who are overweight and obese (Kovacikova et al., 2008). These findings suggest that NAMPT-mediated NAD+ biosynthesis in adipose tissue is responsive to the alterations of environmental input, and could be involved in regulating whole-body glucose metabolism. However, the physiologic importance of adipose tissue NAMPT-mediated NAD+ biosynthesis is not known. In this study, we analyzed adipocyte-specific Nampt knockout (ANKO) mice and tested our hypothesis that defects in NAMPT-mediated NAD+ biosynthesis in adipocytes play a causative role in the pathogenesis of obesity-associated systemic metabolic complications.
RESULTS
Adipocyte-specific deletion of Nampt causes severe insulin resistance in multiple metabolic organs
To determine the metabolic consequence induced by adipocyte-specific Nampt deletion, we have characterized ANKO mice generated by using Nampt-floxed (flox/flox) crossed with adiponectin-Cre transgenic mice (Yoon et al., 2015). The levels of NAMPT protein and NAD+ in visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT) were markedly reduced in ANKO mice compared to flox/flox (control) mice (Yoon et al., 2015). We evaluated glucose metabolism in control and ANKO mice fed a regular chow diet by conducting intraperitoneal glucose tolerance tests (IPGTTs) and insulin tolerance tests (ITTs). We found that female ANKO mice exhibited impaired glucose tolerance and increased plasma insulin concentrations during the IPGTTs compared to control mice (Figs. 1A and B). During the ITTs, insulin injection was not able to decrease blood glucose concentrations in female ANKO mice (Fig. 1C). Although male ANKO mice did not exhibit impaired glucose tolerance (Fig. S1A), they did have increased plasma insulin concentrations during the IPGTTs (Fig. S1B), and failed to respond to insulin injection (Fig. S1C). This disassociation of insulin tolerance with glucose tolerance could be due to the compensatory insulin secretion for insulin resistance. We next conducted hyperinsulinemic-euglycemic clamp procedures (HECP). The glucose infusion rate in ANKO mice was markedly lower than that in control mice (Fig. 1D), even though ANKO mice had slightly increased blood glucose and insulin concentrations (Figs. S1D and S1E). ANKO mice demonstrated a failure of insulin to suppress hepatic glucose production (HGP), whereas control mice suppressed HGP by 90% during the HECP (Figs. 1E and 1F). Insulin clearance rate, another indicator of hepatic insulin sensitivity, was also lower in ANKO mice than in control mice (ANKO=66.2 ± 5.0; control=89.0 ± 7.4 mL/kg-lean body weight/min, P < 0.05). Supporting our in vivo observation, ANKO mice had increased hepatic gene expression of key gluconeogenic enzymes, such as glucose-6-phoshatase (G6pc) and pyruvate dehydrogenase kinase 4 (Pdk4) compared to control mice (Fig. 1G). In addition, whole-body insulin-mediated glucose disposal rates were reduced by 18% in ANKO mice (Fig. 1H), and this was accompanied by reduction in glycolysis rates (Fig. S1F). Consistently, insulin-stimulated glucose uptake in gastrocnemius muscle and heart was reduced by 64% and 50%, respectively (Fig. 1I). Despite such severe multi-organ insulin resistance, ANKO mice had similar total body weight, body composition, oxygen consumption, carbon dioxide production, and respiratory exchange ratio compared to control mice (Figs. S1G and S1H). Taken together, these results demonstrate that adipocyte-specific deletion of Nampt causes severe multi-organ insulin resistance, independent of whole-body adiposity and body weight.
Figure 1. Adipocyte-specific Nampt deletion causes multi-organ insulin resistance.
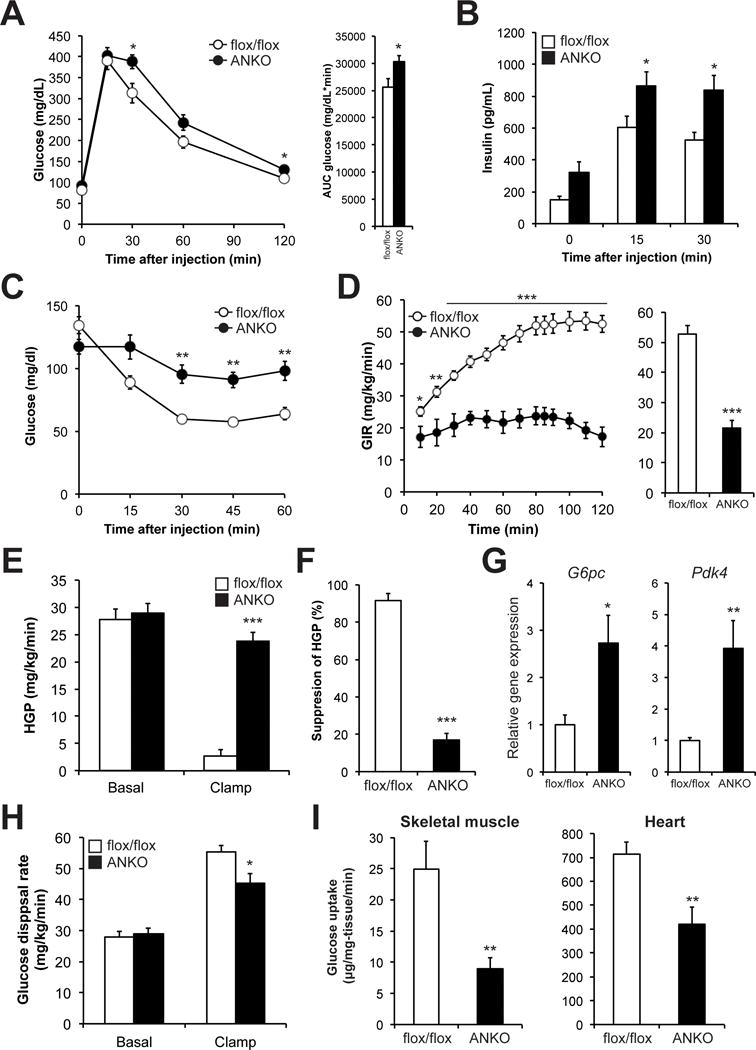
Glucose metabolism in female control (flox/flox) and adipocyte-specific Nampt knockout (ANKO) mice fed a standard chow. Blood glucose (A) and plasma insulin (B) concentrations during the intraperitoneal glucose tolerance tests (IPGTTs) in 3- to 5- month-old mice (n=6–8 per group). The area under the curve (AUC) for glucose is shown next to the glucose tolerance curves. (C) Blood glucose concentrations during the insulin tolerance tests (ITTs) in 2- to 4- month-old mice (n=7–9 per group). (D-I) Hyperinsulinemic euglycemic clamp procedure (HECP) was performed in 3- to 8- month-old mice (n=9–10 per group). (D) Glucose infusion rate (GIR) during the HECP. Average of GIR during the clamp period is shown next to the GIR curves. (E) Basal and insulin-stimulated rates of hepatic glucose production (HGP). (F) Insulin-stimulated percent suppression of basal HGP. (G) Hepatic gene expression of gluconeogenic enzymes, glucose 6-phospahtase (G6pc) and pyruvate dehydrogenase lipoamide kinase isozyme 4 (Pdk4), after the HECP (n=6–7 per group). (H) Whole-body glucose disposal rates. (I) Insulin-stimulated glucose uptake in skeletal muscle and heart. Values are means ± SE. P-values were determined by Student’s t-test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Loss of Nampt causes adipose tissue dysfunction
These findings of systemic insulin resistance prompted us to hypothesize that the defects in NAMPT-mediated NAD+ biosynthesis impair adipose tissue metabolic pathways that can influence whole-body glucose metabolism. Indeed, insulin-stimulated glucose uptake in VAT was markedly reduced by 60% in ANKO mice compared to control mice (Fig. 2A). Similarly, ANKO mice showed impaired insulin-mediated suppression of plasma FFA concentrations (Figs. 2B and C). The insulin-stimulated AKT phosphorylation (serine-473) was also reduced by 70% in VAT obtained from ANKO mice compared to control mice (Fig. 2D). In addition, ANKO mice had increased plasma concentrations of FFA and triglyceride (TG) (Figs. 2E and S2A) and increased hepatic TG content (Fig. 2F). We next asked whether these deleterious alterations are accompanied by adipose tissue inflammation, a hallmark of obesity and its metabolic complications (Guilherme et al., 2008; Scherer, 2006). VAT gene expression of inflammatory markers was up-regulated in ANKO mice compared to control mice (Fig. 2G). Immunodetection of the macrophage-specific antigen F4/80 also suggested increased adipose tissue macrophage accumulation in ANKO mice (Fig. S2B). However, plasma concentrations of major pro-inflammatory cytokines and chemokines were not different between ANKO and control mice (Fig. S2C). ANKO mice did not display altered gene expression of several inflammatory markers in liver (Fig. S2D), suggesting that adipocyte-specific Nampt deletion causes local adipose tissue inflammation, but not systemic inflammation. In contrast, plasma concentrations of key adipokines that regulate hepatic and skeletal muscle insulin sensitivity and glucose homeostasis, adiponectin and adipsin (Kadowaki et al., 2006; Scherer, 2006), were markedly reduced in ANKO mice (Figs. 2H and S2E). Together, these findings demonstrate that loss of Nampt impairs key adipose tissue functions, which could contribute to the development of systemic metabolic complications in ANKO mice.
Figure 2. Loss of Nampt induces adipose tissue dysfunction.
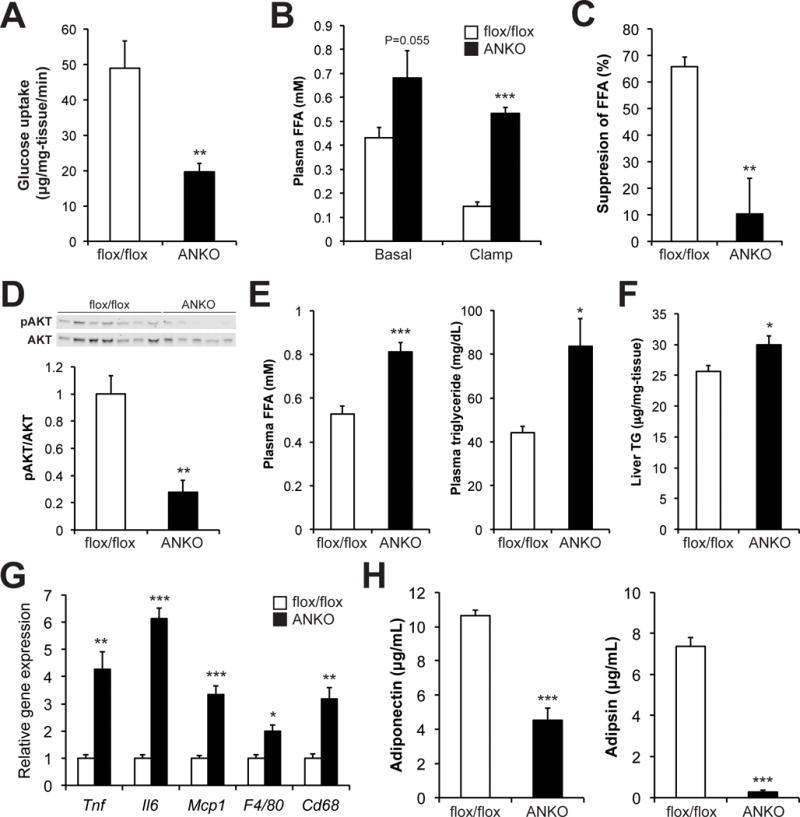
Adipose tissue function in female control and ANKO mice. (A) Insulin-stimulated glucose uptake in visceral adipose tissue (VAT) during the HECP (n=9–10 per group). Plasma free fatty acids (FFA) concentrations in basal and insulin-stimulated conditions (B) and insulin-stimulated percent suppression during the HECP (C) (n=6–7 per group). (D) Western blot and quantification of phosphorylated AKT (serine-473) and total-AKT in VAT obtained after the HECP (n=5–7 per group). (E) Plasma FFA and triglyceride (TG) concentrations at a fed condition in 4- to 5- month-old mice (n=7–9 per group). (F) Hepatic TG concentrations in 2- to 9- month-old mice (n=15–16 per group). (G) VAT gene expression of inflammatory markers in 4- month-old mice (n=4 per group). Il6; interleukin 6, Tnf; tumor necrosis factor, Mcp-1; monocyte chemoattractant protein 1, Cd68; CD68 antigen. (H) Plasma concentrations of adiponectin and adipsin in 2- to 3- month-old mice (n=6–11 per group). Values are means ± SE. P-values were determined by Student’s t-test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
ANKO mice have increased phosphorylation of CDK5 and PPARγ (Ser273) and decreased gene expression of its obesity-linked targets, including adiponectin and adipsin, in adipose tissue
Increased serine-273 (Ser273) phosphorylation of the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) has been found to impair its activity towards a subset of its target genes involved in regulating whole-body glucose metabolism, including a key insulin-sensitizing adipokine, adiponectin. Thus, it is implicated in the pathophysiology of obesity-associated insulin resistance (Banks et al., 2015; Choi et al., 2010; Choi et al., 2011; Choi et al., 2014a; Choi et al., 2014b). Interestingly, ANKO mice had increased protein content of phosphorylated PPARγ (Ser273) in VAT compared to control mice (Fig. 3A). Phosphorylation of cyclin-dependent kinase 5 (CDK5), a critical regulator of PPARγ phosphorylation at Ser273 (Banks et al., 2015; Choi et al., 2010; Choi et al., 2011), was also increased in VAT from ANKO mice (Fig. 3B). Pharmacological inhibition of the NAD+-dependent protein deacetylase SIRT1 significantly increased CDK5 phosphorylation in OP9 adipocytes (Fig. S3A), indicating SIRT1 could be one key mediator of NAMPT-mediated NAD+ biosynthesis in regulating phosphorylation of CDK5. In addition, VAT from ANKO mice displayed increased lysine acetylation of PPARγ (Fig. 3C), indicating that hyperacetylation of PPARγ could also be involved in the observed increase in PPARγ phosphorylation(Mayoral et al., 2015; Qiang et al., 2012). We next evaluated VAT expression of obesity-linked target genes that are known to be the most sensitive to PPARγ phosphorylation (Ser273) (Choi et al., 2010). Nampt deletion caused significant changes in adipose tissue gene expression of 9/12 (75%) of these targets including adiponectin and adipsin (Fig. 3D). Loss of Nampt similarly affected gene expression of these targets in SAT (Fig. S3B). In contrast, ANKO mice did not display alterations in classical PPARγ lipogenic gene targets (Fig. S3C). Importantly, 6 weeks of PPARγ agonist rosiglitazone (RSG) administration (20 mg/kg-body weight/day) improved insulin sensitivity and decreased plasma insulin concentrations in ANKO mice (Figs. 3E and 3F). RSG treatment also restored gene expression of key obesity-linked PPARγ targets including adiponectin and adipsin in VAT and SAT (Fig. 3G), and increased plasma concentrations of adiponectin and adipsin (Fig. 3H). In contrast, RSG treatment did not change body weight, food intake, body temperature, locomotor activity, hypothalamic NAD+ concentrations, and putative RSG targets in liver and skeletal muscle (Figs. 3I and S3D–I), although it is still possible that RSG affected other untested metabolic pathways. Consistent with in vivo observation, a potent NAMPT inhibitor, FK866, reduced adiponectin and adipsin gene expression in OP9 adipocytes and pharmacological inhibition of CDK5 with MRL24 reversed these FK866-induced alterations (Fig. 3J). Altogether, these data suggest that increased phosphorylation of CDK5 and PPARγ (Ser273) are, at least in part, responsible for the metabolic derangements induced by adipocyte-specific Nampt deletion.
Figure 3. ANKO mice have increased phosphorylation of CDK5 and PPARγ (Ser273) and decreased gene expression of obesity-liked phosphorylated PPARγ targets in adipose tissue.
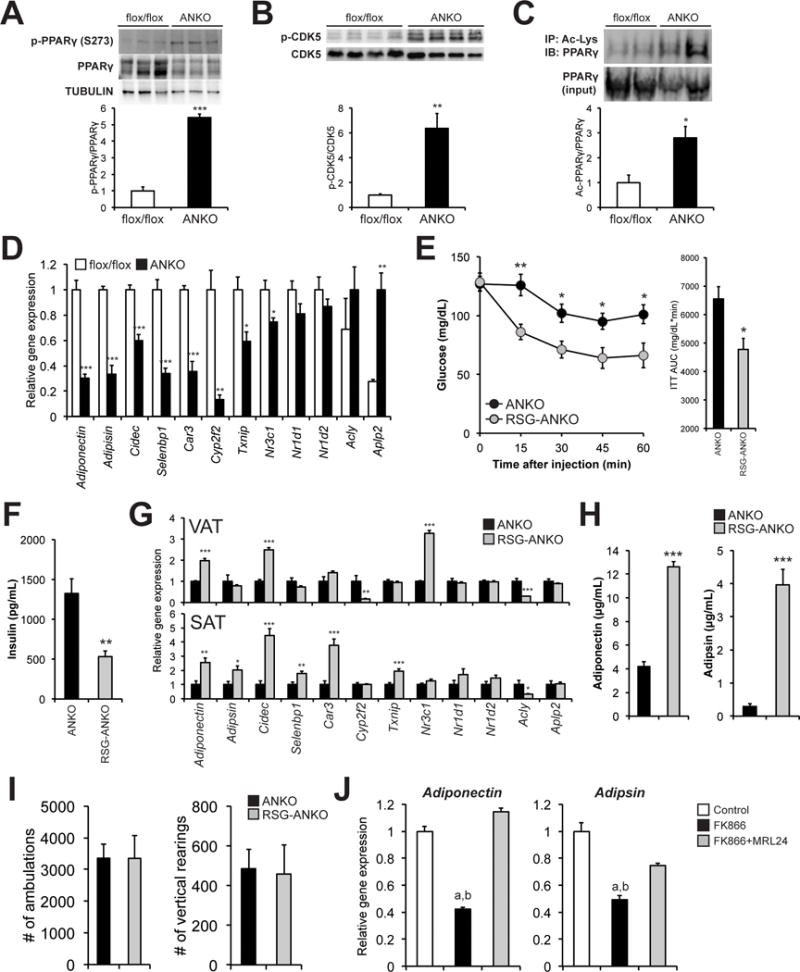
Protein levels of phosphorylated PPARγ (Ser273) (A) and CDK5 (B) in VAT obtained from 2- to 3- month-old female control and ANKO mice after overnight fasting. Phosphorylated protein levels were normalized by total protein content. (C) Immunoprecipitated acetylated PPARγ and PPARγ (inputs) were evaluated in VAT. Acetylated PPARγ levels were normalized by PPARγ protein content (n=4 per group). (D) VAT gene expression of obesity-liked phosphorylated PPARγ (S273) targets in female control and ANKO mice (n=4 per group). Cidec; cell death-inducing DFFA-like effector c, Car3; carbonic anhydrase 3, Cyp2f2; cytochrome P450, family 2, subfamily f, polypeptide 2, Selebbp1; Selenium Binding Protein 1, Txnip; thioredoxin interacting protein, Nr3c1; nuclear receptor subfamily 3, group C, member 1, Nr1d1; nuclear receptor subfamily 1, group D, member 1, Nr1d2; nuclear receptor subfamily 1, group D, member 2, Acly; ATP citrate Lyase, Aplp2; amyloid beta (A4) precursor-like protein 2. (E) Female ANKO mice received a chow containing rosiglitazone (RSG; 20 mg/kg-body weight/day). ITTs were performed after 6 weeks of RSG treatment. ITTs results from RSG-treated and age-matched untreated ANKO mice were shown (n=7–12 per group). The AUC for glucose is shown next to the insulin tolerance curves. (F) Plasma insulin concentrations in RSG-treated and -untreated ANKO mice (n=7–10 per group). (G) VAT and subcutaneous adipose tissue (SAT) gene expression of obesity-liked PPARγ (Ser273) targets in RSG-treated and -untreated ANKO mice (n=4–7 per group). (H) Plasma concentrations of adiponectin and adipsin in RSG-treated and -untreated female ANKO mice (n=7–12 per group). (I) Total number of ambulations and instances of vertical rearing in RSG-treated and -untreated male ANKO mice on the 1 h locomotor test (n=5 per group). (J) OP9 adipocytes were cultured with 0.1% DMSO (control), 500 nM FK866, or FK866 plus 30 nM MRL24 (CDK5 inhibitor) for 48 hours and examined for adiponectin and adipsin gene expression (n=3 per group). Values are means ± SE. P-values were determined by Student’s t-test. a, P < 0.05 (control vs. FK866); b, P < 0.05 (FK866 vs. FK866+MRL24) (ANOVA). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Nicotinamide mononucleotide (NMN), a key NAD+ intermediate, normalizes metabolic derangements in ANKO mice
To determine whether metabolic derangements depend on the defects in NAD+ biosynthesis in ANKO mice, we treated mice with NMN, a key NAD+ intermediate and the product of the NAMPT reaction (Fig. 4A). In this experiment, female ANKO mice were given drinking water containing NMN (500 mg/kg-body weight/day) and metabolic parameters were evaluated after 4–6 weeks of NMN treatment. We determined this dosage based on our previous study (Yoshino et al., 2011). NMN administration significantly increased adipose tissue NAD+ concentrations in ANKO mice (Fig. 4B). Remarkably, NMN administration improved insulin sensitivity in ANKO mice compared to the age-matched untreated ANKO mice (Fig. 4C). The insulin responses in NMN-treated ANKO mice were similar to those in the age-matched control mice, suggesting that NMN administration normalized the insulin resistance phenotype. NMN administration also normalized plasma insulin and FFA concentrations in ANKO mice (Figs. 4D and 4E). However, NMN did not change body weight (ANKO=18.1 ± 0.4; NMN-treated ANKO=18.0 ± 0.5 g), daily food intake (ANKO=0.22 ± 0.01; NMN-treated ANKO=0.21 ± 0.01 g/g-body weight), or body temperature (ANKO=36.7 ± 0.2; NMN-treated ANKO=36.7 ± 0.3 °C) during this relatively short period of treatment. NMN administration decreased phosphorylation of PPARγ (Ser273) and CDK5 in VAT (Figs. 4F and 4G). In addition, NMN-treated ANKO mice reduced global nuclear lysine acetylation (Figs. 4H and S4A), indicating that NMN administration could increase activity of nuclear NAD+-dependent protein deacetylases such as SIRT1. Adipose tissue gene expression and plasma concentrations of adiponectin and adipsin were restored by NMN administration (Figs. 4I and 4J). Additionally, we confirmed that adipose tissue expressed nicotinic acid phosphoribosyltransferase (NAPRT) (Fig. S4B), and tested the effect of 5–7 week administration of NA (500 mg/kg-body weight/day), the precursor in the NAPRT-dependent NAD+ biosynthetic pathway (Fig. 4A). Treatment with NA also normalized the metabolic derangements in ANKO mice (Figs. S4C–G). Although we cannot exclude the possibility that the beneficial effects of NA treatment are partly mediated by the G-protein-coupled receptors (PUMA-G) (Tunaru et al., 2003) and/or other metabolic pathways/organs, these results from NMN and NA treatment support our conclusion that NAMPT-mediated NAD+ biosynthesis in adipocytes is an important physiological regulator of adipose tissue and whole-body metabolic function.
Figure 4. Nicotinamide mononucleotide (NMN), a key NAD+ intermediate, normalizes metabolic deragments in ANKO mice.
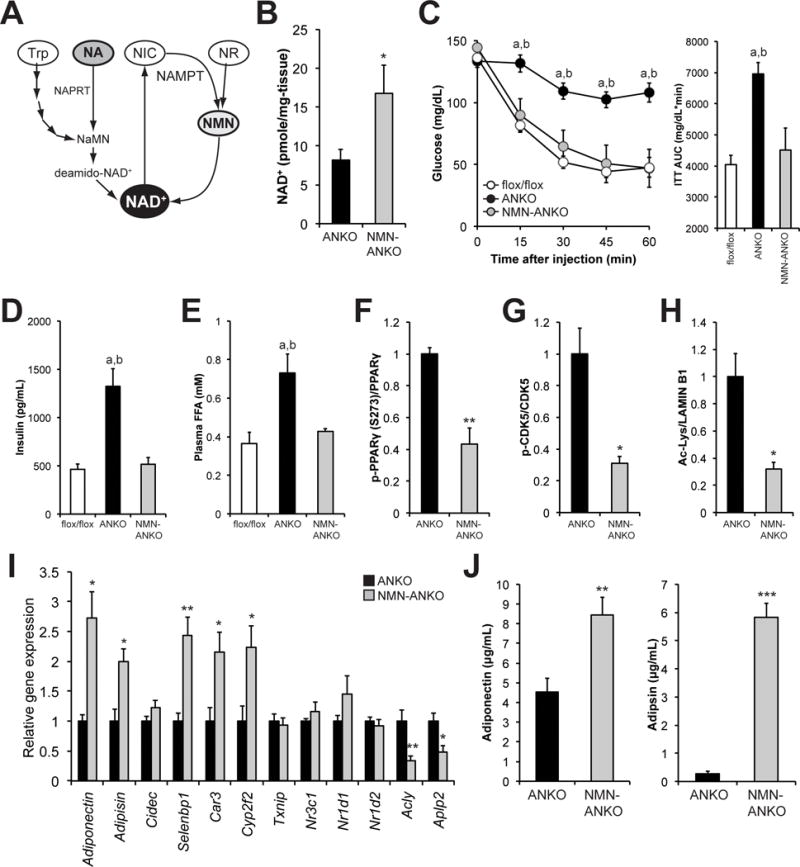
(A) Mammalian NAD+ biosynthetic pathways. Nicotinamide mononucleotide (NMN) is a product of NAMPT-mediated enzymatic reaction and it is directly converted into NAD+. Nicotinic acid (NA) is a precursor for the NAPRT-dependent NAD+ biosynthetic pathway. NIC; nicotinmaide, Trp; tryptophan, NR; nicotinamide riboside, NaMN; nicotinic acid mononucleotide, NAPRT; nicotinic acid phosphoribosyltransferase. (B) Female ANKO mice were given drinking water containing NMN (500 mg/kg-body weight/day). Adipose tissue NAD+ concentrations were determined in female ANKO mice after 4–6 weeks of treatment of NMN and untreated ANKO mice (n=5–7 per group). (C) ITTs were performed after 4 weeks of treatment of NMN. ITTs results from NMN-treated mice (n=7), age-matched 2- to 3- month-old untreated ANKO (n=14), and control mice (n=8) were shown. The AUC for glucose is shown next to the insulin tolerance curves. Plasma concentrations of insulin (D) and FFA (E) in NMN-treated, -untreated ANKO, and control mice (n=5–11 per group). Phosphorylated PPARγ (Ser273) (F), phosphorylated CDK5 (G), and lysine acetylation of nuclear proteins (H), in VAT obtained from NMN-treated and -untreated ANKO mice (n=3–4 per group). (I) VAT gene expression of obesity-liked PPARγ (Ser273) targets in NMN-treated and untreated ANKO mice (n=4–5 per group). (J) Plasma concentrations of adiponectin and adipsin in NMN-treated and -untreated ANKO mice (n=5–12 per group). Values are means ± SE. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (NMN-tread ANKO vs. -untreated ANKO, Student’s t test). a, P < 0.05 (NMN-treated vs. -untreated ANKO); b, P < 0.05 (control vs. NMN-untreated ANKO) (ANOVA).
DISCUSSION
In this study, we provide compelling evidence for the importance of adipose NAMPT-mediated NAD+ biosynthesis in regulating whole-body metabolic function. Our results demonstrate that adipocyte-specific Nampt deletion 1) induces adipose tissue dysfunction, characterized by decreased production of key adipokines, namely adiponectin and adipsin, increased plasma FFA availability, and local adipose tissue inflammation, 2) causes severe insulin resistance in adipose tissue, liver, and skeletal muscle without concomitant increases in body weight or whole-body adiposity, and 3) increases phosphorylation of CDK5 and PPARγ (Ser273) and markedly reduces gene expression of obesity-linked specific targets of phosphorylated PPARγ that include adiponection and adipsin. Furthemore, these deleterious metabolic alterations were completely normalized by administering a PPARγ agonist rosiglitazone, a key NAD+ intermediate NMN, or a NAD+ precursor NA. These results provide important insight into understanding the mechanisms of obesity-associated systemic metabolic complications, including adipose tissue dysfunction and multi-organ insulin resistance.
The present study demonstrates that defects in adipose NAMPT-mediated NAD+ biosynthesis induce various obesity-associated systemic metabolic complications, including severe multi-organ insulin resistance. Previous studies have demonstrated that genetic ablation of adiponectin or its receptor(s) results in the impairment in ability of insulin to suppress glucose production in liver, and to stimulate glucose utilization in skeletal muscle, and that hypoadiponectinemia is related to the degree of insulin resistance in people (Kadowaki et al., 2006; Scherer, 2006). In addition, it has been shown that excessive plasma FFA availability or increased adipose tissue lipolysis can cause insulin resistance in liver and skeletal muscle (Guilherme et al., 2008; Perry et al., 2015). Therefore, it is likely that adipocyte-specific Nampt deletion causes insulin resistance in remote metabolic organs through hypoadiponectinemia and/or excessive FFA release from adipose tissue. It is also possible that the alterations in circulating extracellular NAMPT (eNAMPT) affects neuronal metabolic signals associated with hypothalamus, and is involved in the pathogenesis of systemic metabolic complications observed in ANKO mice (Imai and Yoshino, 2013; Yoon et al., 2015). Given previous findings demonstrating that reduction in adipose NAMPT expression and NAD+ content is associated with obesity and its metabolic complications in people and rodents (Ando et al., 2005; Jukarainen et al., 2015; Kovacikova et al., 2008; Mercader et al., 2008; Xu et al., 2012; Yoshino et al., 2011), our data strongly suggest that impaired NAMPT-mediated NAD+ biosynthesis in adipose tissue plays a causative role in the pathophysiology of obesity-associated metabolic derangements, and thereby could be an important therapeutic target. Indeed, recent studies have shown that systemic administration of NAD+ intermediates, such as NMN and NR, improves insulin resistance, dyslipidemia, NAFLD, and glucose intolerance, in obese mice (Canto et al., 2012; Canto et al., 2015; Gariani et al., 2015; Imai and Yoshino, 2013; Yoshino et al., 2011). Although adipose tissue function was not extensively examined in these studies, it is likely that enhancing adipose NAD+ biosynthesis mediates such metabolic beneficial effects in obese mice. Additional studies are needed to determine whether enhancing NAD+ biosynthesis in adipocytes alone is sufficient enough to counteract the systemic negative effects of obesity on glucose metabolism.
A series of studies has established that PPARγ is an important regulator of adipocyte metabolism, and a molecular target for the treatment of obesity-associated metabolic complications (Lehrke and Lazar, 2005). Post-translational modifications play a critical role in regulating PPARγ function, including recent evidence that increased phosphorylation (Ser273) leads to a selective deactivation of PPARγ towards a subset of its target genes involved in whole-body glucose metabolism, including key adipokines, adiponectin and adipsin (Banks et al., 2015; Choi et al., 2010; Choi et al., 2011; Choi et al., 2014a; Choi et al., 2014b). Our results demonstrate that NAD+ could be an important endogenous physiological regulator of PPARγ in adipocytes. The precise mechanisms by which NAMPT-mediated NAD+ biosynthesis regulates phosphorylation of PPARγ at Ser273 remain unclear. Our results suggest that NAMPT-mediated NAD+ biosynthesis could regulate phosphorylation of CDK5, an important regulator of PPARγ phosphorylation (Banks et al., 2015; Choi et al., 2010; Choi et al., 2011), possibly through the modulation of SIRT1 activity. Given previous findings suggesting that hyperacetylation of PPARγ at lysines 293 also leads to an increase in Ser273 phosphorylation (Qiang et al., 2012), it is likely that NAMPT-mediated NAD+ biosynthesis and SIRT1 together regulate PPARγ Ser273 phosphorylation by modulating CDK5 phosphorylation and PPARγ acetylation. Consistent with this idea, adipocyte-specific Sirt1 knockout mice have increased acetylation and Ser273 phosphorylation of PPARγ in adipose tissue (Mayoral et al., 2015). In addition, several independent groups have reported that adipocyte-specific Sirt1 knockdown/knockout is causatively related to adipose tissue dysfunction and systemic metabolic complications (Chalkiadaki and Guarente, 2012; Gillum et al., 2011; Mayoral et al., 2015). Nonetheless, we cannot exclude the possibility that other sirtuins and NAD+-consuming enzymes, such as poly(ADP-ribose) polymerases (Belenky et al., 2007; Canto et al., 2015; Imai and Yoshino, 2013), also mediate effects induced by the defects in NAPMPT-mediated NAD+ biosynthesis in ANKO mice.
In conclusion, the present study provides important insight into understanding the extraordinary functional capability of adipose tissue to regulate whole-body glucose metabolism. Although future studies are required to determine the effects of genetic and pharmacological activation of adipocyte NAMPT-mediated NAD+ biosynthesis on phosphorylation of CDK5 and PPARγ and metabolic function in obese rodents, it would be of great interest to examine whether enhancing NAD+ biosynthesis in adipocytes provides an effective intervention against obesity-induced multi-organ metabolic derangements without the unfavorable side effect profile associated with PPARγ agonists.
EXPERIMENTAL PROCEDURES
Animal Experimentation
ANKO mice were generated by using adiponectin-Cre transgenic mice and floxed-Nampt mice as previously described (Yoon et al., 2015). Mice were maintained on a standard chow diet (LabDiet 5053; LabDiet, St. Louis, MO) ad libitum. For RSG rescue experiments, ANKO mice received a chow containing RSG (#71740; Cayman Chemical, Ann Arbor, MI) at the approximate dose of 20 mg/kg-body weight/day soon after weaning for up to seven weeks. For NAD+ precursors rescue experiments, we administered NMN (#44500900, Oriental Yeast Co., Tokyo, Japan) and NA (#72309; Sigma, St. Louis, MO) in drinking water, at the approximate dose of 500 mg/kg-body weight, to ANKO mice soon after weaning for up to 2 months. All animal studies were approved by the Washington University Animal Studies Committee.
Hyperinsulinemic-euglycemic clamp procedure
The hyperinsulinemic-euglycemic clamp study was carried out by the University of Michigan Animal Phenotyping Core with modified procedures. See details in the Supplemental Experimental Procedure.
Real time PCR
Gene expression was determined as previously described (Yoshino et al., 2011). See details in the Supplemental Experimental Procedure.
Western blot
Frozen tissue samples were rapidly homogenized in ice-cold lysis buffer. Nuclear protein was extracted by using the Nuclear Extraction kit (#SK-0001, Signosis Inc., Sunnyvale, CA, USA). Antibodies and other details are available in the Supplemental Experimental Procedure.
NAD+ measurement
NAD+ concentrations were determined using an HPLC system (Prominence; Shimadzu Scientific Instruments, Columbia, MD) with a Supelco LC-18-T column (#58970-U; Sigma) as described previously (Yoon et al., 2015; Yoshino and Imai, 2013; Yoshino et al., 2011).
Cell Culture
OP9 mouse stromal cells were generously provided by Dr. Nathan Wolins (Wolins et al., 2006). See details in the Supplemental Experimental Procedure.
Statistical Analyses
Differences between two groups were assessed using Student’s paired or unpaired t test. Comparisons among three groups were performed using one-way ANOVA with the Tukey’s post-hoc test. P values of less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
The authors thank Trey Coleman, Sangeeta Adak (Washington University DRC), and David Wozniak (Animal Behavioral Core), for technical assistance; Kyle McCommis for proof-reading; Brian Finck, Nathan Wolins, Nada Abumrad, and Samuel Klein for insightful discussions.
This study was supported by National Institutes of Health grants DK 56341 (Nutrition and Obesity Research Center), DK 37948 and DK 20579 (Diabetes Research Center), DK52574 (Digestive Diseases Research Core Center), UL1 TR000450 (KL2 Career Developmental Award) and DK104995 to J.Y., AG024150 and AG037457 to S.I., DK089503 (MNORC) and DK020572 (MDRC) (University of Michigan), a Central Society for Clinical and Translational Research Early Career Development Award to J.Y., and a grant from the Longer Lifer Foundation to J.Y. S.Y. is supported by the Sumitomo Life Welfare and Culture Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
J.Y. conceptualized. K.L.S., S.Y., M.J.Y., A.C.M., S.C.K., M.P.F., N.Q., and J.Y. designed and performed experiments. S.Y. and J.Y. performed formal analysis. S.I. and J.Y. provided scientific suggestions and supervisions. J.Y. wrote the manuscript. All authors reviewed and edited the manuscript.
CONFLICT OF INTEREST STATEMENT
S.I. is a co-founder of Metro Midwest Biotech. Dr. Samuel Klein, Division Chief of Nutritional Science at Washington University in St. Louis (WUSTL), has ownership interests with Metro Midwest Biotech. WUSTL may receive royalty income based on a technology licensed by WUSTL to Metro Midwest Biotech. This technology is evaluated in this research.
References
- Ando H, Yanagihara H, Hayashi Y, Obi Y, Tsuruoka S, Takamura T, Kaneko S, Fujimura A. Rhythmic messenger ribonucleic acid expression of clock genes and adipocytokines in mouse visceral adipose tissue. Endocrinology. 2005;146:5631–5636. doi: 10.1210/en.2005-0771. [DOI] [PubMed] [Google Scholar]
- Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, Spiegelman BM. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature. 2015;517:391–395. doi: 10.1038/nature13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton PW, Kieswich J, Yaqoob MM, Holness MJ, Sugden MC. Metformin opposes impaired AMPK and SIRT1 function and deleterious changes in core clock protein expression in white adipose tissue of genetically-obese db/db mice. Diabetes Obes Metab. 2011;13:1097–1104. doi: 10.1111/j.1463-1326.2011.01466.x. [DOI] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. 2012;16:180–188. doi: 10.1016/j.cmet.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Choi SS, Kim ES, Jedrychowski MP, Yang YR, Jang HJ, Suh PG, Banks AS, Gygi SP, Spiegelman BM. Thrap3 docks on phosphoserine 273 of PPARgamma and controls diabetic gene programming. Genes Dev. 2014a;28:2361–2369. doi: 10.1101/gad.249367.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SS, Kim ES, Koh M, Lee SJ, Lim D, Yang YR, Jang HJ, Seo KA, Min SH, Lee IH, et al. A novel non-agonist peroxisome proliferator-activated receptor gamma (PPARgamma) ligand UHC1 blocks PPARgamma phosphorylation by cyclin-dependent kinase 5 (CDK5) and improves insulin sensitivity. J Biol Chem. 2014b;289:26618–26629. doi: 10.1074/jbc.M114.566794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, Ropelle ER, Moullan N, Zhang H, Perino A, Lemos V, et al. Eliciting the mitochondrial unfolded protein response via NAD repletion reverses fatty liver disease. Hepatology. 2015 doi: 10.1002/hep.28245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, Muise ES, Hsiao JJ, Frederick DW, Yonemitsu S, et al. SirT1 regulates adipose tissue inflammation. Diabetes. 2011;60:3235–3245. doi: 10.2337/db11-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab. 2013;15(Suppl 3):26–33. doi: 10.1111/dom.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukarainen S, Heinonen S, Ramo JT, Rinnankoski-Tuikka R, Rappou E, Tummers M, Muniandy M, Hakkarainen A, Lundbom J, Lundbom N, et al. Obesity is associated with low NAD/SIRT pathway expression in adipose tissue of BMI-discordant monozygotic twins. J Clin Endocrinol Metab. 2015:jc20153095. doi: 10.1210/jc.2015-3095. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova M, Vitkova M, Klimcakova E, Polak J, Hejnova J, Bajzova M, Kovacova Z, Viguerie N, Langin D, Stich V. Visfatin expression in subcutaneous adipose tissue of pre-menopausal women: relation to hormones and weight reduction. Eur J Clin Invest. 2008;38:516–522. doi: 10.1111/j.1365-2362.2008.01964.x. [DOI] [PubMed] [Google Scholar]
- Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Mayoral R, Osborn O, McNelis J, Johnson AM, Oh da Y, Izquierdo CL, Chung H, Li P, Traves PG, Bandyopadhyay G, et al. Adipocyte SIRT1 knockout promotes PPARgamma activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol Metab. 2015;4:378–391. doi: 10.1016/j.molmet.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercader J, Granados N, Caimari A, Oliver P, Bonet ML, Palou A. Retinol-binding protein 4 and nicotinamide phosphoribosyltransferase/visfatin in rat obesity models. Horm Metab Res. 2008;40:467–472. doi: 10.1055/s-2008-1065324. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, Rosenbaum M, Zhao Y, Gu W, Farmer SR, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell. 2012;150:620–632. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. 2006;55:1537–1545. doi: 10.2337/db06-0263. [DOI] [PubMed] [Google Scholar]
- Song J, Ke SF, Zhou CC, Zhang SL, Guan YF, Xu TY, Sheng CQ, Wang P, Miao CY. Nicotinamide phosphoribosyltransferase is required for the calorie restriction-mediated improvements in oxidative stress, mitochondrial biogenesis, and metabolic adaptation. J Gerontol A Biol Sci Med Sci. 2014;69:44–57. doi: 10.1093/gerona/glt122. [DOI] [PubMed] [Google Scholar]
- Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9:352–355. doi: 10.1038/nm824. [DOI] [PubMed] [Google Scholar]
- Varma V, Yao-Borengasser A, Rasouli N, Bodles AM, Phanavanh B, Lee MJ, Starks T, Kern LM, Spencer HJ, 3rd, McGehee RE, Jr, et al. Human visfatin expression: relationship to insulin sensitivity, intramyocellular lipids, and inflammation. J Clin Endocrinol Metab. 2007;92:666–672. doi: 10.1210/jc.2006-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Park C, Choi K, Bickel PE. OP9 mouse stromal cells rapidly differentiate into adipocytes: characterization of a useful new model of adipogenesis. J Lipid Res. 2006;47:450–460. doi: 10.1194/jlr.D500037-JLR200. [DOI] [PubMed] [Google Scholar]
- Xu XJ, Gauthier MS, Hess DT, Apovian CM, Cacicedo JM, Gokce N, Farb M, Valentine RJ, Ruderman NB. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. 2012;53:792–801. doi: 10.1194/jlr.P022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MJ, Yoshida M, Johnson S, Takikawa A, Usui I, Tobe K, Nakagawa T, Yoshino J, Imai S. SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD(+) and Function in Mice. Cell Metab. 2015;21:706–717. doi: 10.1016/j.cmet.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Imai S. Accurate measurement of nicotinamide adenine dinucleotide (NAD(+)) with high-performance liquid chromatography. Methods Mol Biol. 2013;1077:203–215. doi: 10.1007/978-1-62703-637-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.