

Abstract
The synthesis and biological evaluation of chromane-containing bryostatin analogues WN-2 to WN-7 and the previously reported salicylate-based analogue WN-8 are described. Analogues WN-2 to WN-7 are prepared through convergent assembly of the chromane-containing Fragment B-I with the “binding domain” Fragment A-I or its C26-des-methyl congener, Fragment A-II. The synthesis of Fragment B-I features enantioselective double C-H allylation of 1,3-propane diol to form the C2-symmetric diol 3 and Heck cyclization of bromodiene 5 to form the chromane core. The synthesis of salicylate WN-8 is accomplished through the union of Fragments A-III and B-II. The highest binding affinities for PKCα are observed for the C26-des-methyl analogues WN-3 (Ki = 63.9 nM) and WN-7 (Ki = 63.1 nM). All analogues, WN-2 to WN-8, inhibited growth of Toledo cells, with the most potent analogue being WN-7. This response, however, does not distinguish between phorbol ester-like and bryostatin-like behavior. In contrast, while many of the analogues contain a conserved C-ring in the binding domain and other features common to analogues with bryostatin-like properties, all analogues evaluated in the U937 proliferation and cell attachment assays displayed phorbol ester-like and/or toxic behavior, including WN-8, for which “bryostatin-like PKC modulatory activities” previously was suggested solely based on PKC binding. These results underscore the importance of considering downstream biological effects, as tumor suppression cannot be inferred from potent PKC binding.
Graphical abstract
Introduction
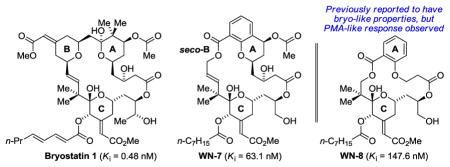
Discovered by Pettit using an assay for inhibitory activity against the P388 leukemia cell system, the bryostatins are a family of structurally complex marine macrolides isolated from the bryozoan Bugula neritina (Figure 1).1 The most abundant and well-studied member of this compound class, bryostatin 1, potently binds the C1 domain of protein kinase C (PKC) isozymes in vitro,2 activating PKC and modulating diverse downstream effects.3 Most notably, although bryostatin 1 potently binds and activates PKC, it antagonizes most biological responses of the phorbol esters, classic PKC activators that are generally tumor promoting, including phorbol 12-myristate 13-acetate (PMA).4 This remarkable behavior triggered a GMP campaign wherein 18 g of bryostatin 1 was isolated from 10,000 gallons of wet bryozoan.5 This material supported dozens of phase I and phase II clinical trials for cancer treatment,3 and led to the identification of bryostatin 1 as a clinical candidate for the treatment of Alzheimer’s disease6 and HIV.7
Figure 1.

PKC binding affinity of bryostatin 1 and 7, selected bryostatin analogues and neristatin 1.a
aBinding affinity to PKCα. See reference 7h for PKCα binding affinity of bryostatin 1 and bryostatin 7. bCompounds I-V prepared by Wender9 were reported to function similarly to bryostatin 1 with regard to the pattern of PKCδ-GFP translocation induced in rat basophilic leukemia cells.9h,i,k Binding affinity refers to a mixture of rat brain PKC isozymes. The initially reported binding affinity of I (0,25 nM) has been revised.9o cFor the indicated Merle bryologs prepared by Keck,10 PMA-like vs bryostatin-like biology was established via U937 attachment and inhibition of proliferation assays.
The biological properties of the bryostatins along with their low natural abundance have inspired heroic efforts toward the synthesis of both natural bryostatins8 and simplified functional analogues.9–12 In the context of cancer therapy, bryostatin-like activity of analogues was assumed based on potent PKC binding and, in certain cases, membrane translocation assays.9 However, as demonstrated by the elegant studies of Keck and Blumberg, bryostatin-like biological activity cannot be anticipated from potent PKC binding and membrane translocation alone, even for compounds that deviate only slightly from the structure of bryostatin 1 itself (Figure 1).10 Downstream biological responses must be assessed to determine whether analogues embody the special properties of bryostatin 1. Here, U937 human histiocytic lymphoma cell attachment and inhibition of proliferation assays have proven diagnostic (vide supra).10e These assays reveal that analogues retaining bryostatin-like activity are relatively intolerant vis-á-vis removal or modification of functional groups in the bryostatin A- and B-rings. In contrast, the bottom portion of bryostatin, which incorporates the C-ring and primarily influences PKC binding, appears to be less important in terms of defining PMA-like or bryostatin-like behavior. The biology of neristatin 1 dramatically illustrates these trends.13 Neristatin 1 incorporates A- and B-rings identical to several bryostatin family members; however, the bottom portion of neristatin is unique. Critically, neristatin 1 displays bryostatin 1-like behavior, not phorbol ester-like behavior, in U937 promyelocytic leukemia cells.13 These results support the hypothesis that the critical mechanistic feature of bryostatin is formation of a cap by the A- and B-rings over the C1 domain, held in position by interaction of the C-region or an equivalent binding group with the binding cleft of the PKC C1 domain. This concept is reflected in recently reported bryostatin analogues that incorporate simple DAG-like substructures in place of the C-region.14 Extensive simplification of the top portion of bryostatin to furnish analogues mimicking the biological profile of bryostatin remains an elusive, unmet challenge.
Using catalytic C-C bond formations developed in our laboratory,15 concise routes to the bryostatin A- and C-rings were devised,16 which, in turn, enabled the total synthesis of bryostatin 78g and the seco-B-ring analogue WN-1.12 Although WN-1 binds PKCα (Ki = 16.1 ± 1.1 nM) and inhibits growth of multiple leukemia cell lines, it displays PMA-like behavior in U937 cell attachment and proliferation assays, and in K562 and MV-4–11 proliferation assays. Such PMA-like behavior is surprising, as the A- and C-rings of WN-1 are shared by analogues that display bryostatin-like behavior in these assays (Figure 1). To assess whether greater conformational rigidity and lipophilicity might restore the desired bryostatin-like behavior in the absence of a B-ring, the synthesis and evaluation of the chromane-based analogues WN-2 to WN-7 and the previously described salicylate-based analogue WN-89m,n was undertaken. Beyond probing the biology of the bryostatins, the development of a novel catalytic asymmetric method for the synthesis of chromanes and chromanones, which are privileged substructures in drug discovery, represents a significant outcome of this work.17
Research Design and Methods
Synthesis of WN-2 to WN-8
Our approach to chromane containing bryostatin analogues WN-2 to WN-8 is illustrated in the retrosynthesis of WN-7 (Figure 2). Macrodiolide WN-7 is assembled from Fragments A-II and B-I via successive ester bond formation. As reported in our synthesis of bryostatin 7,8g Fragment A-I is prepared through hydrogen-mediated reductive coupling of glyoxal 1a and enyne 2a.16a Fragments A-II and A-III are prepared in a similar fashion from glyoxal 1a or 1b and enyne 2b (Scheme 1). Each reductive coupling forms the C20-C21 bond with control C20 carbinol stereochemistry and C21 alkene geometry. The C20 hydroxyl groups of the respective reductive coupling products are converted to the octanoates and then HF•pyridine in methanol is added to the reaction mixtures to furnish Fragment A-I and Fragment A-II in 8 steps from commercially available compounds.
Figure 2.

Retrosynthetic analysis of WN-7 illustrating C-C bonds formed via hydrogenative coupling.
Scheme 1.

Fragments A-I to A-III via H2-mediated reductive coupling of glyoxal 1a and 1b with 1,3-enyne 2a or 2b.a
aThe indicated conditions apply to Fragment A-I. Similar conditions are used for Fragments A-II and A-III. See reference 8g and supporting information for precise experimental details.
The synthesis of chromanone containing Fragment B-I begins with double asymmetric C-H allylation of 1,3-propane diol (Scheme 2).18 The resulting C2-symmetric diol 3 is converted to the mono-TBS ether 4. Deprotonation of 4 using sodium hydride followed by addition of the alkoxide to tert-butyl 3-bromo-2-fluorobenzoate delivers the SNAr product 5.19 Exposure of 5 to conditions for Heck cyclization provided the desired chromane 6,20 which upon concomitant ozonolysis21 of the terminal olefin moieties provides keto-aldehyde 7. Finally, Pinnick oxidation22 followed by treatment with diazomethane and hydrolysis of the tert-butyl ester delivers Fragment B-I.
Scheme 2.

Synthesis of Fragment B-I via transfer hydrogenative double allylation of 1,3-propane diol.a
aSee supporting information for experimental details.
The synthesis of chromanone-containing macrodiolides WN-2 and WN-4 was accomplished as follows (Scheme 3). Fragments A-I and B-I were treated with PyBroP in the presence of Hunig’s base and DMAP to form ester 8 in 85% yield.23 Exposure of 8 to trifluoracetic acid cleaves the acetonide to provide a triol, which is reacted with TBSOTf to form the bis-silyl ether with high levels of chemoselectivity. Trimethyltin hydroxide24 enables chemoselective cleavage of the methyl ester in the presence of the C20 octanoate to form the hydroxy acid 9. Yamaguchi lactonization converts hydroxy acid 9 to macrodiolide 10.25 A step-wise protocol26a for oxidative cleavage of the diene moiety of 10 was more efficient than direct Lemieux-Johnson oxidation.26b Subsequent Pinnick oxidation22 furnished the carboxylic acid, which upon removal of the silyl ethers results in spontaneous closure of the macrodiolide C-ring. This strategy for C-ring closure was not possible for the corresponding methyl ester due to lactonization onto the C23 alcohol. To our knowledge, WN-4 is the first carboxylic acid containing bryostatin analogue. Treatment of WN-4 with TMS diazomethane delivered the methyl ester WN-2.
Scheme 3.

Synthesis of the chromanone-based macrodiolides WN-2 and WN-4.a
aSee supporting information for experimental details.
Syntheses of WN-3 and WN-5, the C26 des-methyl congeners of WN-2 and WN-4, respectively, were developed to determine whether potency could be retained or enhanced through this structural simplification.9o The construction of WN-3 and WN-5 required the synthesis of 1,3-enyne 2b (Scheme 4), the precursor of Fragment A-II. To this end, commercially available (R)-butane-1,2,4-triol acetonide 11 was subjected to PCC-mediated oxidation followed by chelation controlled propargylation of the resulting aldehyde.27 The homopropargyl alcohol 12 was formed with good levels of diastereoselectivity. Conversion of the secondary alcohol to the TBDPS ether followed by Sonogashira coupling provides the 1,3-enyne 2b. As described above (Scheme 1), hydrogen- mediated reductive coupling of 1,3-enyne 2b with glyoxal 1a proceeds in good yield with excellent control of alkene geometry and C20 carbinol stereochemistry. A one-pot octanoylation-desilylation then affords Fragment A-II.
Scheme 4.

Synthesis of 1,3-enyne 2b via chelation controlled propargylation.a
aSee supporting information for experimental details.
With Fragment A-II in hand, the synthesis of C26 des-methyl chromanone-based macrodiolides WN-3 and WN-5 was undertaken (Scheme 5). Although closely related in structure to analogues WN-2 and WN-4, the C26 des-methyl congeners WN-3 and WN-5 required a different protecting group strategy. As in the synthesis of WN-2 and WN-4, Fragments A-II and B-I were treated with PyBroP in the presence of Hunig’s base and DMAP to form ester 13.23 Cleavage of the acetonide using trifluoroacetic acid provides a triol. Treatment with TBSOTf led to selective formation of the bis-TBS ether; however, subsequent saponification using trimethyltin hydroxide24 led to cleavage of the C26-TBS ether. Hence, the more robust C26-TIPS ether was installed and the C3-alcohol was left unprotected. Saponification in the presence of the C26-TIPS ether mediated by trimethyltin hydroxide24 provided the dihydroxy acid 14. Macrolactonization under Shiina conditions28 formed macrodiolide 15. As in the synthesis of WN-2 and WN-4, one-pot diene oxidative cleavage,26a Pinnick oxidation22 and exhaustive silyl deprotection provided WN-5, which upon methylation of the carboxylic acid delivered WN-3.
Scheme 5.

Synthesis of the C26 des-methyl chromanone-based macrodiolides WN-3 and WN-5.a
aSee supporting information for experimental details.
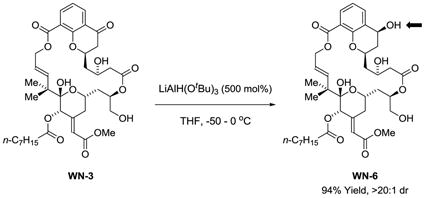
Reduction of chromanone WN-3 at the C7 ketone using LiAl(OtBu)3 occurred with high level of diastereoselectivity to furnish the C7 alcohol WN-6 (eq. 1).29 Direct chemoselective acylation of WN-6 to form the C7 acetoxy compound WN-7 as found in bryostatin 1 was not possible due to competing functionalization of the C26 hydroxyl. Hence, an alternate sequence was devised (Scheme 6). The C26 hydroxyl of WN-3 was converted to the TBS ether and methanolic KBH4 was added to the reaction mixture.29 The resulting secondary alcohol 16 was formed as a single diastereomer as determined by 1H NMR. Acetoxylation of the C7 hydroxyl moiety under conditions developed by Shiina28 followed by removal of the TBS protecting group provided WN-7.
Scheme 6.

Synthesis of the C26 des-methyl chromane-based macrodiolide WN-7.a
aSee supporting information for experimental details.
 |
(1) |
The modularity of our synthetic strategy is highlighted by the synthesis of the salicylate-based analogue WN-8, previously reported by Wender (Scheme 8).9m,n The synthesis of WN-8 begins with the reaction of Fragment A-III with the acid chloride derived from Fragment B-II (Scheme 7, not discussed) to form the neopentyl ester 19. Concomitant removal of the acetonide and tert-butyl ester moieties using trifluoroacetic acid followed by treatment with TBS chloride provided the hydroxy acid 20. Cyclization under Shiina conditions28 delivers the macrodiolide 21. Modified Johnson-Lemieux oxidative cleavage26 of the diene terminus followed by Pinnick oxidation22 and removal of the TBS and TBDPS ethers provides the carboxylic acid 22. Finally, treatment with methyl iodide delivers WN-8 in a total of 14 steps (LLS), where previously 19 steps (LLS) were required for its preparation.9m,n
Scheme 8.

Synthesis of previously reported salicylate-based macrodiolide WN-8.a
aSee supporting information for experimental details.
Scheme 7.

Synthesis of Fragment B-II.a
aSee supporting information for experimental details.
Biological Evaluation of WN-2-WN-8
Determination of Binding Affinity to PKCα
The biological evaluation of WN-2-WN-8 began with the determination of their binding affinities (Ki) toward purified PKCα (Figure 3).30 The C26 des-methyl analogue WN-3 (Ki = 63.9 ± 16.5 nM) has a 3-fold stronger binding affinity than the parent C26 methyl analogue WN-2 (Ki = 213.7 ± 33.1 nM).9o Compared to the methyl esters WN-2 and WN-3, the carboxylic acids WN-4 and WN-5 display a 20–40 fold decrease in potency (Ki = 3988 ± 531 nM and Ki = 2765 ± 738 nM, respectively). The C7-alcohol analogue WN-6 (Ki = 135.2 ± 22.1 nM) is two-fold less potent than the C7-OAc analogue WN-7 (Ki = 63.1 ± 13.6 nM) as well as the C7-ketone analogue WN-3 (Ki = 63.9 ± 16.5 nM). Recently, Wender reported that WN-8 bound to PKCβI and PKCδ with Kis = 24 nM and 18 nM, respectively.9m,n Our studies have shown that WN-8 displays weaker binding affinity toward PKCα (Ki = 147.6 ± 17.5 nM). These differences suggested that WN-8 showed some level of PKC isoform selectivity, as has been previously observed for bryostatin 1.31 The U937 and LNCaP cell lines are the two cell lines in which we have characterized the biological actions of bryostatin analogues in most detail. PKCδ and PKCβII are the major PKC isoforms in the U937 cells; PKCδ and PKCα are the highest expressed PKC isoforms in the LNCaP cells.32 We therefore measured the affinity of WN-8 for PKCβII and PKCδ under comparable conditions to those we used for the measurements with PKCα and obtained Ki values of 82.1 ± 14.9 and 56.2 ± 6.0 nM, respectively. We conclude that there is modest selectivity of WN-8 between various PKC isoforms. The binding affinity of WN-8 is weaker than that of WN-3 and WN-7 and very modestly weaker than that of WN-6. Thus, while the chromanone and salicylate analogues retain PKC binding in the nanomolar regime, WN-1 (Ki = 16.1 nM) remains the most potent compound in the WN-series (Ki = 16.1–3988 nM). These data suggest that the northern region of bryostatin analogues not only plays a critical role in determining bryostatin-like vs phorbol ester-like biological activity but strongly influences preorganization (molecular conformation) of the southern binding region and, ultimately, potency.
Figure 3.

PKC binding affinity of WN-1 to WN-8.a
aBinding affinity to PKCα. See reference 8h for PKCα binding affinity of bryostatin 1 and bryostatin 7. bBinding affinity toward PKCδ and PKCβ, respectively.
Activity in U937 Human Histiocytic Lymphoma Cells
The determination of binding affinity to PKC isozymes represents an initial step in understanding the biological properties of the present analogues. Observing downstream biological responses is crucial to determine whether these compounds capture the unique effects associated with bryostatin 1. With U937 human histiocytic lymphoma cells, bryostatin 1 and PMA induce contrasting cellular responses.10,33 While PMA inhibits the proliferation and promotes attachment of U937 cells, these cells show little response upon treatment with bryostatin 1. Furthermore, coadministration of bryostatin 1 with PMA results in the inhibition of the PMA-like cellular responses, showing that the lack of effect of bryostatin 1 on proliferation and attachment is not due to instability.
In the U937 growth and attachment assays, WN-2 and WN-3 display PMA-like behavior. However, at higher concentrations these analogues display toxicity (Figure 4). In the growth inhibition assay, WN-2 and WN-3 exhibit strong inhibition at 10000, 20000, and 40000 nM. While bryostatin 1 is able to reverse the antiproliferative effects of PMA in U937 cells, it partially reverses the effects of WN-2 and WN-3 at 10,000 nM but not at 40,000 nM. These results are consistent with cell inhibition at 10,000 nM being partially attributable to a PMA-like effect and the further inhibition at higher concentrations being toxicity superimposed on the specific PMA-like inhibition. This trend is also seen in the cell attachment assay for WN-2 and WN-3. The compounds induce the PMA-like response of cell attachment at 10,000 nM and this attachment is antagonized by bryostatin 1. At 20,000 and 40,000 nM, in contrast, the attachment is no longer seen, consistent with toxicity at this higher concentration range.
Figure 4.

Evaluation of WN-2, WN-3, WN-6, WN-7, and WN-8 in U937 Human Histiocytic Lymphoma Cells.a
aSee supporting information for experimental details.
The biological activities of WN-6 and WN-7 in U937 cells are similar to that of WN-2 and WN-3. For WN-6, the toxicity predominates. Growth inhibition is not blocked by bryostatin 1, and the minute induction of attachment caused by WN-6 is also not reversed when coadministered with bryostatin 1. For WN-7, a combination of PMA-like and toxic behavior is observed. It inhibits cell growth like PMA but with only modest reversal from bryostatin 1, suggesting that much of the growth inhibition is due to toxicity. In the cell attachment assay, the PMA-like effect is more prominent, with good inhibition by bryostatin 1.
The salicylate analogue WN-8, first reported by the Wender group, 9m,n also was tested in these cell assays. WN-8 was previously suggested to have “bryostatin-like PKC modulatory activities” solely on the basis of binding.9m,n However, WN-8 behaves like PMA in the U937 growth and attachment assays. Further, in contrast to WN-2, WN-3, WN-6, and WN-7, the PMA-like behavior displayed by WN-8 is not due to a non-specific toxic effect. Analogues WN-4 and WN-5 were not tested in U937 cells given their weak effect relative to WN-2 and WN-3 in the Toledo cells (vide infra) and the marginal effect of WN-2 and WN-3 in the U937 cells.
Effects on TNFα Expression and Activity in Toledo Cells
TNFα secretion from U937 cells was measured after treatment with analogues WN-2, WN-3, WN-6, WN-7 or WN-8 for 60 hours (Figure 5). While bryostatin 1 generally has little effect on TNFα secretion, PMA induces secretion in a dose-dependent manner. Results show that high concentrations (10000 nM) of WN-2 WN-8 are able to induce TNFα secretion even though not to the level induced by PMA. However, this induction is lost at higher concentrations of WN-2, WN-3, WN-6, WN-7, consistent with the higher concentrations being toxic for these analogues.
Figure 5.

TNFα secretion from U937 cellsa
aSee supporting information for experimental details.
Unlike their effects in U937 cells, bryostatin 1 and PMA both induce antiproliferative responses in Toledo cells. Compared to bryostatin 1 and PMA, WN-2 WN-8 had IC50 values for growth inhibition that are significantly shifted to the right, reflecting weaker potency (Figure 6). The most potent of these analogues in Toledo cells is WN-7; WN-2, WN-3, WN-6, and WN-8 are 3-fold less potent than WN-7 and all similar to one another. Lastly, within this assay, the C35-acids WN-4 and WN-5 show only minor growth inhibition until concentrations >10 μM are reached.
Figure 6.

Toledo Cell Growth Assaya
aSee supporting information for experimental details.
Conclusions
In summary, we report the synthesis and biological evaluation of chromane-containing bryostatin analogues WN-2 to WN-7 and the previously reported salicylate-based analogue WN-8.9m,n All WN-series analogues conserve the bryostatin C-ring and A-ring features common to analogues with bryostatin-like properties. Despite this structural homology and the observance of nanomolar binding affinities for PKCα, all analogues evaluated in the U937 proliferation and cell attachment assays displayed PMA-like and/or toxic behavior. These data, along with prior studies by Keck and Blumberg,10 demonstrate the importance of considering downstream biological effects, as potent PKC binding by itself does not predict bryostatin-like biology. Our data further serve as a reminder that the structure of the B-ring region of bryostatin influences PKC binding affinity and profoundly impacts biology, as previously observed.12
Supplementary Material
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM093905) are acknowledged for partial support of this research. Partial support was also provided by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (Z1A BC 005270). The Cancer Prevention Research Institute of Texas (RP101501) is acknowledged for Postdoctoral Fellowship (I.V.). Skilled technical assistance was provided by Kim Wasik. This project was funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E.
Footnotes
Supporting Information. Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra. This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.Pettit GR, Day JF, Hartwell JL, Wood HB. Nature. 1970;227:962. doi: 10.1038/227962a0.Pettit GR, Herald CL, Doubek DL, Herald DL, Arnold E, Clardy J. J Am Chem Soc. 1982;104:6846.Yu HB, Yang F, Li YY, Gan JH, Jiao WH, Lin HW. J Nat Prod. 2015;78:1169. doi: 10.1021/acs.jnatprod.5b00081.It is believed that the bryostatins actually derives from a bacterial symbiont of B. neritina: Sudek S, Lopanik NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. J Nat Prod. 2007;70:67. doi: 10.1021/np060361d.Linneman J, Paulus D, Lim-Fong G, Lopanik NB. PLoS One. 2014;9:e108783. doi: 10.1371/journal.pone.0108783.
- 2.For selected studies on the binding of bryostatin 1 to PKC isozymes, see: Berkow RL, Kraft AS. Biochem Biophys Res Commun. 1985;131:1109. doi: 10.1016/0006-291x(85)90205-0.Kraft AS, Smith JB, Berkow RL. Proc Natl Acad Sci USA. 1986;83:1334. doi: 10.1073/pnas.83.5.1334.Kazanietz MG, Lewin NE, Gao F, Petit GR, Blumberg PM. Mol Pharmacol. 1994;46:374.
- 3.For reviews of the chemistry and biology the bryostatins and their analogues, see: Mutter R, Wills M. Bioorg Med Chem Lett. 2000;8:1841. doi: 10.1016/s0968-0896(00)00150-4.Hale KJ, Hummersone MG, Manaviazar S, Frigerio M. Nat Prod Rep. 2002;19:413. doi: 10.1039/b009211h.Kortmansky J, Schwartz GK. Cancer Invest. 2003;21:924. doi: 10.1081/cnv-120025095.Wender PA, Baryza JL, Hilinski MK, Horan JC, Kan C, Verma VA. In: Drug Discovery Research: New Frontiers in the Post-Genomic Era. Huang Z, editor. Chap 6. Wiley; Hoboken: 2007. p. 127.Hale KJ, Manaviazar S. Chem Asian J. 2010;5:704. doi: 10.1002/asia.200900634.Wender PA, Loy BA, Schrier AJ. Isr J Chem. 2011;51:453. doi: 10.1002/ijch.201100020.Yu L, Krische MJ. In: In Total Synthesis: At the Frontier of Organic Chemistry. Li JJ, Corey EJ, editors. Springer; Heidelberg, Germany: 2013. pp. 103–130.For information on bryostatin 1 in clinical trials for the treatment of cancer, see: (h) http://clinicaltrials.gov.
- 4.(a) Berkow RL, Kraft AS. Biochem Biophys Res Commun. 1985;131:1109. doi: 10.1016/0006-291x(85)90205-0. [DOI] [PubMed] [Google Scholar]; (b) De Vries DJ, Herald CL, Pettit GR, Blumberg PM. Biochem Pharmacol. 1988;37:4069. doi: 10.1016/0006-2952(88)90097-4. [DOI] [PubMed] [Google Scholar]; (c) Wender PA, Cribbs CM, Koehler KF, Sharkey NA, Herald CL, Kamano Y, Pettit GR, Blumberg PM. Proc Nat Acad Sci USA. 1988;85:7197. doi: 10.1073/pnas.85.19.7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaufelberger DE, Koleck MP, Beutler JA, Vatakis AM, Alvarado AB, Andrews P, Marzo LV, Muschik GM, Roach J, Ross JT, Lebherz WB, Reeves MP, Eberwein RM, Rodgers LL, Testerman RP, Snader KM, Forenza S. J Nat Prod. 1991;54:1265. doi: 10.1021/np50077a004. [DOI] [PubMed] [Google Scholar]
- 6.For studies related to the use of bryostatin in treatment of Alzheimer’s disease, see: Etcheberrigaray R, Tan M, Dewachter I, Kuiperi C, Van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozikowski AP, Van Leuven F, Alkon DL. Proc Natl Acad Sci USA. 2004;101:11141. doi: 10.1073/pnas.0403921101.Sun MK, Alkon DL. Eur J Pharmacol. 2005;512:43. doi: 10.1016/j.ejphar.2005.02.028.Alkon DL, Sun MK, Nelson TJ. Trends Pharmacol Sci. 2007;28:51. doi: 10.1016/j.tips.2006.12.002.Hongpaisan J, Sun MK, Alkon DL. J Neurosci. 2011;31:630. doi: 10.1523/JNEUROSCI.5209-10.2011.Williams P, Sorribas A, Howes MJR. Nat Prod Rep. 2011;28:48. doi: 10.1039/c0np00027b.Hongpaisan J, Xu C, Sen A, Nelson TJ, Alkon DL. Neurobiol Dis. 2013;55:44. doi: 10.1016/j.nbd.2013.03.012.Xu C, Liu QY, Alkon DL. Neuroscience. 2014;268:75. doi: 10.1016/j.neuroscience.2014.03.008.Kenmei M, Shigeru S, Hideaki W, Kan S. Am J Phys Med Rehabil. 2015;94:239.Patrizia R, Aliaksei K, Palma L, Rossana M, Giulia P, Massimo F. Marine Drugs. 2016;14:5.
- 7.For studies related to the use of bryostatin in treatment of HIV, see: Perez M, de Vinuesa AG, Sanchez-Duffhues G, Marquez N, Bellido ML, Munoz-Fernandez MA, Moreno S, Castor TP, Calzado MA, Munoz E. Curr HIV Res. 2010;8:418. doi: 10.2174/157016210793499312.Mehla R, Bivalkar-Mehla S, Zhang R, Handy I, Albrecht H, Giri S, Nagarkatti P, Nagarkatti M, Chauhan A. PLoS One. 2010;5:e11160. doi: 10.1371/journal.pone.0011160.DeChristopher BA, Loy BA, Marsden MD, Schrier AJ, Zack JA, Wender PA. Nature Chem. 2012;4:705. doi: 10.1038/nchem.1395.Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, Mau M, Ruelas D, Saleh S, Shirakawa K, Siliciano RF, Singhania A, Soto PC, Terry VH, Verdin E, Woelk C, Wooden S, Xing S, Planelles V. PLoS Path. 2013;9:e1003834. doi: 10.1371/journal.ppat.1003834.Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. Nat Med. 2014;20:425. doi: 10.1038/nm.3489.Archin NM, Margolis DM. Curr Opin Infect Dis. 2014;27:29. doi: 10.1097/QCO.0000000000000026.Martinez-Bonet M, Clemente MI, Alvarez S, Diaz L, Garcia-Alonso D, Munoz E, Moreno S, Munoz-Fernandez MA. Antiviral Res. 2015;123:163. doi: 10.1016/j.antiviral.2015.09.014.Iordanskiy S, Van Duyne R, Sampey GC, Woodson CM, Fry K, Saifuddin M, Guo J, Wu Y, Romerio F, Kashanchi F. Virology. 2015;485:1. doi: 10.1016/j.virol.2015.06.021.Gutierrez C, Serrano-Villar S, Madrid-Elena N, Perez-Elias MJ, Martin ME, Barbas C, Ruiperez J, Munoz E, Munoz-Fernandez MA, Castor T, Moreno S. AIDS. 2016;30:1385. doi: 10.1097/QAD.0000000000001064.
- 8.For total syntheses of naturally occurring bryostatins, see: (a) Bryostatin 1: Keck GE, Poudel YB, Cummins TJ, Rudra A, Covel JA. J Am Chem Soc. 2011;133:744. doi: 10.1021/ja110198y.Bryostatin 2: Evans DA, Carter PH, Carreira EM, Prunet JA, Charette AB, Lautens M. Angew Chem Int Ed. 1998;37:2354. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2354::AID-ANIE2354>3.0.CO;2-9.Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M. J Am Chem Soc. 1999;121:7540.Bryostatin 3: Ohmori K, Ogawa Y, Obitsu T, Ishikawa Y, Nishiyama S, Yamamura S. Angew Chem Int Ed. 2000;39:2290.Ohmori K. Bull Chem Soc Jpn. 2004;77:875.Bryostatin 7: Kageyama M, Tamura T, Nantz MH, Roberts JC, Somfai P, Whritenour DC, Masamune S. J Am Chem Soc. 1990;112:7407.Lu Y, Woo SK, Krische MJ. J Am Chem Soc. 2011;133:13876. doi: 10.1021/ja205673e.Kedei N, Lewin NE, Géczy T, Selezneva J, Braun DC, Chen J, Herrmann MA, Heldman MR, Lim L, Mannan P, Garfield SH, Poudel YB, Cummins TJ, Rudra A, Blumberg PM, Keck GE. ACS Chem Biol. 2013;8:767. doi: 10.1021/cb300671s.Bryostatin 9: Wender PA, Schrier AJ. J Am Chem Soc. 2011;133:9228. doi: 10.1021/ja203034k.Bryostatin 16: Trost BM, Dong G. Nature. 2008;456:485. doi: 10.1038/nature07543.
- 9.For bryostatin analogues prepared in the Wender laboratory, see: Wender PA, De Brabander J, Harran PG, Jimenez JM, Koehler MFT, Lippa B, Park CM, Shiozaki M. J Am Chem Soc. 1998;120:4534.Wender PA, De Brabander J, Harran PG, Jimenez JM, Koehler MFT, Lippa B, Park CM, Siedenbiedel C, Pettit GR. Proc Natl Acad Sci USA. 1998;95:6624. doi: 10.1073/pnas.95.12.6624.Wender PA, Cribbs CM, Koehler KF, Sharkey NA, Herald CL, Kamano Y, Pettit GR, Blumberg PM. Proc Natl Acad Sci USA. 1988;85:7197. doi: 10.1073/pnas.85.19.7197.Wender PA, De Brabander J, Harran PG, Hinkle KW, Lippa B, Pettit GR. Tetrahedron Lett. 1998;39:8625.Wender PA, Lippa B. Tetrahedron Lett. 2000;41:1007.Wender PA, Hinkle KW. Tetrahedron Lett. 2000;41:6725.Wender PA, Baryza JL, Bennett CE, Bi FC, Brenner SE, Clarke MO, Horan JC, Kan C, Lacôte E, Lippa BS, Nell PG, Turner TM. J Am Chem Soc. 2002;124:13648. doi: 10.1021/ja027509+.Baryza JL, Brenner SE, Craske ML, Meyer T, Wender PA. Chem Biol. 2004;11:1261. doi: 10.1016/j.chembiol.2004.06.014.Wender PA, Baryza JL, Brenner SE, Clarke MO, Craske ML, Horan JC, Meyer T. Curr Drug Discovery Technol. 2004;1:1. doi: 10.2174/1570163043484888.Wender PA, DeChristopher BA, Schrier AJ. J Am Chem Soc. 2008;130:6658. doi: 10.1021/ja8015632.Wender PA, Baryza JL, Brenner SE, DeChristopher BA, Loy BA, Schrier AJ, Verma VA. Proc Natl Acad Sci USA. 2011;108:6721. doi: 10.1073/pnas.1015270108.DeChristopher BA, Fan AC, Felsher DW, Wender PA. Oncotarget. 2012;3:58. doi: 10.18632/oncotarget.438.Wender PA, Nakagawa Y, Near KE, Staveness D. Org Lett. 2014;16:5136. doi: 10.1021/ol502491f.Wender PA, Staveness D. Org Lett. 2014;16:5140. doi: 10.1021/ol502492b.(o) Although the des-Me analogue I (Figure 1) was originally reported to have a stronger binding affinity than the parent C26 methyl analogue (ref. 9g), it has since been shown that they share similar binding potencies. For a correction to the initially determined binding affinity of what continues to be referred to as the “picolog,” see reference 9j, Supporting Information (S15, Binding Assay Results table, footnote “c”).
- 10.For bryostatin analogues prepared in the Keck laboratory, see: Keck GE, Kraft MB, Truong AP, Li W, Sanchez CC, Kedei N, Lewin NE, Blumberg PM. J Am Chem Soc. 2008;130:6660. doi: 10.1021/ja8022169.Keck GE, Poudel YB, Welch DS, Kraft MB, Truong AP, Stephens JC, Kedei N, Lewin NE, Blumberg PM. Org Lett. 2009;11:593. doi: 10.1021/ol8027253.Keck GE, Li W, Kraft MB, Kedei N, Lewin NE, Blumberg PM. Org Lett. 2009;11:2277. doi: 10.1021/ol900585t.Keck GE, Poudel YB, Rudra A, Stephens JC, Kedei N, Lewin NE, Peach ML, Blumberg PM. Angew Chem Int Ed. 2010;49:4580. doi: 10.1002/anie.201001200.Kedei N, Lubart ES, Lewin NE, Telek A, Lim L, Mannan P, Garfield SH, Kraft MB, Keck GE, Kolusheva S, Jelinek R, Blumberg PM. ChemBioChem. 2011;12:1242. doi: 10.1002/cbic.201100064.Kedei N, Telek A, Czap A, Lubart ES, Czifra G, Yang D, Chen J, Morrison T, Goldsmith PK, Lim L, Mannan P, Garfield SH, Kraft MB, Li W, Keck GE, Blumberg PM. Biochem Pharmacol. 2011;81:1296. doi: 10.1016/j.bcp.2011.03.018.Keck GE, Poudel YB, Rudra A, Stephens JC, Kedei N, Lewin NE, Blumberg PM. Bioorg Med Chem Lett. 2012;22:4084. doi: 10.1016/j.bmcl.2012.04.073.Kedei N, Telek A, Michalowski AM, Kraft MB, Li W, Poudel YB, Rudra A, Petersen ME, Keck GE, Blumberg PM. Biochem Pharmacol. 2013;85:313. doi: 10.1016/j.bcp.2012.10.028.Kraft MB, Poudel YB, Kedei N, Lewin NE, Peach ML, Blumberg PM, Keck GE. J Am Chem Soc. 2014;136:13202. doi: 10.1021/ja5078188.Kelsey JS, Cataisson C, Chen J, Herrmann MA, Petersen ME, Baumann DA, McGowan KM, Yuspa SH, Keck GE, Blumberg PM. Mol Carcinog. 2016 doi: 10.1002/mc.22460.(k) Also see reference 7h
- 11.For bryostatin analogues prepared in the Trost laboratory, see: Trost BM, Yang H, Thiel OR, Frontier AJ, Brindle CS. J Am Chem Soc. 2007;129:2206. doi: 10.1021/ja067305j.Trost BM, Dong G. J Am Chem Soc. 2010;132:16403. doi: 10.1021/ja105129p.Trost BM, Yang H, Dong G. Chem Eur J. 2011;17:9789. doi: 10.1002/chem.201002932.
- 12.For bryostatin analogues prepared in the Krische laboratory, see: Andrews, Ian P, Ketcham JM, Blumberg PM, Kedei N, Lewin NE, Krische MJ. J Am Chem Soc. 2014;136:13209. doi: 10.1021/ja507825s.
- 13.Kedei N, Kraft MB, Keck GE, Herald CL, Noeleen M, Petit GR, Blumberg PM. J Nat Prod. 2015;78:896. doi: 10.1021/acs.jnatprod.5b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baumann D, McGowan K, Kedei N, Peach M, Blumberg P, Keck G. J Org Chem. 2016;81:7862. doi: 10.1021/acs.joc.6b01516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Dechert-Schmitt AMR, Schmitt DC, Gao X, Itoh T, Krische MJ. Nat Prod Rep. 2014;31:504. doi: 10.1039/c3np70076c.Feng J, Kasun ZA, Krische MJ. J Am Chem Soc. 2016;138:5467. doi: 10.1021/jacs.6b02019.
- 16.(a) Cho CW, Krische MJ. Org Lett. 2006;8:891. doi: 10.1021/ol052976s. [DOI] [PubMed] [Google Scholar]; (b) Lu Y, Krische MJ. Org Lett. 2009;11:3108. doi: 10.1021/ol901096d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For reviews of the synthesis of biological activity of chromanes and chromanones, see: Kamat DP, Tilve SG, Kamat VP, Kirtany JK. Org Prep Proc Int. 2015;47:1.Emami S, Ghanbarimasir Z. Eur J Med Chem. 2015;93:539. doi: 10.1016/j.ejmech.2015.02.048.Wang NX, Xing Y, Wang YJ. Curr Org Chem. 2013;17:1555.Nibbs AE, Scheidt KA. Eur J Org Chem. 2012:449. doi: 10.1002/ejoc.201101228.Shi YL, Shi M. Org Biomol Chem. 2007;5:1499. doi: 10.1039/b618984a.
- 18.Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem Int Ed. 2009;48:5018. doi: 10.1002/anie.200901648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For related SNAr reactions, see: Dandapani S, Lowe JT, Comer E, Marcaurelle LA. J Org Chem. 2011;76:8042. doi: 10.1021/jo2011957.Naidu NB, Patel M, D’Andrea S, Zheng BZ, Connolly TP, Langley DR, Peese K, Wang Z, Walker MA, Kadow JF. Inhibitors of Immunodeficiency Virus Replication. 2014/028384. PCT Int Appl WO. 2014
- 20.For intramolecular Heck reaction of a related system, see: Garcia J, Barluenga S, Beebe K, Neckers L, Winssinger N. Chem Eur J. 2010;16:9767. doi: 10.1002/chem.201001080.
- 21.For a related ozonolysis to form a chromanone ketoaldehyde, see: Hornillos V, Perez M, Fananas-Mastral M, Feringa BL. J Am Chem Soc. 2013;135:2140. doi: 10.1021/ja312487r.
- 22.Bal BS, Childers WE, Jr, Pinnick HW. Tetrahedron. 1981;37:2091. [Google Scholar]
- 23.Coste J, Frerot E, Jouin P. J Org Chem. 1994;59:2437. [Google Scholar]
- 24.Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS. Angew Chem Int Ed. 2005;44:1378. doi: 10.1002/anie.200462207. [DOI] [PubMed] [Google Scholar]
- 25.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989. [Google Scholar]
- 26.(a) Keck GE, Giles RL, Cee VJ, Wager CA, Yu T, Kraft MB. J Org Chem. 2008;73:9675. doi: 10.1021/jo802215v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu W, Mei Y, Kang Y, Hua Z, Jin Z. Org Lett. 2004;6:3217. doi: 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- 27.Almendros P, Rae A, Thomas EJ. Tetrahedron Lett. 2000;41:9565. [Google Scholar]
- 28.Shiina I, Kubota M, Oshimumi H, Hasizume M. J Org Chem. 2004;69:1822. doi: 10.1021/jo030367x. [DOI] [PubMed] [Google Scholar]
- 29.See Supporting Information for stereochemical assignments associated with the LiAl(OtBu)3 reduction to form WN-6 and the KBH4/MeOH reduction to form 16. Stereoselective reductions of structurally related chromanones using both these reagents have been reported: Buckle DR, Eggleston DS, Houge-Frydrych CSV, Pinto IL, Readshaw SA, Smith DG, Webster RAB. J Chem Soc Perkin Trans 1. 1991:2763.Ishikawa T, Oku Y, Kotake KI. Tetrahedron. 1997;53:14915.Tanaka T, Kumamoto T, Ishikawa T. Tetrahedron Lett. 2000;41:10229.
- 30.For experimental details regarding binding affinity of WN-series to PKCα see supporting information.
- 31.Kazanietz MG, Lewin NE, Gao F, Pettit GR, Blumberg PM. Mol Pharmacol. 1994;46:374. [PubMed] [Google Scholar]
- 32.Chen JQ, Heldman MR, Herrmann MA, Kedei N, Woo W, Blumberg PM, Goldsmith PK. Anal Biochem. 2013;442:97. doi: 10.1016/j.ab.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vrana JA, Saunders AM, Chellappan SP, Grant S. Differentiation. 1998;63:33. doi: 10.1046/j.1432-0436.1998.6310033.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.