

Abstract
Background and purpose
Chronic traumatic encephalopathy (CTE) is a neurodegenerative disease occurring most commonly in athletes and is caused by repeated concussive or subconcussive blows to the head. The main purpose of this review is to evaluate the published literature on chronic traumatic encephalopathy (CTE) in athletes participating in high-impact sports. In particular, we highlight the significance of concussive and subconcussive impacts in multiple sports, elucidate the differences between clinical/pathological features of CTE and related neurodegenerative diseases, and provide an explanation for the variation in clinical presentation between athletes of different sports.
Methods
A review targeting relevant publications to CTE was performed. The PubMed/MEDLINE index was searched for keywords such as “chronic traumatic encephalopathy,” “repetitive traumatic brain injury,” “mild traumatic brain injury,” and “concussion” from year 1924 through March 1, 2016.
Results
A consensus panel’s recent identification of a pathognomonic pathology in CTE, characterized by an irregular distribution of phosphorylated tau deposits, is an important step in developing consensus diagnostic criteria and clinicopathological studies. After review of major clinical studies, evidence suggests that there are clear differences in neuropathological features, clinical progression, and manifestation of symptoms between CTE and other neurodegenerative diseases. The literature suggests boxers tend to have more severe symptoms than other athletes due to more frequent rotational and shearing impacts. Data regarding genetic predispositions of CTE have been inconsistent in part due to low subject populations. Positron emission tomography imaging involving tau-binding ligands has recently proven effective in differentiating CTE from control groups and other neurodegenerative diseases.
Conclusions
Further longitudinal studies should be conducted to correlate the number of suffered concussive/subconcussive forces to the likelihood of developing chronic traumatic brain injury symptoms. Research striving for a reliable antemortem CTE diagnosis would be immensely beneficial, leading to more accurate estimates of prevalence, allowing clinicians to assess future risk of athletes’ continued participation in sports, and enabling clinicians to make appropriate preventive recommendations.
Keywords: Chronic traumatic encephalopathy, brain injury, traumatic brain injury, concussion, neurological injury, pathology
Abbreviations
- 18F-FDDNP
2-(1-{6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene) malononitrile
- Aβ
Amyloid beta
- AD
Alzheimer’s disease
- AGD
Argyrophilic grain disease
- Apo
Apolipoprotein
- ATP
Adenosine triphosphate
- BBB
Blood brain barrier
- C9ORF72
Chromosome 9 open reading frame 72
- CBD
Corticobasal degeneration
- CBI
Chronic brain injury
- CCI
Controlled cortical impact
- CPCS
Chronic postconcussive syndrome
- CSF
Cerebrospinal fluid
- CT
Computed tomography
- CTE
Chronic traumatic encephalopathy
- DP
Diffuse plaques
- FTD
Frontotemporal dementia
- GRN
Progranulin
- GT
Glial tangles
- Kd
Kilodaltons
- MAPT
Microtubule-associated protein tau
- MND
Motor neuron disease
- MRI
Magnetic resonance imaging
- mTBI
Mild traumatic brain injury
- NFT
Neurofibrillary tangles
- NIBIB
National Imaging of Biomedical Imaging and Bioengineering
- NINDS
National Institute of Neurological Disorders and Stroke
- NN
Neuropil neurites
- NT
Neuropil threads
- p-tau
Phosphorylated tau
- PART
Primary age-related tauopathy
- PD
Parkinson’s disease
- PET
Positron emission tomography
- PSP
Progressive supranuclear palsy
- rTBI
Repetitive traumatic brain injury
- TBI
Traumatic brain injury
- TDP-43
Tar-DNA-binding-protein of approximately 43 kilodaltons
- TMEM106B
Transmembrane protein 106B
INTRODUCTION
Chronic traumatic encephalopathy (CTE) is characterized as a progressive, neurodegenerative disease caused by repetitive mild traumatic brain injury (mTBI) [1]. The vast majority of confirmed CTE cases have been in athletes competing in high-impact sports such as football and boxing, where the frequency of concussive and subconcussive forces is substantial [2].
Currently, the only established method to confirm the diagnosis of CTE is histopathological analysis of the brain postmortem. Distinct neuropathological changes are evident at both the gross and microscopic levels on autopsy, characterized by a prominent tauopathy in a pattern that differentiates CTE from other neurodegenerative diseases [3,4]. Recently, the first consensus meeting for CTE neuropathological criteria was held, substantiating CTE as a distinct disease with pathognomonic pathology [4]. Although currently no method exists for diagnosing definite CTE in living patients, nascent research is being performed to identify clinical biomarkers that would allow for accurate diagnosis antemortem. This would provide further insight into true incidence and prevalence rates [5] and may eventually lead to earlier intervention to improve clinical outcomes.
Neurological injury associated with participation in high-impact sports has been apparent for decades, but CTE is gaining increased attention in the media and academia more recently, due to heightened awareness, or perhaps an increasing prevalence [6]. CTE has a heterogeneous clinical presentation. Some people who suffer from frequent repetitive mTBI do not develop symptoms of CTE, while others experience early onset and rapid progression of the disease. Though development of neurodegenerative disease is usually multifactorial, the differences in symptom severity between those individuals who endure similar exposure of mTBI suggest that some athletes who are involved with high-impact sports are more susceptible to developing CTE than others. The pathophysiologic mechanisms of CTE are still being elucidated, and no consensus has been reached to date [7–9].
In this review, we have provided a summary of the most relevant research on the following aspects of CTE: (1) epidemiology of CTE, (2) clinical manifestations between athletes of different sports, (3) genetic predispositions of CTE, (4) discuss the existing in vivo models that simulate CTE, (5) review clinical studies regarding CTE, and (6) highlight the potential for clinical biomarkers and imaging techniques that aid in the diagnosis of CTE in vivo.
SEARCH STRATEGY
We reviewed the published literature relevant to CTE both in preclinical and in clinical settings. The PubMed/MEDLINE index was searched for keywords such as “chronic traumatic encephalopathy,” “repetitive traumatic brain injury,” “mild traumatic brain injury,” “neurodegenerative diseases,” “tauopathy,” “neurofibrillary tangles,” “apolipoprotein (Apo)E4,” and “concussion.” The search was conducted from the year 1924 through March 1, 2016.
EPIDEMIOLOGY OF CTE
It is estimated that between 1.6 and 3.8 million sports-related mTBIs occur annually, and sport-related head injuries make up 20% of TBI cases in the United States each year [10,11]. This may be an underestimate, as mTBI frequently goes unreported at multiple levels in a variety of sports [12–16]. As the number of athletes at the high school, collegiate, and professional levels continues to increase, related concussive injuries may rise proportionally [17,18]. Incidence of reported mTBI is significant in high-impact sports such as American football, boxing, ice hockey, and rugby [19,20]. The incidence of concussion in American football is particularly alarming due to the large number of total participants across a variety of levels [20]. Broglio et al. [21] suggest that, on average, high school football players sustain 24 head impacts per game that exceed 14 g-force. Similar results have been shown in male collegiate hockey players, indicating that a variety of athletes endure hundreds of significant head impacts per season [21,22]. Recent advances in protective equipment act to mitigate the effects of high-impact forces; however, an athlete’s perceived lower level of risk may spur more risky behavior during games, leading to more severe mTBI impacts [23].
The incidence and prevalence of CTE in athletes due to repetitive mTBI is largely unknown, as most data regarding the disease have been analyzed retrospectively after patients are deceased [2]. A lack of clearly defined neuropathological and clinical diagnostic criteria has inhibited large epidemiological studies from going into effect [24]. Among longitudinal studies linking TBI to neurodegenerative symptoms in living patients, it is unclear whether patients’ clinical presentation fully resembles CTE, Alzheimer’s disease (AD), frontotemporal dementia (FTD), Parkinson’s disease (PD), motor neuron disease (MND), or a combination of these diseases [24], even though the average age of symptom onset for these neurodegenerative diseases is different. Still, previous research has found a significant association between a single mTBI event and chronic cognitive impairment, which is frequently seen in those presenting with CTE [25–27]. Additionally, a retrospective analysis of over 3400 American professional football players indicates three times as many deaths from neurodegenerative disease compared to the general population, and recent research shows that CTE may be the primary or secondary cause of many of these deaths [28]. In professional boxers, it has been suggested that mTBI leads to CTE at least 17% of the time; longer careers and higher number of bouts is associated with higher CTE incidence [7].
It is important to note the limitations in estimating incidence and prevalence of CTE within the current research climate. Currently, CTE can only be diagnosed through histopathological analysis postmortem after athletes agree to donate their brains for neurodegenerative disease research. Performing autopsy on all athletes exposed to repetitive mTBI is not practical; between 1954 and 2013, only 153 athletes were confirmed to have CTE by neuropathological criteria [2]. A recent study by McKee et al. found 80% of examined athletes’ brains showing evidence of CTE [1]. However, any estimates of prevalence from such a subject group are unwarranted, as they may not represent the general athlete population and may be inflated by referral bias [24]. A validation of consensus diagnostic criteria could be the first step to reporting accurate incidence and prevalence rates.
CLINICAL PRESENTATION
Core Symptoms
The number of confirmed CTE cases is greatest among boxers and football players; however, CTE has also been diagnosed in soccer, ice hockey, wrestling, and rugby players [2]. Case reports suggest differences in clinical presentation of CTE between various types of athletes, suggesting that mechanisms inherent to a sport may influence the rapidity of onset and severity of illness [29]. Recently, there has been an increased interest in the clinical features thought to spur CTE, particularly in professional football players. In the past, boxers who were diagnosed with CTE were reported to have severe neuropsychiatric symptoms, pseudobulbar signs (dysarthria and dysphagia), motor and language disturbances, and a Parkinsonian-like syndrome [5]. More recently, likely due to increased awareness, athletes are more frequently being diagnosed with milder CTE symptoms [5].
Symptoms associated with CTE are varied, nonspecific, and present insidiously, which leads to a delayed diagnosis occurring years after trauma [30]. Clinical symptoms of CTE (see Table 1 for details) manifest themselves in four main areas: cognition, mood, behavior, and motor. By 2009, the average age of onset for 46 athletes with histopathologically confirmed CTE cases was 42.8 years (range 25–76 years, SD=12.7) [31]. A recent review conducted by Montenigro et al. [29] analyzed athletes with repetitive TBI (rTBI). They characterized core cognitive features appearing in over 70% of confirmed cases of CTE, including memory impairment, executive dysfunction, and reduced attention span. Common behavioral symptoms include physical and verbal abuse, and mercurial behavior (possibly due to pseudobulbar affect). Regarding mood, core symptoms involve depression and feelings of hopelessness. No motor symptoms were found in over 70% of confirmed CTE cases, but severe cases involved ataxia, dysarthria, abnormal gait, Parkinsonism, and tremor [29]. The clinical manifestations of CTE can be separated into two areas: one primarily affecting mood/behavior and the other affecting primarily cognitive function [32]. Based on analysis of 36 male athletes with confirmed CTE from the Boston University brain bank, those who had behavior and mood dysfunction had much earlier onset of symptoms than those with cognitive impairment [32]. Studies with larger sample sizes are needed to see if this characterization is representative of all athletes diagnosed with CTE.
Table 1. Clinical and pathological features of CTE and CTE mimics.

McKee et al. [1] recently characterized four main pathological stages of CTE, with symptoms progressively worsening with extent of pathological changes. Stage I is characterized by headache, loss of concentration and reduced attention span; Stage II includes additional symptoms of depression, short term memory loss, and impulsive behavior; Stage III adds clinical presentation of further cognitive impairment; Stage IV involves language dysfunction and increased aggression [1].
Mimics of CTE
Clinical diagnosis of CTE remains challenging due to symptomatic overlap with other neurodegenerative diseases, such as AD, chronic postconcussive syndrome (CPCS), and FTD (Table 1). Although CTE cases show prolonged progression, the average age at death is younger than other neurodegenerative diseases. In CPCS, similar symptoms such as headache, poor memory, reduced concentration, personality changes, and depressive behavior arise. Up to 15% of these individuals have symptoms that persist after one year [32]. However, CTE can be differentiated from CPCS from the patient’s comprehensive history, which includes repetitive incidence of mTBI. CPCS generally occurs after one TBI incident and symptoms manifest early, whereas in those patients with CTE, symptoms are insidious and typically present 8 to 10 years after rTBI [1]. In FTD, symptoms manifest themselves around age 45-65 and progress quite rapidly [33]. The analysis of McKee et al. [1] shows progression of CTE between each stage of CTE to be 11 to 14 years. Similar to CPCS, FTD, and AD patients generally do not have history of rTBI, which is a hallmark for those diagnosed with CTE. Other mimics of CTE include Parkinson’s disease, vascular dementia, and psychiatric disease (Table 1).
CTE PATHOLOGY
Gross Neuropathology
Neuropathology varies based on the stage of the disease. In mild CTE, the brain appears grossly intact in some cases. Other cases may show enlargement of the anterior and inferior portions of the lateral ventricles. It is possible that concussive forces are transmitted through the ventricular system. In more advanced CTE, additional gross features include septal fenestrations with associated cavum septum pellucidum, further enlargement of the lateral and third ventricles, atrophy in the frontal and temporal lobes, mammillary body atrophy, and thinning of the hypothalamic floor and corpus callosum. Additionally, the locus coeruleus and substantia nigra may lose their characteristic dark color [34]. In severe cases, there may be atrophy of the hippocampus and amygdala [32]. The marked atrophy throughout many areas may result in an overall decrease in brain mass [34]. Though these changes can act as supporting evidence of CTE, macroscopic changes alone cannot bring a definitive, distinctive diagnosis.
Microscopic Neuropathology
Though microscopic pathological findings are similar between CTE and other neurodegenerative diseases, McKee et al. [1] has found characteristics that make CTE distinguishable. The characteristic most indicative of CTE is the presence of neurofibrillary tangles (NFTs) formed in specific areas by aggregates of hyperphosphorylated tau protein, a protein normally responsible for stabilizing microtubules. Other inclusions form in both neurons and glial cells in the form of neuropil threads (NTs), neuropil neurites (NNs), and glial tangles (GTs), mainly in the frontal and temporal cortices [30,34,35]. Thus, CTE is one of the many neurodegenerative diseases, such as AD and some forms of FTD, in which a marked feature is progressive tauopathy. The structures of tau deposition in CTE and AD are virtually indistinguishable, as observed isoform ratios are comparable in both diseases [36]. However, the distribution of tau aggregation in CTE is distinctive. The tau NFTs in CTE are found in outer layers of the cerebral cortex—layers II and III. In contrast, distribution of tau in AD is mainly in layers III and V, which contains an increased proportion of larger pyramidal neurons [37]. Also unique to CTE is the nonuniform regional distribution of NFTs, found preferentially in frontal, temporal, and insular cortices and prone to aggregate close to small blood vessels and near the depths of sulci [30]. In AD, however, tau deposition is more uniform and evenly dispersed across all cortical regions [34]. CTE tangles are frequently found in the olfactory bulb, hippocampus, amygdala, entorhinal cortex, mammillary bodies, the substantia nigra, and the locus coeruleus [35]. Further, tau aggregates in cases of CTE are usually of significantly higher density than tau deposition in severe AD [34].
Multifocal axonal varicosities in both the cortex and subcortical white matter are bolstered by neurofibrillary inclusions and are manifest in many cases of CTE. In mild cases, axonal damage is restrained to the frontal cortex, subcortical white matter, and deep white matter [1]. Neuronal loss is notable in more severe cases, where it is diffuse and conspicuous in the hippocampus, entorhinal cortex, and the amygdala [31].
Along with widespread tau aggregation and neuronal loss, many cases of CTE exhibit deposits of Tar-DNA-binding protein of approximately 43 Kd (TDP-43), a protein important in the regulation of transcription and neuronal development. Elevated TDP-43 levels are common in patients diagnosed with MND. The presence of TDP-43 proteinopathy may explain the motor impairment symptoms that are frequently seen in boxers. Similar findings are present in other neurodegenerative diseases such as FTD and MND [38–41]. McKee et al. [42] analyzed brains of 12 former professional athletes diagnosed with CTE postmortem. They found 10 out of 12 to have extensive inclusion of TDP-43 in the frontal and temporal cortices, basal ganglia, diencephalon, and brainstem. Three of these athletes developed MND, in part due to TDP-43 extending to the spinal cord. Unlike FTD-TDP 43/MND, which is affiliated with ubiquitin-positive tau-negative inclusions, TDP-43 proteinopathy in CTE is coupled with tauopathy [42].
In contrast to AD, amyloid beta (Aβ) deposition is not a hallmark of CTE but is often present. Numerous groups have found differences between AD and CTE in how Aβ is dispersed.
Though neuritic Aβ plaques are essential for diagnosis of AD, they are present only in a minority of CTE cases. Rather, diffuse plaques (DP), deposits that are non-neuritic and do not disrupt the neuropil, are more common in CTE. Stein et al. [43] has recently found 52% of 60 deceased athletes and war veterans to have Aβ deposition in either diffuse or neuritic form, indicating that though there is an association of Aβ with CTE, it is by no means necessary for diagnosis.
McKee et al. [1] characterized a spectrum of neuropathology associated with the four stages of CTE. In stage I, localized tauopathy was restricted to the depths of sulci in the frontal cortex. In stage II, there is a diffuse spread of tau aggregation and neurofibrillary inclusions occurring throughout multiple layers of the cerebral cortex. Stage III shows tauopathy to be widespread, affecting the frontal, temporal, parietal, and insular cortices, with most damage done to the frontal and temporal cortices at the depths of sulci. The amygdala and hippocampus experience neurofibrillary degeneration in this stage. Stage IV involves further expansion of tauopathy into the medial temporal lobe, affecting most regions of the cerebral cortex and accompanied by neuronal loss [1]. Though there are no validated pathological consensus criteria for diagnosing CTE, multiple groups have proposed guidelines [1,30,44].
In the first National Institute for Neurological Disorders and Stroke and National Imaging of Biomedical Imaging and Bioengineering (NINDS/NIBIB) consensus meeting for neuropathology of CTE in early 2015 [4], a group of seven neuropathologists blindly examined 25 brains, each resembling one or more of the following tauopathies: CTE, AD, progressive supranuclear palsy (PSP), argyrophilic grain disease (AGD), corticobasal degeneration (CBD), primary age-related tauopathy (PART), and parkinsonism-dementia complex of Guam. Neuropathologists were not given any information about patients before examination. Using preliminary criteria [1] based on review of previous literature, there was strong agreement among neuropathologists when diagnosing CTE; 90% of responses that indicated CTE as the diagnosis agreed with the presumptive diagnosis [4]. Through analysis of results, the neuropathologists established a preliminary standard requirement for the diagnosis of CTE: a lesion composed of “p-tau aggregates in neurons, astrocytes, and cell processes around small vessels in an irregular pattern at the depths of the cortical sulci” [4]. The neuropathologists also established supportive neuropathological features of CTE, involving phosphorylated-tau-related and nonphosphorylated-tau-related pathologies. These supporting features, though present in many cases of CTE, were not solely sufficient to make a definitive diagnosis. Though results from the first consensus meeting are promising in demonstrating the distinctiveness of CTE, they should be substantiated in future meetings by increasing the number of reviewing neuropathologists and using a larger sample size of diseased brains.
Clinical and Imaging Biomarkers
Currently, there is no definite method to diagnose CTE while patients are living; the disease can only be confirmed postmortem after histological analysis of the brain. Multiple groups are evaluating serum and cerebrospinal fluid (CSF) biomarkers that may be specific to CTE [45–51]. CSF biomarkers are currently used to help confirm early diagnosis of AD, so it is likely a similar approach will eventually aid in the diagnosis of CTE. The rationale for isolating CTE biomarkers in CSF is based on the known mechanisms of cerebral ventricular injury in mTBI, which leads to alteration of CSF composition. Therefore, the biochemical makeup of CSF may represent underlying pathological changes in the parenchyma [30]. It is unknown whether the CSF biomarkers would remain elevated for extended periods of time after the patient is exposed to repetitive mTBI [48]. Neselius et al. [48] studied CSF proteins in 30 Olympic boxers 1 to 6 days after a bout and after a rest period of at least 14 days. Although phosphorylated tau (p-tau) was elevated in CSF immediately after bouts, p-tau concentrations in CSF declined to baseline after the rest period. In contrast, neurofilament light polypeptide, a protein thought to provide structure to axons as a component of the cytoskeleton, was elevated in boxers both immediately after bouts and after the rest period. These results indicate that neurofilament light polypeptide as a CSF biomarker may have the most promise, as it can be detected long after injury onset.
CSF biomarkers, even if diagnostic, require collection via an invasive lumbar puncture. Therefore, it would be more practical if the chosen biomarkers were also present in serum. Serum biomarkers have a number of problems, however. First, concentrations in plasma are typically very low, as protein diffusion from the brain to outside the blood brain barrier (BBB) is restricted, and due to a large dilution factor arising from plasma volume [52]. Additionally, serum biomarkers may not be specific to brain injury. Thus, extremely sensitive assays are needed to differentiate levels of biomarkers in control patients versus those with mTBI. A clinical challenge is to identify whether CSF and serum biomarkers are highly specific to CTE rather than other neurodegenerative diseases.
CTE Imaging
Computed tomography (CT) and magnetic resonance imaging (MRI) studies are frequently used to evaluate mTBI. However, routine CT and MRI studies often fail to elucidate definite presence of brain injury due to CTE (Figure 1). Positron emission tomography (PET) imaging has emerged as a potential imaging tool that can differentiate between CTE and other neurodegenerative diseases. Multiple studies have utilized 2-(1-{6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene) malononitrile (18F-FDDNP) [53–55], a radioactive ligand administered intravenously that crosses the BBB and binds selectively, but not exclusively, to tau deposits, a hallmark of CTE. 18F-FDDNP also binds to amyloid plaques [56], and is only for investigational use at this point. Yet, researchers have found that distributions of bound 18F-FDDNP can differentiate CTE from other neurodegenerative diseases such as AD. When imaging brains of 14 retired professional football players, Barrio et al. [53] found 18F-FDDNP to be concentrated in different areas compared to controls and patients with AD. Specifically, 18F-FDDNP signal in those with suspected CTE was stronger than AD in the mesencephalon, extending to the amygdala and subcortical areas. Imaging findings coincided with previous histopathological findings post-mortem of those with confirmed CTE [53]. Figure 2 displays the distribution of 18F-FDDNP signal in suspected CTE patients versus AD patients [53].
Figure 1. Samples head CT images of possible CTE cases. (A) Axial noncontrast head CT scan of a 76-year-old male, retired football player (in college and briefly in NFL), with a medical history of severe cardiac disease presented to the hospital with shortness of breath. The CT scan is remarkable. However, during hospitalization the patient had his pacemaker replaced, and then developed severe postprocedural agitation. The patient’s family stated that approximately 1 month prior to admission, he had an episode of getting lost and was unaware of his surroundings. Since then, his cognition status had continued declining. He was previously diagnosed with frontotemporal lobe dementia. Patient’s family stated that for the last 20 years, he has been extremely agitated. Patient also noted some type of jerking movement of the extremities, and hallucinating (seeing animals) and having delusions. (B) Postmortem CT imaging of a professional boxer who died in his 40s with symptoms of CTE. This coronal slice shows a cavum septum pellucidum (the authors would like to acknowledge Dr. Gary Hatch for his input in generating this neuroradiographic image).
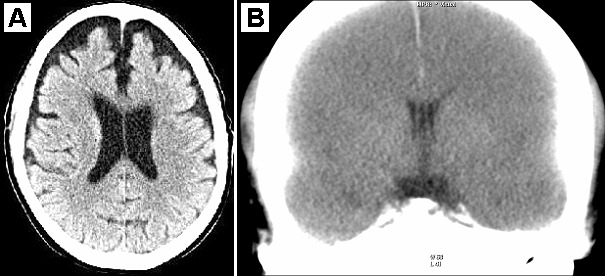
Figure 2. Differences in 18F-FDDNP signal in patients with suspected CTE versus patients with AD. Note the higher density regions in the CTE case (Adapted, with permission, from Barrio et al. [53]).
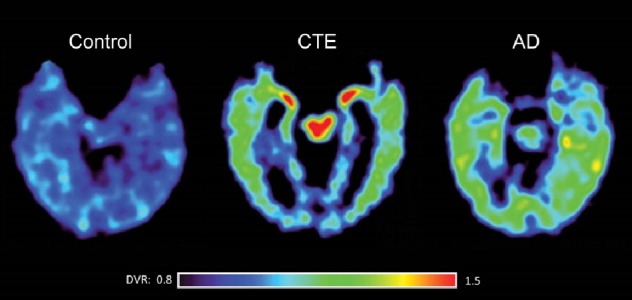
There is yet to be a marker that binds selectively to tau only, though numerous groups are working towards developing one [57]. Once such a marker emerges, PET imaging findings may be more precise and offer definitive evidence that the distribution of tau in CTE is consistent and distinctive. Overall, PET imaging has immense potential in helping diagnose CTE ante-mortem. However, imaging by itself is not sufficient to warrant a CTE diagnosis. Although treatments for CTE are virtually nonexistent as of now, early imaging results can lead physicians to make appropriate recommendations to athletes to minimize and impede progression of the disease.
Type of Athletic Activity versus Clinical Presentation and Pathology of CTE
Clinical and pathological features of CTE can manifest differently between sports, as rTBI exposure and mechanisms of impact can vary considerably. In fact, an analysis of previously reported CTE cases by Montenigro et al. [29] showed a vast difference in clinical presentation. 83% (5/6) of professional boxers, who had more debilitating motor impairments, compared to 18.8% (3/16) of professional football players. In addition, severe dentate neurofibrillary tangles were present in 17% (2/12) and 80% (4/5) of professional football players and boxers, respectively, indicating a more pernicious progression in boxers [29]. The difference in symptoms and neuropathology may be explained through the frequency of linear and rotational impact forces that occur in both sports.
Rotational forces causing angular accelerations are frequent in boxing. Boxers face their greatest danger when their opponent lands a hook punch, where impact near the lateral side of the head cause rapid outward rotation of the skull and twisting forces the brain [29]. Lateral bending of the neck can also occur, but linear forces from a punch are often below the mTBI threshold [58]. The rotational movement of the brain causes shearing forces that can lead to axonal damage [59]. Shearing forces are most prominent near areas such as the midbrain section, where glial and axonal injury could result in severely debilitating consequences [29,58].
As opposed to punches, helmet-to-helmet or helmet-to-ground contact forces cause the majority of mTBI injuries in professional football players. Viano et al. [58] have shown that in professional football concussions, inertial forces can be up to 30% greater than inertial forces in professional boxers who endure a hook punch. The greater inertial forces correlate with a higher linear acceleration endured by football players, suggesting that linear forces are prominent in causing concussive and subconcussive impacts in professional football players. In support of this mechanism, brain modeling shows that rotational accelerations from uppercuts or hook punches are much greater than rotational accelerations in professional football helmet-to-helmet impacts [58]. The linear to rotational force ratio difference between boxers and football players could explain the differences in clinical presentation between the two sports.
In professional football, helmet-to-helmet collisions can cause the head to move in the anterior or posterior direction. The incidence rates of mTBI have been shown to vary depending on position, with running backs and wide receivers suffering from mTBI more than linemen [60]. Neck musculature acts to stabilize the position of the head, and a more developed musculature is directly correlated to lowered mTBI risk [61]. Linemen have been found to have stronger necks and larger girth compared to running backs, which could act to slow linear accelerations of the head and reduce risk of mTBI [62]. The differences in neck strength between positions may explain the varying incidence rates of mTBI. Additionally, it should be noted that different player positions may be more prone to certain types of impacts—linemen may experience more frequent subconcussive helmet-to-helmet impact, while wide receivers could endure more threatening forces while being tackled. The pathological repercussions of variations in impact type and frequency between boxing and football have yet to be elucidated in full detail, but they may partially explain the difference in clinical presentation between different types of athletes.
CTE GENETIC RISK FACTORS
It is unclear why only some people exposed to repetitive mTBI do not develop CTE while others do. One possible explanation is that genetic factors play a role in the disease pathogenesis. ApoE4, a protein important in cholesterol transport, is a well-established major risk factor for developing AD. Saunders et al. [63] first linked ApoE4 to late-onset AD after analysis of 176 patients postautopsy. Roses et al. [64] propose that an inheritance of an ApoE4 allele leads to an 80% chance of developing late-onset AD by age 75, while Corder et al. [65] reported homozygous ApoE4 allele presentation almost always causes AD by age 80. ApoE4 expression has been linked to hyperphosphorylation of the tau protein, allowing the protein to aggregate in neurofibrillary tangles and increase risk of AD [63]. There are multiple proposed mechanisms of how ApoE4 results in an increased susceptibility to neurodegenerative disease. Numerous studies have shown a strong association between ApoE and aggregation of Aβ-peptide and plaque formation [66]. ApoE4 in particular has been found to bind with high affinity to Aβ-peptide, potentially due to its relative instability compared to ApoE2 and ApoE3 and the presence of arginine, containing a positive charge, in a specific position [66].
Given the association of ApoE4 with other neurodegenerative diseases, and as tau aggregation is also common in traumatic brain injury, researchers have investigated the effect of ApoE on susceptibility to developing CTE [56]. The literature regarding ApoE4 and CTE is inconsistent with conflicting results [1,32,44,67,68]. Though ApoE4 has been studied quite extensively, other genes linked to neurodegeneration may also be associated with susceptibility to CTE, including the microtubule-associated protein tau (MAPT) gene, the progranulin (GRN) gene, the chromosome 9 open reading frame 72 (C9ORF72) gene, and the transmembrane protein 106B (TMEM106B) gene [32,69]. A recent study did not find statistically significant differences in MAPT or TMEM106B genotypes between athletes with CTE pathology (n=17) and control groups (n=408) [70]. Studies with larger sample sizes, particularly for the group harboring CTE pathology, are needed to substantiate the results.
A study by Casson et al. [71] analyzed a sample of 45 retired professional football players. They found that the ApoE4 allele was present in 38% of all the players, many who were suffering from symptoms of CTE. Interestingly, the 38% who expressed ApoE4 was considerably greater than the estimated 23% in the general population who harbor the ApoE4 allele. [71] Another study by Jordan et al. [68] looked for a relationship between ApoE genotype and CTE in thirty professional boxers. Neurological and behavioral assessments evaluated severity and extent of disease in the athletes through a chronic brain injury (CBI) score. Boxers with many bouts harboring ApoE4 had significantly increased CBI scores than those without ApoE4 expression. Additionally, all boxers with severe CBI harbored at least one ApoE4 allele. The results suggest there is an interaction between number of boxing matches and ApoE4 carrier status in which ApoE4 expression is linked to more severe indications of traumatic encephalopathy [68]. Another study conducted by Kutner et al. [67] evaluated the relationship between ApoE genotype and cognitive function. Fifty-three active professional football players underwent genotypic analysis and several neuropsychological assessments to evaluate cognitive performance. Older athletes carrying the ApoE4 gene scored significantly lower on cognitive tests compared to younger athletes with ApoE4 status or those without this variant. In fact, the older ApoE4 carriers often scored two standard deviations below the mean of the general population, suggesting there may be a time dependent effect between ApoE4 expression and cognitive decline in those with repetitive mTBI [67].
Conversely, a study by Omalu et al. [44] questioned the presumed importance of ApoE4 in CTE. Performing neuropathological and ApoE genotyping analysis in ten professional athletes and two high school athletes with CTE revealed that only three harbored the ApoE4 allele. In contrast, 90% of the professional athletes sampled had at least one ApoE3 allele, and 100% of athletes diagnosed with CTE postmortem exhibited at least one ApoE3 allele. Yet, both high school athletes who never exhibited histologic signs of CTE also harbored at least one ApoE3 allele [44]. However, the small sample size should be taken into account when considering the importance of the ApoE4 allele in CTE.
Analogous populations may provide insight into CTE pathology, as there are many overlapping characteristics between mTBI and more severe forms. Teasdale et al. [72] conducted a prospective cohort study analyzing 984 patients admitted to the neurosurgery unit with acute brain injury symptoms. Patient outcomes were tracked over time, and there was no association between ApoE genotype and severity of outcome. However, their data suggested that children and young adults who express ApoE4 are at increased risk of an unfavorable outcome [72]. It should be noted that many of the patients in the sample presented with either moderate or severe TBI. The heterogeneity of TBI included within the study introduces a limitation, as CTE arises strictly from repeated mTBI. Perhaps the most comprehensive study regarding associations between ApoE4 and TBI comes from Zhou et al., who conducted a meta-analysis of 14 prospective cohort studies that studied the possible association between ApoE4 expression and outcomes of TBI. A total of 2527 participants were analyzed—736 harboring the ApoE4 allele and 1791 without the allele. Though ApoE4 expression did not correlate with initial severity of TBI, those with the ApoE4 allele significantly fared worse six months after injury than those without the ApoE4 allele [73]. The results from a study including a wide spectrum of TBI severity, however, may not translate directly to athletes struck by repeated mTBI.
The conflicting results with ApoE genotyping analysis suggests the need for further research, ideally utilizing a large, healthy subject population to establish more definitive evidence of the relationship between ApoE4 expression and CTE. Athletes exposed to repetitive head injuries such as football, soccer, boxing, and ice hockey should be monitored as a cohort as opposed to those with general TBI to assess whether the associations between ApoE4 and repetitive mTBI continue to exist. Such cohort studies would reduce the heterogeneity of the sample populations and eliminate related confounding variables.
Hypotheses for Pathogenesis of CTE
There have been numerous proposed hypotheses to explain the neuropathology and associated clinical presentation connected to CTE. Though many hypotheses are worth investigating, the molecular pathogenesis is largely speculative at this point, and the details of hypotheses are not supported by clinical data. It is generally agreed upon that repeated impact forces to the brain are the causal factor of CTE. One of the beginning hypotheses was developed after the observation of “punch-drunk” syndrome in boxers, where Martland [7] believed repeated blunt forces induce intracerebral hemorrhages that lead to gliosis or lesions in the associated area.
Giza and Hovda [8] proposed that concussive and subconcussive impact forces lead to significant shear forces to the axons, causing the axons to have increased efflux of potassium ions and increased influx of calcium ions. The calcium influx further depolarizes the neuron and changes functional physiology. Attempting to recalibrate neuron potential, the Na+-K+ pump is over activated and consumes more adenosine triphosphate (ATP). Due to higher amounts of ATP needed, the affected brain areas increase the demand for more glucose, which can be considerably higher than the supply of glucose. The authors [8] proposed that the discrepancy in glucose supply and demand resulted in a “cellular energy crisis”, which may exacerbate the effects of future blows to the head by inhibiting an adequate recovery response. Ionic flux has been shown to occur in animal models recapitulating concussive brain injury [74–76]. Calcium influx has been shown to be correlated with the release of caspase-7 and caspase-12, both of which can each play roles in calcium-dependent apoptosis [77]. The influx could also spur tau hyperphosphorylation and formation of neurofibrillary tangles [34].
Blaylock [9] proposed an alternate hypothesis that immunoexcitotoxicity is the central mechanism driving CTE, caused by microglial activation from mTBI. Activated microglia release high levels of both pro and anti-inflammatory cytokines and chemokines, along with three excitotoxins: glutamate, aspartate, and quinolinic acid. Pro-inflammatory signals are more prevalent directly following mTBI, while anti-inflammatory signals are released later. The presence of pro-inflammatory signals can be ominous—if blunt concussive or subconcussive forces continually occur, microglia stay in an activated state. During this activated state, microglia continue to release excitotoxins while recruiting astrocytes, the main reservoir of glutamate and aspartate. The excitotoxins released from both astrocytes and microglia further ‘excite’ neurons, leading to neurotoxic concentrations, greater vulnerability to neuronal injury, and larger propensity to develop neurofibrillary tangles from hyperphosphorylated tau inclusions [9]. Both animal and clinical studies have observed microglial activation as a key event occurring after TBI [78–84]. One study involving rat models indicates that glutamate excitotoxins are removed by related transporters in between 24 and 72 hours after a single TBI incident [85], pointing to the possibility that excitotoxins do not reach neurotoxic concentrations unless repeated concussive forces continually occur. The Blaylock hypothesis [9] could also explain the progressive nature of CTE, as aging has been shown to be associated with increased microglial activity and related neurodegeneration [86].
McKee et al. [31] approached pathogenesis from a gross rather than molecular level. They proposed mTBI leads to fluid waves within the cerebral ventricles and disturbs the flow of cerebrospinal fluid (CSF). The disrupted CSF induces atypical shear stresses, which transmits to the intraventricular septum, resulting in an enlarged cavum septum and septal fenestrations. Additionally, they proposed the release of various neurotoxins and damage to the BBB may contribute to the unique distribution of tau aggregates near blood vessels in CTE [31]. It is likely that multiple pathways lead to the observed macro- and microscopic neuropathological changes.
Animal Models of CTE
To further gain insight into relationships between concussive injury and CTE symptoms, multiple in vivo models have been proposed, each with their respective advantages and disadvantages [81]. There have been preclinical models, specifically in mice, which have aimed to mimic repetitive mTBI injury [81,82,87–91]. Different pathological markers in humans, including increased microglial activation [81,90,91], astrogliosis [81,82,91], and tauopathy [81,82,91] have been replicated in murine models to some extent after repeated concussive impact exposure. Kane et al. [82] developed a novel method of inducing repetitive mTBI in mice utilizing a modified Marmarou [92] weight drop method. Pathology was assessed through histochemical staining 30 days after the last impact. Damage to the BBB and microglia activation was not evident, but there was mild astrocytic response and increased phosphorylated tau present, the hallmark of CTE. Mild balance and coordination deficits were observed. Interestingly, these mice developed an increase in locomotive activity, which potentially could be linked with CTE symptoms of reduced attention span observed in humans [82]. The relatively mTBI symptoms while inducing rapid linear accelerations of the head make this model distinctive.
Petraglia et al. [81] recently characterized a novel mouse model that allows for controlled closed head injury in unanesthetized mice via a controlled cortical impact (CCI) device. For the study, 12-week-old mice received six concussive impacts daily for seven days while wearing a thin helmet, so the injury could spread over a wider surface area, aiming to accurately emulate post-concussion like symptoms. Animals were sacrificed at 7 days, 1 month, and 6 months post-injury with neuropathological examinations performed at each study time point. Though no gross pathology was evident at any of the time points, increased microglia activation, astrogliosis, and tau inclusions were conspicuous in the cortex and amygdala in all three delayed time points [81]. Significant microscopic features of CTE were thus recapitulated in this model.
Though preclinical models can certainly enhance our understanding of neurodegenerative disorders such as CTE, one should be wary of directly translating the results to humans. First, lifespan of mice is approximately 2–3 years, and CTE symptoms in humans normally manifest 8-10 years after TBI injury. Thus, “chronic” effects in mice may not fully capture symptoms over the same duration. Nonhuman primates would be desirable to recapitulate human symptoms, yet this would substantially increase number of resources, training, and cost associated with studies. Further, brain size and cell density differ considerably between humans and mice. Human brains are approximately 2700 times larger than a mouse brain and contain 1200 times as many neurons with significantly more myelin [93]. The massively greater number of neurons and their respective interconnections in humans explain the white matter/gray matter ratio of 60:40 in humans rather than 10:90 found in mice. Additionally, humans have a higher glial/neuron ratio. A combination of these factors may explain the unique metabolic needs of the human brain and its distinctive susceptibility to neurodegenerative disease [93]. Further, CTE in humans has shown neurofibrillary tangles to originate in the depths of sulci. However, mouse models cannot demonstrate this neuropathological feature, as rodent brains are smooth and lack the sulcal pattern found in the human brain [93]. Other factors that prove difficult to model include age at onset of injury and duration of mTBI. Therefore, researchers should be cautious when interpreting data from experimental models and extrapolating it to humans.
Conclusions and Future Research
CTE research has burgeoned over the last decade, as the repercussions of high-impact sports are beginning to reach the American public. Though genetic risk factors have been proposed, many of the nontrauma-related risk factors are unknown or have yet to be substantiated. We suggest that the underlying pathophysiology of the ApoE4 protein and its mechanisms contributing to symptoms of CTE should be studied in more detail, so results of cohort studies can be substantiated. Further epidemiological and longitudinal research should be conducted to address associations between number and severity of injuries and propensity to develop chronic injury, as well as correlations between neuropathological findings and changes in neurobehavior/cognition. Neuropathological findings have been elucidated over the last few years, with a spectrum of pathology corresponding to severity of clinical presentation. The first NINDS/NIBIB consensus meeting has taken an important step to clearly define neuropathology. Additional clinicopathological correlation studies would be useful in determining pathognomonic symptoms for CTE. Currently, though estimates have been proposed, the incidence and prevalence of CTE is uncertain because in vivo diagnosis is not considered accurate or reliable. The emergence of clinical biomarkers is exciting, as PET imaging has been utilized with tau-specific ligands and holds great promise in early diagnosis. We believe the most important field of research should be focused on developing an accurate in vivo diagnostic method in the early stages of the disease. Reliable antemortem diagnoses can lead to more accurate incidence and prevalence rates, direct evaluation of clinical and pathological progression, and therapeutic studies. CTE, like other consequences of TBI, poses a great clinical challenge in both diagnosis and treatment. Future research must take into consideration analysis of optimal helmets with adequate shock absorbing mechanisms, modifications to rules regarding some of the sports, and other preventive measures. Furthermore, more sensitive neurobehavioral diagnostic tools/assessment scales, including routine cognitive tests (tailored for TBI) and eye movement tracking techniques [94], combined with clinicopathologic data can help physicians and athletes assess their future risk of continued participation in sports. CTE is a complex disease, and there are no published studies that have investigated the role of steroids in the pathophysiology. As far as we know, steroid use is associated with increased risk for depression, suicide, rhabdomyolysis and corticosteroid induced myopathy. However, the presence of depression is associated with worse performance on cognitive tests and may contribute to poor clinical outcome in CTE patients that also needs to be investigated in future studies.
Table 2. Summary of major clinical studies identifying non-duplicate, neuropathologically confirmed [2] CTE cases in athletes.

REFERENCES
- McKee AC, Stein TD, Nowinski CJ, Stern RA, Daneshvar DH, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroon JC, Winkelman R, Bost J, Amos A, Mathyssek C, Miele V. Chronic traumatic encephalopathy in contact sports: a systematic review of all reported pathological cases. Lewis P, editor. PLoS One. 2015;10:e0117338. doi: 10.1371/journal.pone.0117338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Program S, HR . Report on the neuropathology of chronic traumatic encephalopathy Workshop. Bethedsa: 2012. [Google Scholar]
- McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Dirk Keene C, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O’Connor K, Alvarez VE, Gordon WA, TBI/CTE group The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131:75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson GL, Gardner AJ, McCrory P, Zafonte R, Castellani RJ. A critical review of chronic traumatic encephalopathy. Neurosci Biobehav Rev. 2015:276–293. doi: 10.1016/j.neubiorev.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Carone DA, Bush SS. Mild traumatic brain injury: Symptom validity assessment and malingering. In: Carone D, Bush S, editors. Mild Trauma Brain Inj Symptom Validity Assess Malingering. New York City: Springer Publishing Company; 2013. p. 15. [Google Scholar]
- Martland H. Punch Drunk. J Am Med Assoc. 1924;91:1103–1107. [Google Scholar]
- Giza CC, Hovda DA. The new neurometabolic cascade of concussion. Neurosurgery. 2014;75:S24–S33. doi: 10.1227/NEU.0000000000000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaylock RL, Maroon J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-A unifying hypothesis. Surg Neurol Int. 2011;2:107. doi: 10.4103/2152-7806.83391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois J a, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- Sosin DM, Sniezek JE, Thurman DJ. Incidence of mild and moderate brain injury in the United States, 1991. Brain Inj. 1996;10:47–54. doi: 10.1080/026990596124719. [DOI] [PubMed] [Google Scholar]
- McCrea M, Hammeke T, Olsen G, Leo P, Guskiewicz K. Unreported concussion in high school football players: implications for prevention. Clin J Sport Med. 2004;14:13–17. doi: 10.1097/00042752-200401000-00003. [DOI] [PubMed] [Google Scholar]
- Llewellyn T, Burdette GT, Joyner AB, Buckley TA. Concussion reporting rates at the conclusion of an intercollegiate athletic career. Clin J Sport Med. 2014;24:76–79. doi: 10.1097/01.jsm.0000432853.77520.3d. [DOI] [PubMed] [Google Scholar]
- Mulligan I, Boland M, Payette J. Prevalence of neurocognitive and balance deficits in collegiate aged football players without clinically diagnosed concussion. J Orthop Sports Phys Ther. 2012;42:625–632. doi: 10.2519/jospt.2012.3798. [DOI] [PubMed] [Google Scholar]
- Fraas MR, Coughlan GF, Hart EC, McCarthy C. Concussion history and reporting rates in elite Irish rugby union players. Phys Ther Sport. 2014;15:136–142. doi: 10.1016/j.ptsp.2013.08.002. [DOI] [PubMed] [Google Scholar]
- Broglio SP, Vagnozzi R, Sabin M, Signoretti S, Tavazzi B, Lazzarino G. Concussion occurrence and knowledge in italian football (soccer) J Sport Sci Med. 2010;9:418–430. [PMC free article] [PubMed] [Google Scholar]
- Associations NF. High School Sports Participation Increases for 26th Consecutive Year [Internet] [2015 Dec 22;];2015 Available from: https://nfhs.org/articles/high-school-sports-participation-increases-for-26th-consecutive-year/ [Google Scholar]
- Brown G. NCAA student-athlete participation hits 450,000 [Internet] [2015 Dec 22;];2012 Available from: http://www.ncaa.org/about/resources/media-center/news/ncaa-student-athlete-participation-hits-450000. [Google Scholar]
- Tommasone BA, McLeod TCV. Contact sport concussion incidence. J Athl Train. 2006:470–472. [PMC free article] [PubMed] [Google Scholar]
- Clay MB, Glover KL, Lowe DT. Epidemiology of concussion in sport: A literature review. J Chiropr Med. 2013;12:230–251. doi: 10.1016/j.jcm.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broglio SP, Martini D, Kasper L, Eckner JT, Kutcher JS. Estimation of head impact exposure in high school football: implications for regulating contact practices. Am J Sports Med. 2013;41:2877–2884. doi: 10.1177/0363546513502458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox BJ, Beckwith JG, Greenwald RM, Chu JJ, McAllister TW, Flashman LA, Maerlender AC, Duhaime AC, Crisco JJ. Head impact exposure in male and female collegiate ice hockey players. J Biomech. 2014;47:109–114. doi: 10.1016/j.jbiomech.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagel B, Meeuwisse W. Risk compensation: a “side effect” of sport injury prevention? Clin J Sport Med. 2004;14:193–196. doi: 10.1097/00042752-200407000-00001. [DOI] [PubMed] [Google Scholar]
- Gardner RC, Yaffe K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci. 2015:75–80. doi: 10.1016/j.mcn.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll L, Cassidy JD, Peloso P, Borg J, von Holst H, Holm L, Paniak C, Pépin M, WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury Prognosis for mild traumatic brain injury: results of the who collaborating centre task force on mild traumatic brain injury. J Rehabil Med. 2004;36:84–105. doi: 10.1080/16501960410023859. [DOI] [PubMed] [Google Scholar]
- Lee YK, Hou SW, Lee CC, Hsu CY, Huang YS, Su YC. Increased risk of dementia in patients with mild traumatic brain injury: a nationwide cohort study. PLoS One. 2013;8:e62422. doi: 10.1371/journal.pone.0062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbolt AK, Cancelliere C, Hincapie CA, Marras C, Boyle E, Kristman VL, Coronado VG, Cassidy JD. Systematic review of the risk of dementia and chronic cognitive impairment after mild traumatic brain injury: results of the International Collaboration on Mild Traumatic Brain Injury Prognosis. Arch Phys Med Rehabil. 2014;95:S245–S256. doi: 10.1016/j.apmr.2013.06.036. [DOI] [PubMed] [Google Scholar]
- Lehman EJ, Hein MJ, Baron SL, Gersic CM. Neurodegenerative causes of death among retired national football league players. Neurology. 2012;79:1970–1974. doi: 10.1212/WNL.0b013e31826daf50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenigro PH, Bernick C, Cantu RC. Clinical features of repetitive traumatic brain injury and chronic traumatic encephalopathy. Brain Pathol. 2015;25:304–317. doi: 10.1111/bpa.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mez J, Stern RA, McKee AC. Chronic traumatic encephalopathy: where are we and where are we going? Curr Neurol Neurosci Rep. 2013;13:9–11. doi: 10.1007/s11910-013-0407-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, Fritts NG, Stamm JM, Robbins CA, McHale L, Simkin I, Stein TD, Alvarez VE, Goldstein LE, Budson AE, Kowall NW, Nowinski CJ, Cantu RC, McKee AC. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81:1122–1129. doi: 10.1212/WNL.0b013e3182a55f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh CM, Stamm JM, Riley DO, Gavett BE, Shenton ME, Lin A, Nowinski CJ, Cantu RC, McKee AC, Stern RA. Chronic traumatic encephalopathy: Neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav. 2012;6:244–254. doi: 10.1007/s11682-012-9164-5. [DOI] [PubMed] [Google Scholar]
- Gavett BE, Stern RA, McKee AC. Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med. 2011;30:179–188. doi: 10.1016/j.csm.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern R a, Riley DO, Daneshvar DH, Nowinski CJ, Cantu RC, McKee AC. Long-term consequences of repetitive brain trauma: chronic traumatic encephalopathy. PM R. 2011;3:460–467. doi: 10.1016/j.pmrj.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 2001;101:518–524. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- Hof PR, Bouras C, Bue L, Delacourte A, Perp DP, Morrison JH. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol. 1992;85:23–30. doi: 10.1007/BF00304630. [DOI] [PubMed] [Google Scholar]
- Hu WT, Grossman M. TDP-43 and frontotemporal dementia. Curr Neurol Neurosci Rep. 2009:353–358. doi: 10.1007/s11910-009-0052-3. [DOI] [PubMed] [Google Scholar]
- Mackenzie IRA, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol . 2010:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- Fang YS, Tsai KJ, Chang YJ, Kao P, Woods R, Kuo PH, Wu CC, Liao JY, Chou SC, Lin V, Jin LW, Yuan HS, Cheng IH, Tu PH, Chen YR. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat Commun. 2014;5:4824. doi: 10.1038/ncomms5824. [DOI] [PubMed] [Google Scholar]
- Geser F, Lee VMY, Trojanowski JQ. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology . 2010:103–12. doi: 10.1111/j.1440-1789.2009.01091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Daneshvar DH, Mez J, Solomon T, Meng G, Kubilus CA, Cormier KA, Meng S, Babcock K, Kiernan P, Murphy L, Nowinski CJ, Martin B, Dixon D, Stern RA, Cantu RC, Kowall NW, McKee AC. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–929. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y, et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. 2015;130:21–34. doi: 10.1007/s00401-015-1435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, Fitzsimmons R. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. 2011;69:173–183. doi: 10.1227/NEU.0b013e318212bc7b. [DOI] [PubMed] [Google Scholar]
- Franz G, Beer R, Kampfl a, Engelhardt K, Schmutzhard E, Ulmer H, Deisenhammer F. Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology. 2003;60:1457–1461. doi: 10.1212/01.wnl.0000063313.57292.00. [DOI] [PubMed] [Google Scholar]
- Ost M, Nylén K, Csajbok L, Ohrfelt AO, Tullberg M, Wikkelsö C, Nellgård P, Rosengren L, Blennow K, Nellgård B. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology. 2006;67:1600–1604. doi: 10.1212/01.wnl.0000242732.06714.0f. [DOI] [PubMed] [Google Scholar]
- Zetterberg H, Smith DH, Blennow K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat Rev Neurol. 2013;9:201–210. doi: 10.1038/nrneurol.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neselius S, Brisby H, Theodorsson A, Blennow K, Zetterberg H, Marcusson J. CSF-biomarkers in Olympic boxing: diagnosis and effects of repetitive head trauma. PLoS One. 2012;7:e33606. doi: 10.1371/journal.pone.0033606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kövesdi E, Lückl J, Bukovics P, Farkas O, Pál J, Czeiter E, Szellár D, Dóczi T, Komoly S, Büki A. Update on protein biomarkers in traumatic brain injury with emphasis on clinical use in adults and pediatrics. Acta Neurochir. (Wien) 2010;152:1–17. doi: 10.1007/s00701-009-0463-6. [DOI] [PubMed] [Google Scholar]
- Naeimi ZS, Weinhofer A, Sarahrudi K, Heinz T, Vécsei V. Predictive value of S-100B protein and neuron specific-enolase as markers of traumatic brain damage in clinical use. Brain Inj. 2006;20:463–468. doi: 10.1080/02699050600664418. [DOI] [PubMed] [Google Scholar]
- Neselius S, Zetterberg H, Blennow K, Randall J, Wilson D, Marcusson J, Brisby H. Olympic boxing is associated with elevated levels of the neuronal protein tau in plasma. Brain Inj. 2013;27:425–433. doi: 10.3109/02699052.2012.750752. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Blennow K, Ikonomovic MD, Gandy S. Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nat Rev Neurol. 2013;9:192–200. doi: 10.1038/nrneurol.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrio JR, Small GW, Wong K-P, Huang S-C, Liu J, Merrill DA, Giza CC, Fitzsimmons RP, Omalu B, Bailes J, Kepe V. In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc Natl Acad Sci. 2015;201409952 doi: 10.1073/pnas.1409952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenigro PH, Corp DT, Stein TD, Cantu RC, Stern RA, Barrio JR, et al. In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Acta Neuropathol. 2015;11:309–30. [Google Scholar]
- Small GW, Kepe V, Siddarth P, Ercoli LM, Merrill DA, Donoghue N, Bookheimer SY, Martinez J, Omalu B, Bailes J, Barrio JR. PET scanning of brain tau in retired national football league players: preliminary findings. Am J Geriatr Psychiatry. 2013;21:138–144. doi: 10.1016/j.jagp.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Baugh CM, Robbins CA, Stern RA, McKee AC. Current understanding of chronic traumatic encephalopathy. Curr Treat Options Neurol. 2014;16:306. doi: 10.1007/s11940-014-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer ER, Leuzy A, Gauthier S, Rosa-Neto P. Developments in Tau PET Imaging. Can J Neurol Sci. 2014;41:547–553. doi: 10.1017/cjn.2014.15. [DOI] [PubMed] [Google Scholar]
- Viano DC, Casson IR, Pellman EJ, Bir C a, Zhang L, Sherman DC, Boitano MA. Concussion in professional football: comparison with boxing head impacts – Part 10. Neurosurgery. 2005;57:1154–1170. doi: 10.1227/01.neu.0000187541.87937.d9. [DOI] [PubMed] [Google Scholar]
- Levin HSE, Eisenberg HM, Benton AL. Mild head injury. 19891989 [Google Scholar]
- Nathanson JT, Connolly JG, Yuk F, Gometz A, Rasouli J, Lovell M, Choudhri T. Concussion incidence in professional football: position-specific analysis with use of a novel metric. Orthop J Sport Med. 2016;4 doi: 10.1177/2325967115622621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs K, Nichols J, Holmes B, Buono M. Isometric cervical extension strength of recreational and experienced cyclists. Can J Appl Physiol = Rev Can Physiol Appl. 1995;20:230–239. doi: 10.1139/h95-017. [DOI] [PubMed] [Google Scholar]
- Franco JL, Herzog W. A comparative assessment of neck muscle strength and vertebral stability. J Orthop Sports Phys Ther. 1987;8:351–356. doi: 10.2519/jospt.1987.8.7.351. [DOI] [PubMed] [Google Scholar]
- Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roses AD, Strittmatter WJ, Pericak-Vance MA, Corder EH, Saunders AM, Schmechel DE. Clinical application of apolipoprotein E genotyping to Alzheimer’s disease. Vol. 343. Lancet; London, England: 1994. pp. 1564–1565. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders a M, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Kutner KC, Erlanger DM, Tsai J, Jordan B, Relkin NR. Lower cognitive performance of older football players possessing apolipoprotein E epsilon4. Neurosurgery. 2000;47:651–658. doi: 10.1097/00006123-200009000-00026. [DOI] [PubMed] [Google Scholar]
- Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA. 1997;278:136–140. [PubMed] [Google Scholar]
- Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, Busch JI, Akle S, Grossman M, Van Deerlin V, Trojanowski JQ, Lee VM. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci. 2012;32:11213–11227. doi: 10.1523/JNEUROSCI.0521-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniek KF, Ross OA, Cormier KA, Walton RL, Soto-Ortolaza A, Johnston AE, DeSaro P, Boylan KB, Graff-Radford NR, Wszolek ZK, Rademakers R, Boeve BF, McKee AC, Dickson DW. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. 2015;130:877–889. doi: 10.1007/s00401-015-1502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casson IR, Viano DC, Haacke EM, Kou Z, LeStrange DG. Is there chronic brain damage in retired NFL players? Neuroradiology, neuropsychology, and neurology examinations of 45 retired players. Sports Health. 2014;6:384–395. doi: 10.1177/1941738114540270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teasdale G, Murray GD, Nicoll JAR. The association between APOE epsilon4, age and outcome after head injury: A prospective cohort study. Brain. 2005;128:2556–2561. doi: 10.1093/brain/awh595. [DOI] [PubMed] [Google Scholar]
- Zhou W, Xu D, Peng X, Zhang Q, Jia J, Crutcher KA. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008;25:279–290. doi: 10.1089/neu.2007.0489. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Becker DP, Tamura T, Hovda DA. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889–900. doi: 10.3171/jns.1990.73.6.0889. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Hillered L, Pontén U, Ungerstedt U. Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J Cereb Blood Flow Metab. 1990;10:631–637. doi: 10.1038/jcbfm.1990.115. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Manaka S, Sano K. Changes in extracellular potassium concentration in cortex and brain stem during the acute phase of experimental closed head injury. J Neurosurg. 1981;55:708–717. doi: 10.3171/jns.1981.55.5.0708. [DOI] [PubMed] [Google Scholar]
- Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A, Wu Q, Zhang J, Hong Y. Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Int J Biol Sci. 2014;10:1072–1083. doi: 10.7150/ijbs.9719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel S, Schluesener H, Mittelbronn M, Seid K, Adjodah D, Wehner HD, Meyermann R. Dynamics of microglial activation after human traumatic brain injury are revealed by delayed expression of macrophage-related proteins MRP8 and MRP14. Acta Neuropathol. 2000;100:313–22. doi: 10.1007/s004019900172. [DOI] [PubMed] [Google Scholar]
- Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34:1397–1411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petraglia AL, Plog BA, Dayawansa S, Dashnaw ML, Czerniecka K, Walker CT, Chen M, Hyrien O, Iliff JJ, Deane R, Huang JH, Nedergaard M. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int. 2014;5:184. doi: 10.4103/2152-7806.147566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MJ, Angoa-Pérez M, Briggs DI, Viano DC, Kreipke CW, Kuhn DM. A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods. 2012;203:41–49. doi: 10.1016/j.jneumeth.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Smith C, Gentleman SM, Leclercq PD, Murray LS, Griffin WST, Graham DI, Nicoll JA. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol. 2013;39:654–666. doi: 10.1111/nan.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Landeghem FKH, Stover JF, Bechmann I, Brck W, Unterberg A, Bhrer C, von Deimling A. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia. 2001;35:167–179. doi: 10.1002/glia.1082. [DOI] [PubMed] [Google Scholar]
- Von Bernhardi R, Tichauer JE, Eugen J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J Neurochem. 2010:1099–1114. doi: 10.1111/j.1471-4159.2009.06537.x. [DOI] [PubMed] [Google Scholar]
- Petraglia AL, Plog Ba, Dayawansa S, Chen M, Dashnaw ML, Czerniecka K, Walker CT, Viterise T, Hyrien O, Iliff JJ, Deane R, Nedergaard M, Huang JH. The spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: a novel mouse model of chronic traumatic encephalopathy. J Neurotrauma. 2014;31:1211–1224. doi: 10.1089/neu.2013.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols JN, Deshane AS, Niedzielko TL, Smith CD, Floyd CL. Greater neurobehavioral deficits occur in adult mice after repeated, as compared to single, mild traumatic brain injury (mTBI) Behav Brain Res. 2016;298:111–124. doi: 10.1016/j.bbr.2015.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Teng Z, Song Y, Hu M, Chen C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J Cereb blood flow Metab. 2015;35:443–453. doi: 10.1038/jcbfm.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Bolton AN. Regional Neurodegeneration and Gliosis Are Amplified by Mild Traumatic Brain Injury Repeated at {24-Hour} Intervals. J. Neuropathol. Exp. Neurol. 2014;73:933. doi: 10.1097/NEN.0000000000000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo JO, Mouzon B, Greenberg MB, Bachmeier C, Mullan M, Crawford F. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J Neuropathol Exp Neurol. 2013 doi: 10.1097/NEN.0b013e3182814cdf. [DOI] [PubMed] [Google Scholar]
- Marmarou CR, Prieto R, Taya K, Young HF, Marmarou A. In: Animal models of acute neurological injuries. Chen J, Xu ZC, Xu X-M, Zhang JH, editors. Totowa, NJ: Humana Press; 2009. pp. 393–407. [Google Scholar]
- Ojo JO, Mouzon BC, Crawford F. Repetitive head trauma, chronic traumatic encephalopathy and tau: Challenges in translating from mice to men. Exp Neurol. 2016;275:389–404. doi: 10.1016/j.expneurol.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Fischer TD, Red SD, Chuang AZ, Jones EB, McCarthy JJ, Patel SS, Sereno AB. Detect of subtle cognitive changes after mTBI using a novel tablet-based task. J Neurotrauma. 2015 doi: 10.1089/neu.2015.3990. Ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- Hazrati LN, Tartaglia MC, Diamandis P, Davis KD, Green RE, Wennberg R, Wong JC, Ezerins L, Tator CH. Absence of chronic traumatic encephalopathy in retired football players with multiple concussions and neurological symptomatology. Front Hum Neurosci. 2013;7:222. doi: 10.3389/fnhum.2013.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]