

Abstract
Outcome varies among patients with subarachnoid hemorrhage but known prognostic factors explain only a small portion of the variation in outcome. We hypothesized that individual genetic variations influence brain and vascular responses to subarachnoid hemorrhage and investigated this using inbred strains of mice.
Subarachnoid hemorrhage was induced in seven inbred and a chromosome 7 substitution strain of mouse. Cerebral blood flow, vasospasm of the middle cerebral artery, and brain injury were assessed. After 48 h of subarachnoid hemorrhage, mice showed significant middle cerebral artery vasospasm that correlated positively with reduction in cerebral blood flow at 45 min. Mice also had increased neuronal injury compared to sham controls; A/J and C57BL/6 J strains represented the most and least severe, respectively. However, brain injury did not correlate with cerebral blood flow reduction at 45 min or with vasospasm at 48 h. Chromosome 7 substitution did not influence the degree of vasospasm or brain injury.
Our data suggested that mouse genetic background influences outcome of subarachnoid hemorrhage. Investigations into the genetic factors causing these inter-strain differences may provide insight into the etiology of the brain damage following subarachnoid hemorrhage. These findings also have implications for animal modeling of disease and suggest that genetic differences may also modulate outcome in other cardiovascular diseases.
Keywords: Genetic variation, mouse, neuronal cell death, subarachnoid hemorrhage
Introduction
Subarachnoid hemorrhage (SAH) accounts for only 5% of stroke but for more than one quarter of potential life years lost to stroke.1 Outcome has improved over the last decades, probably due to administration of nimodipine, early repair of the ruptured aneurysm, and better intensive care.2,3 However, the mortality is still 35%, and half of the survivors cannot return to their pre-morbid level of functioning.4 The etiology of brain injury after SAH is probably multifactorial, although multivariable analysis of a large number of patients shows that only a small proportion of the variation in outcome is explained by identifiable factors.5,6 We hypothesized that an important amount of the variation is due to genetic differences between individuals. This has not been widely investigated in humans and is not addressed in studies of genetically manipulated animals because they do not model the natural response to disease.7 In order to determine the role of genetic variation in the response to SAH, we induced SAH in seven inbred strains of mice and a chromosome 7 substitution strain of mouse. The latter strain was used because chromosome 7 has been shown to contain a locus, Civq1, which affects infarct volume in mice, putatively by affecting both collateral blood vessel number and susceptibility of neurons to ischemia.8–10 The aims were to determine if there is variability in the degree of vasospasm of the middle cerebral artery (MCA, “angiographic vasospasm,” in this manuscript termed “vasospasm”) and brain injury among genetically different mouse strains, and if brain injury and vasospasm correlate.
Materials and methods
Animals
Animal protocols were approved by the Animal Care Committee at St. Michael’s Hospital and complied with the regulations of the Canadian Counsel on Animal Care and with the ARRIVE guidelines (Animal Research: Reporting in Vivo Experiments). The animals were kept in a 12-h light-dark cycle and had access to food and water ad libitum. Seventy-two male mice weighing 20–25 g from seven inbred strains were used: C57BL6/J (C57), DBA/2 J (DBA), FVB/NJ (FVB), A/J (AJ), 129S1/SvImJ (129S1), KK/HIJ (KK), BALB/c (BALB) (all obtained from Jackson Laboratories, Bar Harbor, ME). The strains were operated on in random order and were randomly allocated to sham or SAH groups. Strains were selected to span the spectrum of cerebral infarct volumes reported after permanent MCA occlusion.7 In a second set of experiments, 10 male C57BL/6 J-Chr7A/J/NaJ (C57-Chr7AJ) mice weighing 20–25 g were used (Jackson Laboratories); in this chromosome substitution strain, chromosome 7 from an AJ mouse is substituted into a C57 mouse. Mice were randomized to sham or SAH groups (Supplementary Table 1).
SAH model
SAH was induced as reported in Sabri et al.11 Animals were anesthetized with inhalation of isofluorane (5% induction; 2–3% maintenance) carried by oxygen (1 L/min). Body temperature was maintained at 37.0 ± 0.5℃ with a homeothermic heating pad and a rectal probe (Harvard Apparatus, Holliston, MA). Oxygen saturation was monitored using a MouseSTAT pulse oximeter (Kent Scientific, Torrington, CT). The head was fixed in a stereotactic frame equipped with a mouse adaptor (Harvard apparatus). A 0.9-mm burr hole was drilled 4.5 mm anterior to the bregma and angled ventrally at 40°. A Doppler flow meter (BLT21, Transonics Systems, New York, NY) was used to monitor relative cerebral blood flow (CBF) for 15 min before and 45 min after blood injection or needle insertion (see Supplementary Figure 1 for a set of representative traces for sham and SAH mouse).
For SAH (n = 5 per strain), 60 µL of donor blood from a mouse of the same strain (anesthetized with 120/30 mg/kg ketamine/xylazine) was injected into the pre-chiasmatic cistern with a 27-gauge spinal needle (BD Biosciences, San Jose, CA) over 10 s. The sham-operated group (n = 5 per strain) had the needle inserted but nothing was injected. Buprenorphine (0.4 mg/kg) was administered subcutaneously as a post-operative analgesic twice daily for 48 h in both SAH- and sham-operated animals.
Animals were sacrificed 48 h after surgery and perfused through the left cardiac ventricle with 0.9% NaCl (1 mL), followed by 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS, 5 mL), both at physiologic blood pressure. Brains were removed and fixed in 4% PFA for 48 h then sectioned in the coronal plane using a mouse brain matrix (Zivic Instruments, Pittsburgh, PA). Brains were cut into three pieces in the coronal plane: the groove between the forebrain and cerebellum (−4.5 mm from bregma), 2 mm posterior to the groove (−6.5 mm from bregma), and 3 mm anterior to the groove (−1.5 mm from bregma). The remaining anterior portion of the forebrain was rotated 90° to make two sagittal cuts 1 mm lateral to the midline at the crossing of the olfactory tract with the MCA (MCA cross sections). Blocks were embedded in paraffin and trimmed in 20 µm increments until tissue was evenly distributed (∼160 µm). Samples were then cut into 5 µm ribbons consisting of six sequential sections using a microtome (Leica, Wetzlar, Germany). Each section was mounted on a glass slide. Hematoxylin and eosin, fluoro-jade B, TUNEL, and caspase-3 staining was conducted on the first, second, third, and fourth slides, respectively.
Hematoxylin and eosin staining
In all mouse and chromosome substitution strains, brain sections were deparaffinized in xylene and rehydrated with decreasing concentrations of ethanol solutions. Slides were stained with hematoxylin and eosin and coverslipped with a xylene-based mounting medium (Permount, Sigma-Aldrich, St. Louis, MO).
TUNEL and fluoro-jade B staining
Apoptosis was assessed using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL, DeadEnd Fluorometric kit, Promega, Fitchburg, WI). Fluoro-jade B (Histo-Chem Inc., Jefferson, AR) was used to assess neuronal degeneration. Brain sections were deparaffinized and rehydrated. Following incubation with deionized water, the slides were incubated in 0.06% potassium permanganate (Sigma-Aldrich) for 15 min. Slides were then rinsed in deionized water and immersed for 30 min in 0.001% fluoro-jade B working solution (0.1% acetic acid). Slides were washed and dried (60℃) for 15 min, then cleared in xylene and coverslipped with a non-aqueous, low fluorescence, styrene-based mounting media (DPX, Sigma-Aldrich). Slides were viewed under a fluorescent light microscope (Olympus BX50, Olympus, Richmond Hill, ON, Canada), and images were taken using constant parameters (exposure time and contrast values).
Caspase-3/anti-neuronal nuclei immunohistological staining
In the first set of animals, after deparaffinization and rehydration, antigen was retrieved by heating tissue sections for 30 min in 0.01 mmol/L sodium citrate (pH 6.0) at 98℃. Sections were permeabilized with 0.3% Triton X-100 for 1 h and then incubated with 10% normal goat serum and 1% bovine serum albumin (BSA) in PBS for 30 min. Slides were incubated with primary rabbit anti-human active caspase-3 (1:300 in BSA, BD Pharmingen, Franklin Lakes, NJ) and mouse anti-neuronal nuclei (NeuN) (1:200 in BSA, Invitrogen, Carlsbad, CA) for 24 h. This was followed by a wash and incubation with secondary antibodies (goat anti-rabbit Alexa Fluor 488 (1:700 in BSA) and goat anti-mouse Alexa Fluor 568 (1:700 in BSA, Invitrogen)) for 1 h. Slides were counter-stained with 4',6-diamidino-2-phenylindole (DAPI) and viewed with a fluorescence microscope.
Data quantification and statistical analysis
Raw CBF data were standardized to baseline and expressed as percent change from an averaged baseline for each independent strain. All images for vasospasm and histology were taken with cellSens 1.7 image capture software (Olympus) and a 12.8 megapixel color camera (DP72, Olympus). To assess vasospasm, images of the MCA were taken at 200× magnification and the lumen area and thickness of the MCA wall were quantified using Image J (NIH, Bethesda, MD). For fluoro-jade B staining, images were taken at 200× magnification of the entire cerebral cortex (∼20 images), hippocampus (dentate gyrus, CA3 and CA1; ∼12 images), and basal ganglia, thalamus, and midbrain (∼16 images). TUNEL-positive cells that co-localized with DAPI and caspase-3-positive cells that co-localized with NeuN and DAPI were quantified in the cerebral cortex at 200× magnification (∼20 images). Two blinded observers counted the number of degenerated/apoptotic neurons and measured the degree of MCA vasospasm; these values were averaged.
Data are presented as means ± standard error of the mean (SEM). Fold changes ± SEM were calculated by normalizing SAH values to sham controls. Student’s t-test was performed when two groups were compared. Analysis of variance with post-hoc Fisher least squares difference was used when assessing significance across multiple groups. Correlations were identified using the Pearson product moment correlation coefficient. A p < 0.05 was considered significant.
Results
No difference in mortality across strains
Mortality occurred in 3% (2 of 72) of mice undergoing SAH in the first protocol. Death occurred after blood injection and did not happen in any strain-related fashion. Mice that did not survive to tissue collection (48 h) were not included in analyses. No mortality was observed in sham-operated animals or in the second set of C57-Chr7AJ mice.
Variable degree of CBF recovery across strains
The CBF recorded during induction of SAH was consistent with previous descriptions.11 There was no difference between strains in CBF in sham-operated animals (Figure 1(a)). In SAH animals, CBF dropped more than 80% right after blood injection (t = 0) and ranged from 9.5% to 18.3% of respective baseline control levels (p < 0.01, Figure 1(a)). At 45 min after blood injection, CBF of the SAH groups slowly recovered but was significantly lower than corresponding sham controls (p < 0.05), except for C57 and FVB strains (Figure 1(a)). Across strains, there was variation in the degree of recovery of CBF post-SAH (Figure 1(a)). For example, at 45 min post-SAH, KK mice recovered to only 60.0 ± 11.0%, while C57 mice recovered to a relative CBF of 88.7 ± 2.7% of baseline. CBF of each strain stabilized at their respective levels after approximately 10–20 min. Additionally, CBF in C57 mice recovered significantly faster than other strains, except for 129S1 at 10 and 12.5 min post-SAH induction (p < 0.05). At 20 min after blood injection, C57 mice had significantly higher CBF than BALB, AJ, and KK mice (p < 0.01, Figure 1(a)). Arterial oxygen saturation remained at normal levels (80–100%).
Figure 1.

Differential acute CBF change and correlation with vasospasm. (a) CBF measurements in seven inbred mouse strains after experimental SAH. Blood was injected at t = 0 min. CBF was monitored for 15 min pre-injection (baseline) and 45 min post-injection. Data expressed as mean ± SEM, n = 5 control and 5 SAH per strain. (b) A correlation was seen between CBF and vasospasm across seven mouse strains. CBF and vasospasm were measured at 45 min and 48 h post-SAH, respectively. Data expressed as mean of normalized samples ± SEM, Pearson product moment correlation, p < 0.05, n = 5 control and 5 SAH per strain.
CBF: cerebral blood flow; SAH: subarachnoid hemorrhage.
MCA vasospasm varied across strains and inversely correlated with CBF
Vasospasm was assessed 48 h after sham or SAH surgery. Sham-operated mice showed normal morphology with no evidence of vasospasm (Figure 2(a)). SAH mice had a reduction in the lumen of the MCA and an increase in artery wall thickness with a corrugated appearance (Figure 2(a)). The raw MCA lumen/wall thickness ratio in SAH animals was: 2.72 ± 0.53 for FVB, 3.44 ± 0.73 for BALB, 3.62 ± 1.41 for AJ, 3.72 ± 0.71 for DBA, 4.30 ± 0.56 for C57,4.44 ± 0.95 for KK, and 4.71 ± 1.21 for 129S1. The MCA lumen/wall thickness ratio was also significantly smaller in SAH mice compared to their sham counterparts (p < 0.05) but there was no significant difference among strains with SAH. To account for observable differences in baseline MCA measurements, all SAH animals were normalized to sham controls within each strain. After normalization, FVB mice showed the greatest and KK mice showed the least vasoconstriction with a 76.1% and 34.8% reduction in MCA lumen/wall thickness ratio, respectively (Figure 2(b)). There was a significant difference in vasospasm between KK and C57, BALB, FVB, and AJ mice (p < 0.05, Figure 2(b)) and between 129S1 and FVB strains (p < 0.05, Figure 2(b)). Surprisingly, mice with greater CBF recovery after SAH also exhibited greater vasospasm; the converse was also evident. A positive correlation between CBF and vasospasm was observed (p < 0.05, Figure 1(b)). It must be noted that CBF was measured 45 min while vasospasm was measured 48 h after SAH.
Figure 2.

Different degrees of vasospasm across strains. (a) Representative images of the MCA from sham and SAH mice of various strains. A reduction in MCA diameter and an increase in wall thickness are observed in SAH animals compared to sham controls (hematoxylin and eosin). (b) Fold change in MCA radius/wall thickness ratio after SAH in various mouse strains. Each strain was normalized to their respective sham control. The degree of vasospasm varies across strains. Data expressed as mean fold change ± SEM, *p < 0.05 compared to KK, †p < 0.05 compared to 129S1 (ANOVA, Fisher LSD), n = 5 control and 5 SAH per strain.
SAH: subarachnoid hemorrhage.
Differential neuronal cell death across strains
All SAH animals exhibited an increase in fluoro-jade B-positive cells, with the majority evident in the cerebral cortex (Figure 3(a)). Fewer fluoro-jade B-positive cells were observed in the hippocampus, midbrain, thalamus, and basal ganglia. Sham animals had few to no positive cells. All SAH animals differed significantly from sham controls (p < 0.05). The raw cell counts of SAH animals in increasing order were 142 ± 15 for C57, 608 ± 152 for DBA, 631 ± 145 for FVB, 682 ± 211 for 129S1, 759 ± 136 for BALB, 1066 ± 273 for AJ, and 1239 ± 121 for KK mice. Comparing strains, KK mice had the most fluoro-jade B positive cells, which was significantly different from all strains, except AJ mice (p < 0.05). C57 mice had the least fluoro-jade B positive cells, which was significant compared to DBA, FVB, and 129S1 (p < 0.05) and AJ and KK strains (p < 0.001). After normalizing cell counts to sham controls, the order of severity changed for some strains; DBA mice and FVB mice demonstrated the highest and lowest neuronal injury, respectively, while C57 and AJ mice remained at the same position on the injury spectrum (Figure 3(b)). Across strains, only DBA and FVB strains showed a significant difference (p < 0.05, Figure 3(b)).
Figure 3.

Neurodegeneration as detected by fluoro-jade B staining in different strains. (a) Fluoro-jade B positive neurons were present in the cerebral cortex of all SAH mice (white arrows) but not sham controls (except KK mice). (b) Fold change in fluoro-jade B positive neurons in coronal sections of SAH mice of various strains. Each strain was normalized to their respective sham control. Data expressed as mean fold change ± SEM. ANOVA with post-hoc Fisher LSD test was used, n = 5 control and 5 SAH per strain.
SAH: subarachnoid hemorrhage.
Caspase-3 and TUNEL staining was performed to further verify the strains with the most (AJ) and least (C57) brain injury after SAH, as compared to fluoro-jade B staining. In caspase-3 staining, both AJ (Figure 4(a)) and C57 strain (Figure 4(b)) SAH animals had numerous caspase-3-positive neurons in the cerebral cortex. Immunoreactivity was most evident in the cytoplasm with little nuclear localization, indicating early apoptosis (Figure 4(c)). Sham animals showed little to no caspase-3-positive neurons. Both C57 and AJ strains showed a significant number of apoptotic neurons after SAH compared to sham controls (Figure 4(d), p < 0.05), and AJ mice showed significantly more apoptotic neurons compared to C57 mice (Figure 4(d), p < 0.05). No TUNEL-positive cells were detected, and there were no statistically significant differences between sham and SAH groups for any of the strains.
Figure 4.

Differential neuronal cell death across strains. (a) and (b) Representative images of caspase-3 staining in the cerebral cortex of AJ and C57 mice. Caspase-3-positive cells indicate apoptotic neurons and were present in AJ mice (a) and C57 (b) mice. (c) Magnification of boxed regions from (a) and (b) in SAH mice demonstrates the predominant cytoplasmic localization of caspase-3, with few neurons showing nuclear localization (white arrows). (d) Quantification of caspase-3 positive cells in coronal sections of AJ and C57 mice. Significantly more apoptotic cells are present in AJ mice. Data expressed as mean ± SEM, Student’s t-test, n = 5 control and 5 SAH per strain.
SAH: subarachnoid hemorrhage; NeuN: anti-neuronal nuclei; DAPI: 4',6-diamidino-2-phenylindole.
Correlations between brain injury, CBF, and vasospasm
Mouse strains were ranked from most severe/worst to least severe/best in terms of brain injury, vasospasm, and CBF reduction and compared to results found in ischemic stroke (Figure 5(a)).7 C57 and AJ mice consistently had low and high brain injury, respectively, in raw and normalized data. FVB and BALB strains demonstrated the most vasospasm while KK and 129S1 strains showed little vasospasm in raw and normalized data. CBF recovery was worst in KK and best in C57 mice. The order of brain injury severity after SAH was similar to that found in ischemic stroke.7 There was no correlation between vasospasm and brain injury (p > 0.05, Figure 5(b)) or between CBF and brain injury (p > 0.05, Figure 6). The correlations between brain injury, CBF, and vasospasm were summarized in three dimension (Figure 7).
Figure 5.

Injury severity across strains and correlation of brain injury with vasospasm. (a) Mice were ranked in terms of brain injury, vasospasm, and CBF and were also compared to findings in ischemic stroke.7 The blue arrows between strains indicate that their values were similar. (b) No correlation was found between brain injury and vasospasm across the seven mouse strains. Data expressed as mean of normalized samples ± SEM, Pearson product moment correlation, n = 5 control and 5 SAH per strain.
CBF: cerebral blood flow.
Figure 6.
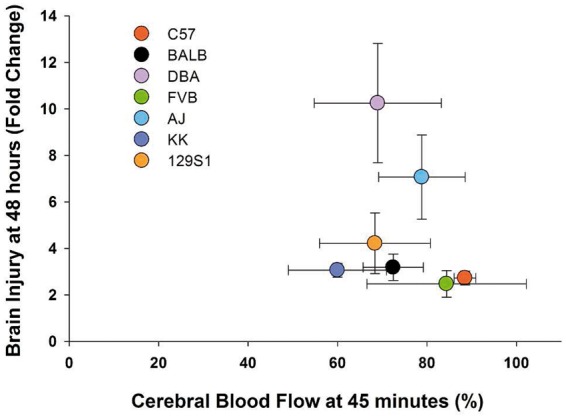
Correlation of brain injury with CBF. No correlation was found between brain injury at 48 h and CBF reduction at 45 min after SAH across the seven mouse strains. Data expressed as mean of normalized samples ± SEM, Pearson product moment correlation, n = 5 control and 5 SAH per strain.
Figure 7.

AJ chromosome 7 substitution into C57 mice does not alter vasospasm or brain injury. (a) The degree of vasospasm was not significantly different between C57, C57-Chr7AJ, and AJ mice. (b) The number of fluoro-jade positive cells were significantly higher in AJ mice, but not C57-Chr7 AJ mice, compared to C57. Data expressed as mean ± SEM, Student’s t-test, n = 5 control and 5 SAH per strain.
SAH: subarachnoid hemorrhage.
Chromosome 7 is not responsible for inter-strain variation in vasospasm and brain injury
C57-Chr7AJ mice demonstrated a reduced MCA radius/wall thickness ratio after SAH (Figure 7(a)). However, this reduction was not significantly different from C57 or AJ mice after SAH (p > 0.05); in fact, the degree of vasospasm was similar across the three groups. There was no difference between the three strains in sham-operated animals. In SAH groups, the number of fluoro-jade B positive cells was significantly different between C57 and AJ (p < 0.05) but not significantly different compared to the C57-Chr7AJ group (p > 0.05, Figure 7(b)). Sham-operated animals showed no differences across groups. Overall, chromosome 7 did not influence the severity of vasospasm or brain injury after SAH. This surprising finding stands in contrast to work from Keum et al.9 and Chu et al.,8 indicating that chromosome 7 harbors a genetic locus mediating infarct size after MCA occlusion.
Discussion
These experiments discover that vasospasm and brain injury after SAH vary among inbred mouse strains. This shows that genetic factors are fundamentally important in determining the response of the brain and vasculature to SAH. Furthermore, the lack of effect of chromosome 7 substitution on brain injury and vasospasm, in contrast to its large effect on ischemic stroke in mice, suggests that mechanisms of brain injury after SAH may be very different from ischemic stroke.
The findings of this study are fundamentally important in that mouse strain, or inherent genetic background, affects acute CBF reduction, vasospasm, and brain injury after SAH and furthermore, the effects of background are different depending on the outcome assessed. We found that AJ mice demonstrated the most neuronal injury and C57 demonstrated the least. Since these mouse strains are highly inbred and animals in each strain are theoretically and practically genetically identical, the variations observed between strains must be due to genetic variation. It is important that the strains were maintained in a shared environment. This control is important given the emerging view that environmental differences may modify disease propensity, perhaps by epigenetic mechanisms.12
Second, we found no correlation between brain injury and vasospasm or CBF, but acute CBF reduction 45 min after SAH was positively correlated with vasospasm 48 h later. Finally, we determined that unlike ischemic stroke, the variation in brain injury and vasospasm after SAH is not primarily controlled by loci on chromosome 7.7 These findings suggest that genetic variation affects the brain response to SAH. Also, the mechanism by which SAH causes neurological changes is mediated by factors that differ from ischemic stroke.
Mouse genetic background also has been shown to influence other phenomena such as spatial learning, brain and vascular anatomy, inflammation, and susceptibility to experimental allergic encephalitis and excitotoxicity.13–20 Thus, differences in sensitivity to injury, caused in part by genetic background, highlight the potential role of different genes in aggravating, or protecting against SAH-induced brain injury, as well as the potential need for personalized treatment for SAH. Genome wide association studies have not been conducted to assess effects of SAH on outcomes in humans, although some genes have been suggested to modify susceptibility to several complications of SAH. Patients with the APOE4 allele may have increased risk of delayed cerebral ischemia (DCI) and poor outcome after SAH.21 Patients with one or two alleles of the haptoglobin 2 genotype have a greater risk of angiographic vasospasm and DCI after SAH, although the effect on outcome is unclear.22–24 In a Finnish population, risk of fatal SAH was linked to genetic polymorphisms in human leukocyte antigen linked genes.25 Serum tumor necrosis factor-alpha (TNF-α) and non-wild type TNF-α genotype were not associated with DCI and poor outcome after SAH and were suggested to be unlikely to be important in SAH.26
Similarly, these findings may also be an important consideration for other cardiovascular diseases where genetic factors may also influence presentation of the disease in both a clinical and laboratory setting.
Variation in infarct volume in response to focal cerebral ischemia has been demonstrated across different mouse strains.19,20,27–30 Some of this variation is due to obvious differences in collateral circulation, such as the size of the posterior communicating artery, but other differences in anastomotic circulation could play a role.19,27 In terms of CBF, we report that C57 mice were able to almost fully recover after SAH (Figure 1(a)) and exhibited less brain injury 48 h later. The C57 strain has increased collateral density in the pial circulation compared to other strains.27 Studies using chromosome substitution strains found a locus on chromosome 7, possibly related to vascular endothelial growth factor that contributed to variation in infarct size across diverse mouse strains after permanent MCA occlusion.7,27 Further studies demonstrated the locus on chromosome 7 contains genes that influence both collateral circulation and neuronal susceptibility to ischemia.9 Chromosome 7 did not significantly influence vasospasm or brain injury after SAH (Figure 7), which suggests that ischemic stroke and SAH induce neurological changes through different molecular mechanisms. Although we did not identify the specific loci that influence neuronal cell death and vasospasm after SAH, our results demonstrate that these two outcomes are not primarily controlled by chromosome 7.
Vasospasm can also affect CBF and is a major cause of morbidity after SAH.31,32 It is interesting to note that CBF 45 min and vasospasm 48 h after SAH had a positive relationship; the strains with the worst CBF recovery, such as KK, had the least vasospasm (Figure 1(b)). This unexpected finding possibly connects the early and late vascular responses within this model of SAH; however, we recognize that the two were measured at different time points. The degree of vasospasm we observed at 48 h did not appear to be enough to reduce CBF, and we did not measure CBF at 48 h, so the contribution of CBF changes to brain injury, as opposed to metabolic differences, is uncertain. It is also possible that C57, and other strains, may have microvascular responses mediated by variations in gene expression; this may protect them from SAH-induced vascular damage. Differences in metabolism and neurons could also affect the degree of brain injury. Furthermore, changes in regional CBF in humans are not static and they vary regionally as well as over time (probably over a spectrum from second-to-second and day-to-day).33 Changes in CBF are dynamic and may be mediated by cortical spreading depolarization.33
Brain injury and neuronal injury, which we assessed by fluoro-jade B, most directly affect patient outcome.34 We identified the strains with the most and least neuronal degeneration to be AJ and C57 strains, respectively. Although DBA mice had the greatest fold change in brain injury, this was due to fewer positive cells in sham mice; the raw number of degenerated neurons was quite low in sham-operated DBA mice compared to other strains (Figures 3(b) and 5(a)). Therefore, the DBA strain was not selected as the strain with highest brain injury. Additionally, KK mice develop diabetes mellitus with age, which might predispose them to more brain injury.35 Male KK strain mice can develop weight- and diet-dependent features of non-insulin dependent diabetes. This adds a confounding factor to their assessment so we did not examine them further in our selection of low- and high-severity groups.
TUNEL and caspase-3 staining was conducted on representative strains (AJ and C57). Caspase-3 staining was localized to the cytoplasm (Figure 4(c)). In order for caspase-3 to cause deoxyribonucleic acid degradation it would need to translocate to the nucleus,36 which explains the lack of TUNEL-positive cells. It has been suggested that these results are consistent with early apoptosis.37
Finally, the degree of brain injury was not related to the severity of vasospasm (Figure 5(b)) or acute reduction in CBF (Figure 6). Given that our findings demonstrate a disparity between vasospasm and neurological outcome in mice, this would suggest that distinct genes and molecular pathways mediate these processes. It also suggests the need for therapeutic interventions that target a multitude of pathways, not solely angiographic vasospasm, in treatment of SAH.
There are some limitations to this work, including how well the effects of SAH in humans are reproduced in mice. In particular, the mouse model used here has not been clearly shown to develop DCI with focal infarctions. For example, since there may be no DCI in mice, the finding that chromosome 7 substitution did not affect brain injury after SAH in mice could not prove that this chromosome does not harbor genes that influence DCI.
Overall, these findings have implications for choice of strain for experimental SAH as well as other cardiovascular diseases. More importantly, identification of factors that make individuals more or less susceptible to deleterious responses to a disease may lead to better understanding of pathophysiology and to new treatments.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: RLM receives grant support from the Physicians Services Incorporated Foundation, Brain Aneurysm Foundation, Canadian Stroke Network, and the Heart and Stroke Foundation of Ontario. RLM is Chief Scientific Officer of Edge Therapeutics, Inc.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JAD, RLM, PAM, and JA conceived and designed the experimental study. JAD executed all experiments, collected data, and analyzed the results. EL assisted in data collection. HW, SB, MKT, and CKL assisted in data analysis. JAD, RLM, and JA constructed the figures and wrote the manuscript. All authors assisted in reviewing and revising the manuscript.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Taylor TN, Davis PH, Torner JC, et al. Lifetime cost of stroke in the United States. Stroke 1996; 27: 1459–1466. [DOI] [PubMed] [Google Scholar]
- 2.Lovelock CE, Rinkel GJ, Rothwell PM. Time trends in outcome of subarachnoid hemorrhage: population-based study and systematic review. Neurology 2010; 74: 1494–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alleyne CH., Jr Aneurysmal subarachnoid hemorrhage: have outcomes really improved? Neurology 2010; 74: 1486–1487. [DOI] [PubMed] [Google Scholar]
- 4.Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 2010; 41: e519–e536. [DOI] [PubMed] [Google Scholar]
- 5.Rosengart AJ, Schultheiss KE, Tolentino J, et al. Prognostic factors for outcome in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38: 2315–2321. [DOI] [PubMed] [Google Scholar]
- 6.Lo BW, Macdonald RL, Baker A, et al. Clinical outcome prediction in aneurysmal subarachnoid hemorrhage using Bayesian neural networks with fuzzy logic inferences. Comput Math Methods Med 2013; 2013: 904860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keum S, Marchuk DA. A locus mapping to mouse chromosome 7 determines infarct volume in a mouse model of ischemic stroke. Circ Cardiovasc Genet 2009; 2: 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu PL, Keum S, Marchuk DA. A novel genetic locus modulates infarct volume independently of the extent of collateral circulation. Physiol Genomics 2013; 45: 751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keum S, Lee HK, Chu PL, et al. Natural genetic variation of integrin alpha L (Itgal) modulates ischemic brain injury in stroke. PLoS Genet 2013; 9: e1003807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Chen G, Zhu WW, et al. Influence of simvastatin on microthrombosis in the brain after subarachnoid hemorrhage in rats: a preliminary study. Ann Clin Lab Sci 2010; 40: 32–42. [PubMed] [Google Scholar]
- 11.Sabri M, Jseon H, Ai J, et al. Anterior circulation mouse model of subarachnoid hemorrhage. Brain Res 2009; 1295: 179–185. [DOI] [PubMed] [Google Scholar]
- 12.Matouk CC, Marsden PA. Epigenetic regulation of vascular endothelial gene expression. Circ Res 2008; 102: 873–887. [DOI] [PubMed] [Google Scholar]
- 13.Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A 1997; 94: 4103–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laghmouch A, Bertholet JY, Crusio WE. Hippocampal morphology and open-field behavior in Mus musculus domesticus and Mus spretus inbred mice. Behav Genet 1997; 27: 67–73. [DOI] [PubMed] [Google Scholar]
- 15.Nawashiro H, Tasaki K, Ruetzler CA, et al. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab 1997; 17: 483–490. [DOI] [PubMed] [Google Scholar]
- 16.Bernard CC. Experimental autoimmune encephalomyelitis in mice: genetic control of susceptibility. J Immunogenet 1976; 3: 263–274. [DOI] [PubMed] [Google Scholar]
- 17.Lathe R. Mice, gene targeting and behaviour: more than just genetic background. Trends Neurosci 1996; 19: 183–186. [DOI] [PubMed] [Google Scholar]
- 18.Upchurch M, Wehner JM. Differences between inbred strains of mice in Morris water maze performance. Behav Genet 1988; 18: 55–68. [DOI] [PubMed] [Google Scholar]
- 19.Barone FC, Knudsen DJ, Nelson AH, et al. Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab 1993; 13: 683–692. [DOI] [PubMed] [Google Scholar]
- 20.Majid A, He YY, Gidday JM, et al. Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke 2000; 31: 2707–2714. [DOI] [PubMed] [Google Scholar]
- 21.Lanterna LA, Ruigrok Y, Alexander S, et al. Meta-analysis of APOE genotype and subarachnoid hemorrhage: clinical outcome and delayed ischemia. Neurology 2007; 69: 766–775. [DOI] [PubMed] [Google Scholar]
- 22.Borsody M, Burke A, Coplin W, et al. Haptoglobin and the development of cerebral artery vasospasm after subarachnoid hemorrhage. Neurology 2006; 66: 634–640. [DOI] [PubMed] [Google Scholar]
- 23.Ohnishi H, Iihara K, Kaku Y, et al. Haptoglobin phenotype predicts cerebral vasospasm and clinical deterioration after aneurysmal subarachnoid hemorrhage. J Stroke Cerebrovasc Dis 2013; 22: 520–526. [DOI] [PubMed] [Google Scholar]
- 24.Kantor E, Bayir H, Ren D, et al. Haptoglobin genotype and functional outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg 2014; 120: 386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frosen J, Pitkaniemi J, Tulamo R, et al. Association of fatal aneurysmal subarachnoid hemorrhage with human leukocyte antigens in the Finnish population. Hum Immunol 2007; 68: 100–105. [DOI] [PubMed] [Google Scholar]
- 26.Beeftink MM, Ruigrok YM, Rinkel GJ, et al. Relation of serum TNF-alpha and TNF-alpha genotype with delayed cerebral ischemia and outcome in subarachnoid hemorrhage. Neurocrit Care 2011; 15: 405–409. [DOI] [PubMed] [Google Scholar]
- 27.Chalothorn D, Clayton JA, Zhang H, et al. Collateral density, remodeling, and VEGF-A expression differ widely between mouse strains. Physiol Genomics 2007; 30: 179–191. [DOI] [PubMed] [Google Scholar]
- 28.Fujii M, Hara H, Meng W, et al. Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke 1997; 28: 1805–1810. [DOI] [PubMed] [Google Scholar]
- 29.Kitagawa K, Matsumoto M, Yang G, et al. Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery. J Cereb Blood Flow Metab 1998; 18: 570–579. [DOI] [PubMed] [Google Scholar]
- 30.Yang G, Kitagawa K, Matsushita K, et al. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res 1997; 752: 209–218. [DOI] [PubMed] [Google Scholar]
- 31.Crowley RW, Medel R, Dumont AS, et al. Angiographic vasospasm is strongly correlated with cerebral infarction after subarachnoid hemorrhage. Stroke 2011; 42: 919–923. [DOI] [PubMed] [Google Scholar]
- 32.Dankbaar JW, Rijsdijk M, van der Schaaf IC, et al. Relationship between vasospasm, cerebral perfusion, and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neuroradiology 2009; 51: 813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dreier JP, Major S, Manning A, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009; 132: 1866–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vergouwen MD, Etminan N, Ilodigwe D, et al. Lower incidence of cerebral infarction correlates with improved functional outcome after aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab 2011; 31: 1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikeda H. KK mouse. Diabetes Res Clin Pract 1994; 24 Suppl: S313–S316. [DOI] [PubMed] [Google Scholar]
- 36.Chaitanya GV, Steven AJ, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal 2010; 8: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramuz O, Isnardon D, Devilard E, et al. Constitutive nuclear localization and initial cytoplasmic apoptotic activation of endogenous caspase-3 evidenced by confocal microscopy. Int J Exp Pathol 2003; 84: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.