

Abstract
Although tight junctions between human brain microvascular endothelial cells in the blood–brain barrier prevent molecules or cells in the bloodstream from entering the brain, in Alzheimer’s disease, peripheral blood monocytes can “open” these tight junctions and trigger subsequent transendothelial migration. However, the mechanism underlying this migration is unclear. Here, we found that the CSF2RB, but not CSF2RA, subunit of the granulocyte-macrophage colony-stimulating factor receptor was overexpressed on monocytes from Alzheimer’s disease patients. CSF2RB contributes to granulocyte-macrophage colony-stimulating factor-induced transendothelial monocyte migration. Granulocyte-macrophage colony-stimulating factor triggers human brain microvascular endothelial cells monolayer tight junction disassembly by downregulating ZO-1 expression via transcription modulation and claudin-5 expression via the ubiquitination pathway. Interestingly, intracerebral granulocyte-macrophage colony-stimulating factor blockade abolished the increased monocyte infiltration in the brains of APP/PS1 Alzheimer’s disease model mice. Our results suggest that in Alzheimer’s disease patients, high granulocyte-macrophage colony-stimulating factor levels in the brain parenchyma and cerebrospinal fluid induced blood–brain barrier opening, facilitating the infiltration of CSF2RB-expressing peripheral monocytes across blood–brain barrier and into the brain. CSF2RB might be useful as an Alzheimer’s disease biomarker. Thus, our findings will help to understand the mechanism of monocyte infiltration in Alzheimer’s disease pathogenesis.
Keywords: Alzheimer’s disease, CSF2RB, granulocyte-macrophage colony-stimulating factor, monocyte infiltration
Introduction
The blood–brain barrier (BBB) is a highly specialized structural and biochemical barrier regulating the entry of blood-borne molecules into the brain and preserving ionic homeostasis within the brain microenvironment. Junctional complexes between cerebral endothelial cells, comprising tight and adherens junctions, primarily determine BBB properties.1 Tight junctions reportedly seal gaps between adjacent epithelial cells for a continuous blood vessel wall, whereas adherens junctions maintain communication between endothelial cells.2 The former increases trans-epithelial electric resistance (TEER) and reduces paracellular permeability.1
Although BBB prevents bloodstream molecules or cells from entering the brain, more studies reported transendothelial cell migration under pathological and normal circumstances; for example, peripheral blood monocytes3,4 and T lymphocytes5,6 in Alzheimer’s disease (AD), small-cell lung cancer cells,7 and mesenchymal stem cells3 can “open” the tight junctions between human brain microvascular endothelial cells (HBMECs), thus triggering transendothelial migration.
AD is pathologically characterized by neurofibrillary tangles and extracellular deposits of beta amyloid (Aβ) or senile plaques (SP). Aβ production and deposition are believed to drive AD pathogenesis.4 Aβ deposition in the brain parenchyma could trigger peripheral cell entry into the brain.3,5,6,8–11 Monocytes can infiltrate the injured brain and subsequently differentiate into activated macrophages.12 To date, microglia have either beneficial (e.g., Aβ clearance12–16) or detrimental (e.g., neurotoxic factor secretion) effects in AD.5 It is important to address the mechanism by which microglia are involved in monocyte transendothelial migration and AD pathogenesis. We previously reported that monocytes derived from AD patients overexpress chemokine (C-X-C motif) ligand 1 (CXCL1), which interacts with CXC chemokine receptor 2 (CXCR2) on HBMECs.6 The chemokine receptor CCR2, expressed on monocytes, microglia, astrocytes, and neurons, is reportedly instrumental in microglial accumulation in the APP and APP/presenilin 1 (PS1) mouse models of AD;17,18 however, the underlying molecular mechanism remains unknown.
Entry of circulating cells into the central nervous system initially requires the passage of the cells across the vascular endothelial cell layer. High granulocyte-macrophage colony-stimulating factor (GM-CSF) levels are found in the brain parenchyma and cerebrospinal fluid (CSF) of AD patients19 GM-CSF is instrumental in either monocyte/macrophage activation or T cell function maintenance.20,21 The heterodimeric GM-CSF receptor comprises a major binding subunit, GMRα,22 and major signaling subunit, βc.23,24 To determine the factors contributing to transendothelial monocyte migration, we found that the GM-CSF/CSF2RB (human GM-CSF receptor beta-chain) axis affects monocyte migration through BBB.
Materials and methods
Cells and chemicals
HBMECs, obtained from surgical resections of 4 to 7-year-old children,25 were cultured in complete RPMI-1640 medium containing 10% fetal bovine serum (FBS), 10% Nu-serum (BD Biosciences, Franklin Lakes, NJ, USA), 2 mM glutamine, 1 mM sodium pyruvate, 1 × nonessential amino acids, and 1 × MEM vitamins. THP-1 (TIB-202, acute monocytic leukemia; ATCC, Manassas, VA, USA) and U937 (CRL-1593.2, histiocytic lymphoma; ATCC) cell lines were cultured in RPMI-1640 medium containing β-mercaptoethanol (0.05 mM for THP-1, 10 mM for U937; Invitrogen, Carlsbad, CA, USA) FBS to a final concentration of 10%. All cells were incubated at 37℃ in a humidified atmosphere with 5% CO2 and 95% air. HEK293T cells (CRL-11268, human embryonic kidney cells; ATCC) were maintained in DMEM supplemented with 10% FBS.
Human subjects
Twenty-three AD patients (13 men and 10 women, aged 60–88 years; mean age: 75.3 ± 3.5 years) and 21 age-matched controls (10 men, 11 women, mean age: 70.6 ± 8.6 years) were recruited from the First and Second Affiliated Hospitals, China Medical University, under an Institutional Review Board-approved human study protocol. AD patients had no immunological diseases or vascular risk factors (e.g., hypertension, cardiac disease, diabetes). AD diagnosis was based on National Institute of Neurological Disorders and Stroke-Alzheimer Disease and Related Disorders Association criteria7,26 and included the Mini Mental State Exam.8,27 Subjects were not experiencing systemic infection or taking nonsteroidal anti-inflammatory drugs at the time of phlebotomy. All participants’ next-of-kin or guardians provided written informed consent for study participation. The above study and consent procedure were reviewed and approved by the China Medical University Review Committee and followed ethical guidelines of the Helsinki Declaration of 1964 (revised 2013).
Animals and experimental groups
Wistar rats (male, 250–300 g) were obtained from the Lab Animal Center of China Medical University. Human Aβ1-42 or reverse peptide Aβ42-1 was prepared by dissolving the peptide in a freshly prepared 35% acetonitrile solution, which was diluted to a final concentration of 500 µM in 0.1 M phosphate-buffered saline (PBS, pH 7.4).6,28 Experimental AD was induced in rats as described.6 Briefly, the Aβ1-42 or Aβ42-1 solution was incubated at 37℃ and 2.5 µl Aβ1-42 solution (0.5 mM in PBS) was bilaterally cannulated into CA1 region of the hippocampus (posterior from bregma, 3.0 mm; lateral, 2.2 mm; and depth 2.8 mm) 24 h after the incubation.
The APP/PS1 double transgenic mice (B6C3-Tg (APPswe, PSEN1dE9) 85Dbo/J mice) were housed in cages in a controlled environment (22–25℃, 50% humidity). Original breeding pairs were obtained from the Jackson Laboratory (West Grove, PA, USA). To block GM-CSF, intracerebroventricular injection (i.c.v.) was performed as described.11 APP mice were anaesthetized and stereotaxically implanted with a cannula (RWD Life Science, Shenzhen, China) aimed at the right lateral ventricle (posterior from bregma, 0.6 mm; lateral, 1.1 mm; depth, 2.5 mm). The cannula was fixed to the skull with dental cement. After the surgical procedure, a dummy cannula was inserted into the cannula to prevent blockage. Mice were allowed to recover from surgery for five days. Nine-month-old mice, four male and eight female, were randomly assigned to 1 of 2 groups that were injected i.c.v. on days 1 and 4: anti-GM-CSF neutralization antibody (25 µg in 5 µl; R&D systems, Minneapolis, MN, USA) and isotypic IgG (25 µg in 5 µl; R&D systems). Injections were given over a 5-min period, after which the cannula was left in place for an additional 5 min to allow diffusion. Seven days post-injection, mice were re-anesthetized and monocytes were isolated.4,29
Experimental procedures were conducted in accordance with the regulations of the animal protection laws of China and approved by the animal ethics committee of China Medical University (JYT-20060948). Efforts were made to minimize animal suffering and the number of animals used. The above protocols were reviewed and approved by the China Medical University Review Committee. All studies involving animals were performed in accordance with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines.
Monocyte isolation
Fresh heparinized blood was collected from AD patients and healthy subjects. Human peripheral blood monocytes were isolated using CD14 Dynabeads (Invitrogen) within 3 h of phlebotomy, according to the manufacturer’s protocol. Monocytes were resuspended at 5 × 105 cells/ml in TEM buffer (RPMI-1640 without phenol red + 1% bovine serum albumin + 1 mM Hepes).
Rats were anesthetized with an intraperitoneal injection of 10% chloral hydrate (30 mg/kg; Sigma-Aldrich, St. Louis, MO, USA). Monocytes were isolated from whole blood behind the bulbus oculi via Ficoll-Hypaque (Amersham Pharmacia/GE Healthcare, Little Chalfont, UK) density gradient centrifugation seven days after Aβ peptide injection.
Mice were anesthetized and perfused with ice-cold 1 × HBSS. Brain mononuclear cells from APP/PS1 mice were isolated via Ficoll-Hypaque gradient.6
Flow cytometric analysis
Followed by percoll gradient centrifugation, fresh brain cells were prepared and incubated with allophycocyanin (APC)-labeled antibodies to CD45 (1.25 µg/ml) and phycoerythrin (PE)-labeled antibodies to CD11b (1.25 µg/ml) (both from eBioscience, USA), or with isotype-matched control antibodies on ice for 30 min. Fluorescence intensity was measured using a FACScalibur (BD) flow cytometer.
Real-time PCR
Total RNA was prepared using Trizol (Invitrogen) according to the manufacturer’s protocol, treated with RNase free DNase I (TaKaRa Bio Inc., Kusatsu, Japan), and reverse transcribed with M-MLV reverse transcriptase (Promega, Madison, WI, USA). Real-time PCR was performed on an ABI 7500 real-time PCR system (Applied BioSystems, Foster City, CA, USA) with SYBR Premix Ex Taq (Takara Biotechnology (Dalian) Co. Ltd, Dalian, China) according to the manufacturer’s protocol. cDNAs were amplified in a two-step amplification scheme (94℃, 2 min; 40 cycles of 94℃, 15 s and 60℃, 40 s). Recombinant PMD 18 T-vectors (Takara Biotechnology (Dalian) Co. Ltd) containing CSF2RB or GAPDH cDNAs were constructed and used to establish standard curves from which the initial target mRNA copy numbers were calculated. CSF2RB transcript amounts were normalized to GAPDH expression. CSF2RB and GAPDH primers and probes are listed in Table 1.
Table 1.
Primers for human and rat CSF2RB detection by real-time RT-PCR.
| Target | Oligonucletide sequence forward, reverse | 5′–3′ Nucleotide position | Genbank accession no. |
|---|---|---|---|
| Human CSF2RB | GCAGCATGTCGGCCTTCACTA | 1710–1730 | NM_000395.2 |
| GTCCCCGAATCCTACAGGGAA | 1790–1810 | ||
| Human GAPDH | GAAGGTGAAGGTCGGAGTC | 81–99 | NM_002046 |
| GAAGATGGTGATGGGATTTC | 287–306 | ||
| Rat CSF2RB | AGAAATGCTCTCCAGTGGTGAAG | 1031–1053 | NM_133555 |
| CTTGTTCCAGCCGCTTAACAG | 1136–1156 | ||
| Rat GAPDH | CCCTTCATTGACCTCAACTACAT | 949–971 | NM_017008 |
| GCCAGTAGACTCCACGACATA | 1123–1143 |
Plasmid construction
To construct Lenti-CSF2RB-shRNA, siRNA corresponding to human CSF2RB was designed using siRNA Target Finder software (http://www. Genscript.com/ssl-bin/app/rnai); shRNAs (AACCGTGCGATACGAACACATAGATTCAAGAGATCTATGTGTTCGTATCGCATTTTTTCCAAC; TCGAGTTGGAAAAAATGCGATACGAACACATAGATCTCTTGAATCTATGTGTTCGTATCGCACGGTT) were synthesized by Invitrogen. CSF2RB-shRNA was inserted into the lentiviral shRNA expression plasmid pll3.7 (Addgene, Cambridge, MA, USA). A non-silencing siRNA sequence (5′-TTACTGCTGTATGGACCGAGG-3′) was used as a control.
To construct a ZO-1 promoter-reporter fusion plasmid pGL3-ZO-1, the ZO-1 promoter was amplified as a 2787-bp fragment directly from HBMEC genomic DNA using the following primers: sense, 5′-GGTACCGCAACTTGTAAAAGTGACAA-3′ (pGL3-ZO-full, −2787 to −2768 bp); anti-sense, 5′-CTCGAGCTTGTCTCTCTCCAGCGCCG-3′ (−17 to −1 bp). The fragment was cloned in-frame into the pGL3 basic vector (Promega) at the KpnI and XhoI sites, and the resulting construct was sequence-verified. A series of 5′-deleted constructs were derived from pGL3-ZO-1 by PCR using the following sense primers: 5′-GGTACCTGCCAGACCTTGGATAAAAAC-3′ (pGL3-ZO-2112, −675 to −655 bp), 5′-GGTACCTTGGAGGGACAGCATTGGAATG-3′ (pGL3-ZO-1560, −1227 to −1206 bp), and 5′-GGTACCTTCAAGCGATTCTCCTGC-3′ (pGL3-ZO-451, −1998 to −1974 bp). The antisense primer was the same as that used for pGL3-ZO-1 construction.
Lentiviral infection of THP-1
Lenti-CSF2RB-shRNA and Lenti-non-silencing RNAs were linearized and transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen) for virus packaging and propagation. Recombinant CSF2RB-shRNA-expressing lentivirus particles were used to infect THP-1 cells. More than 90% of lentivirus-transduced THP-1 cells expressed GFP during the first 48 h post-infection.
Cell fractionation and Western blotting
Cell fractionation experiments were performed as described previously. Briefly, confluent HBMEC were pretreated with 20 ng/ml rhGM-CSF (R&D Systems) for different time intervals, washed, extracted in Triton X-100 lysis buffer (25 mM HEPES, 150 mM NaCl, 4 mM EDTA, 1% Triton X-100), and centrifuged to obtain the soluble fraction. Pellets were dissolved in sodium dodecyl sulfate (SDS) lysis buffer (25 mM HEPES, 4 mM EDTA, 1% SDS) to obtain the insoluble fraction. Equal portions of the soluble and insoluble fractions were analyzed for occludin expression (Abcam, Cambridge, UK) by Western blotting.
In other experiments, HBMECs pretreated with rhGM-CSF or rhGM-CSF + MG − 132 were collected at different time points, washed twice with ice-cold PBS, and prepared in radioimmunoprecipitation assay buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS) containing a protease inhibitor cocktail (Roche, Basel, Switzerland). The samples were separated by SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA) using a semi-dry transfer cell apparatus (Bio-Rad, Hercules, CA, USA). The PVDF membrane was blocked with 5% non-fat milk and incubated separately with polyclonal antibodies against claudin-5, VE-cadherin, occludin (Abcam), or ZO-1 (Invitrogen) at 4℃ overnight. The blots were incubated with a horseradish peroxidase (HRP) conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at room temperature. Protein bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL, USA). The relative signal densities were analyzed using a LAS-3000 imaging system (Fujifilm, Tokyo, Japan), and GAPDH densities were used as an internal control when comparing expression levels between samples.
Immunoprecipitation assay samples were subjected to Western blot analysis for ubiquitin (Abcam), myc (ABMART, Shanghai, China), or Flag (ABMART, Shanghai, China) after eluting the immune complex proteins in SDS sample buffer.
Immunofluorescence
GM-CSF-pretreated HBMEC monolayers grown on glass coverslips were fixed with 4% paraformaldehyde, then permeabilized with 0.1% Triton X-100. The cells were incubated with mouse anti-ZO-1 (Zymed Laboratories) after blocking with 5% BSA in PBS. Counterstaining was performed with DAPI (Sigma-Aldrich, St Louis, MO). The coverslips were mounted and analyzed using immunofluorescence microscopy (Olympus BX51, Tokyo, Japan).
Immunoprecipitation
This procedure was performed as previously reported.30 Cells were washed and lysed with lysis buffer (50 mM Tris, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1% Triton X-100, 1 mM sodium orthovanadate, 25 mM b-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride) containing a protease inhibitor cocktail. After centrifugation, the supernatants from cell lysates were collected. The protein contents were determined using the Bradford method. Subsequently, 1 mg of protein was incubated overnight with an anti-claudin 5 antibody at 4℃, followed by a 2 h-incubation with Protein A/G-agarose (Santa Cruz Biotechnology). Immune complex proteins were eluted in SDS sample buffer prior to Western blot analysis.
Transendothelial migration and endothelial cell permeability assay
HBMECs (1 × 105) were seeded in the upper chambers of 24-well Transwell plates with 0.4-µm pore size (Corning Inc., Corning, NY, USA). Serum-free media (800 μl) containing rhGM-CSF (0, 10, 20 ng/ml) was added to the lower chambers for 24 h. TEER was measured using a Millicell-ERS endothelia volt-ohmmeter (World Precision Instruments Inc., Sarasota, FL, USA) to monitor HBMEC monolayer integrity.
In the transendothelial migration assay, 1 × 105 HBMECs were seeded in the upper chambers of 24-well Transwell plates with 8-µm pore size (Corning). Four days later, 5 × 105 U937 or THP-1 or 5 × 105 Lenti-CSF2RB or Lenti-NS infected THP-1 cells were loaded into the HBMEC-precultured Transwell inserts. Serum-free media (800 μl) containing rhGM-CSF (0, 10, 20 ng/ml) was added to the lower chambers. Four hours later, cells that transmigrated into the lower chamber were harvested and counted in a hemocytometer.
For HRP flux measurements, medium was removed from both the upper and lower Transwell chambers after rhGM-CSF (0, 10, 20 ng/ml) stimulation for 24 h. Subsequently, serum-free RPMI-1640 was placed in both chambers of insert containing the HBMECs for 1 h, followed by serum-free RPMI-1640 containing chloroquine (50 μM) for 30 min. Next, HRP dissolved in chloroquine-containing serum-free RPMI-1640 was added to the Transwell insert to a final concentration of 0.5 mM. The medium in the lower chamber was collected after 1 h, and the HRP content was evaluated spectrophotometrically at 492 nm by assaying the peroxidase activity in buffer containing 0.5 mM orthopenylenediamine.
Transfections and luciferase assays
The luciferase reporter assay is a sensitive technique used to investigate gene promoter regulation. In our study, HBMECs in 24-well plates were incubated with GM-CSF for 20 h and cotransfected with pGL3-ZO-1 and pRL-TK. After 24 h, the cells were harvested, washed, and lysed in 80 μl of lysis buffer. Luciferase activities were analyzed using a dual-luciferase reporter assay system (Promega), and luciferase activity was normalized to the control Renilla luciferase activity. Relative luciferase activity was read using a Lumat LB 9507 luminometer (Berthold Technologies, Bad-Wildbad, Germany).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was assessed using two-tailed Student’s t-test (α = 0.05) between two groups. One-way ANOVA followed by post hoc Tukey test for multiple comparisons. Two-way ANOVA was used to compare transmigration rates between U937 and THP-1. Differences were considered statistically significant at a p value < 0.05.
Results
CSF2RB overexpression in monocytes from AD patients
To identify the factors that contributed to monocyte migration from the blood to the brain in AD, we subjected mRNA isolated from the monocytes of 23 AD patients and 21 aged-matched elderly controls to microarray analysis. The biological functions of GM-CSF are initiated by interactions with its dimeric cell surface receptor, which comprises the α (CSF2RA) and β (CSF2RB) subunits. The α subunit is ligand-specific, with low affinity for GM-CSF. The β subunit does not directly bind GM-CSF but forms a high-affinity receptor with the α subunit.31 Real-time RT-PCR was used to detect CSF2RB mRNA expression in the patient monocytes and confirm the transcriptional differences. The results indicated significantly higher CSF2RB expression in patient monocytes than in cells from age-matched elderly controls (Figure 1(a)). However, CSF2RA expression levels did not differ (Figure1(b)). Similarly, significantly higher CSF2RB expression was observed in experimental AD rats than in controls (Supplemental data 1), indicating a link between CSF2RB and AD pathogenesis.
Figure 1.
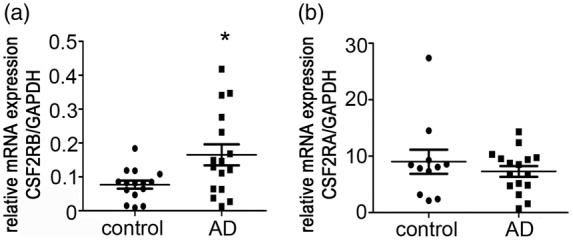
CSF2RB overexpression in monocytes from AD patients. (a) CSF2RB expression levels in monocytes from AD patients. Peripheral blood monocytes were isolated from 16 AD patients and 15 age-matched elderly controls; total RNA was extracted for real-time RT-PCR. Two-tailed t-test (α = 0.05) was used to compare monocytes CSF2RB expression in AD patients and age matched controls. *p < 0.05 vs. age-matched controls. (b) CSF2RA expression levels in peripheral blood monocytes from 16 AD patients and 11 age-matched elderly controls were detected by real-time RT-PCR. CSF2RB/GAPDH and CSF2RA/GAPDH indicate the relative expression levels of CSF2RB and CSF2RA, respectively, after normalization to the expression level of GAPDH. Two-tailed t-test (α = 0.05) was used to compare CSF2RA expression in AD patients and age-matched controls. Data are shown as means ± standard deviations.
CSF2RB affects GM-CSF-induced transendothelial monocyte migration
HBMEC monolayers cultured on Transwell inserts have been broadly used as a BBB model.5,6 To mimic AD brain microenvironment, a GM-CSF containing in vitro BBB model was constructed in this study. As shown in Figure 2(a), the HBMEC monolayer cultured on Transwell inserts was placed on the 24-well plate. GM-CSF in 20 ng/ml was added into the lower chamber. The monocytic cell lines THP-132 and U93733 were used in in vitro experiments to study the CSF2RB effect. THP-1 was found to express significantly higher levels of CSF2RB than U937 (Figure 2(b)).
Figure 2.

CSF2RB expression is required for monocyte transendothelial migration. (a) Schematic diagram of in vitro BBB model. The HBMEC monolayer cultured on the Transwell insert was placed on a 24-well plate. GM-CSF (0, 20 ng/ml) was present in the lower chamber. U937 or THP-1 was added to the upper chamber. (b) CSF2RB expression in U937 and THP-1 cells was detected by RT-PCR. Two-tailed t-test (α = 0.05) was used to compare CSF2RB expression. (c) CSF2RB expression is associated with monocyte transendothelial migration. In this Transwell experiment, the lower chamber was filled with 20 ng/ml GM-CSF in serum-free medium, and 5 × 105 U937 or THP-1 cells were loaded in the upper chamber, which had been plated with an HBMEC monolayer. After a 4 h-incubation, the cells that had transmigrated to the lower chamber were counted. The results represent three independent experiments. Two-way ANOVA was performed to compare U937 and THP-1 transendothelial migration. Data are shown as means ± standard deviations (SD). ***p < 0.001 vs. the control, the experiment was independently performed three times (n = 3). (d) CSF2RB silencing decreased the number of THP-1 cells that transmigrated to the lower chamber. A total of 5 × 105 Lenti-CSF2RB- or Lenti-NS-infected THP-1 cells were loaded in the upper chamber, and the cells transmigrated to the lower chamber were counted after 4 h. The results represent three independent experiments. Two-tailed t-test (α = 0.05) was used to compare THP-1 transmigration. Data are shown as means ± SD. ***p < 0.001 vs. the control.
In accordance with the above-mentioned differential CSF2RB expression, THP-1 cells exhibited increased migration across the Transwell to the lower chamber (Figure 2(c)), indicating that CSF2RB affects transendothelial migration in monocytes. Furthermore, lentiviral vector-mediated CSF2RB gene silencing (Supplemental data 2(a) to (c)) significantly reduced the transendothelial migration ability of THP-1 cells (Figure 2(d)). These results indicated that CSF2RB is instrumental in monocyte migration through BBB.
GM-CSF triggers tight junction disassembly in a HBMEC monolayer
The restrictive nature of BBB is due to tight junctions between adjacent endothelial cells at the apical membrane, resulting in high TEER and low paracellular permeability.1 To investigate the mechanism underlying GM-CSF-induced transendothelial monocyte migration, HBMEC monolayer integrity was initially examined. GM-CSF significantly increased HRP flux and decreased TEER in a dose-dependent manner (Figure 3(a) and (b)). The high HRP flux is not caused by HBMECs death induced by chloroquine (Supplemental data 3(a)).
Figure 3.

The effect of GM-CSF on endothelial permeability. (a) Dose-dependent changes in HRP flux. One-way ANOVA was used for repeated measurements. (b) Dose-dependent changes in TEER. HBMECs were plated in 24-well fibronectin-coated Transwell insert plates (pore size: 0.4 µm) and grown for four days. Next, GM-CSF (0, 10, 20 ng/ml) was added to the HBMEC monolayer for 24 h. Dose-dependent changes in the TEER and HRP fluxes of the HBMEC were subsequently measured. One-way ANOVA was used for repeated measurements. * p < 0.05 vs. normal HBMEC, the experiment was independently performed three times (n = 3). (c) ZO-1 expression in GM-CSF-stimulated HBMECs. HBMECs were stimulated with GM-CSF (0, 20 ng/ml) for the indicated time, after which tight junction protein expression was examined. The results represent three independent experiments. Data are shown as means ± standard deviations. One-way ANOVA was used to compare ZO-1 expression. *p < 0.05 vs. the 48 h-GM-CSF-treated HBMECs. (d) Changes in the location of ZO-1 (green) in HBMECs and GM-CSF-stimulated HBMECs were visualized by immunofluorescence. DAPI was used to visualize cell nuclei (blue). The disassembly of ZO-1 (not continuous) was indicated by white arrow. Scale bar: 50 µm. (e) Claudin-5 expression in GM-CSF-stimulated HBMECs. (f) Occludin distribution in GM-CSF stimulated HBMECs. Western blotting detected a shift in endothelial occludin protein from the insoluble to soluble phase. One-way ANOVA was used for repeated measurements. * p < 0.05 vs. normal HBMEC, the experiment was independently performed three times.
Tight junctions mainly comprise three transmembrane proteins, claudins, occludin, and junction adhesion molecules, that associate with cytoplasmic accessory proteins (zonula occludens (ZO) -1, -2, -3, cingulin) and bind across or “glue” adjacent cell membranes.34–37 Changes in BBB permeability were associated with altered tight junction protein expression.10,38 GM-CSF reduced ZO-1 and claudin-5 expressions in HBMECs in a time-dependent manner (Figure 3(c) and (e)). Meanwhile, ZO-1 distribution became discontinuous, segmented, and punctate (Figure 3(d)) compared with untreated HBMECs.
Tight junction protein distribution between detergent-soluble and -insoluble cell fractions can be used to measure tight junction integrity.39 Furthermore, occludin distribution was detected in GM-CSF-treated HBMECs. Here, we observed an obvious shift in occludin distribution from the insoluble to soluble fraction (Figure 3(f)). However, the total occludin expression level remained unchanged (Supplemental data 3(b)); expression of the tight junction proteins claudin-3 (Supplemental data 3(c)) and VE-cadherin,40 the main adhesive proteins at adherens junctions,11 also remained unchanged (Supplemental data 3(d)), indicating that GM-CSF induces tight junction protein disassembly that is unrelated to adherens junction disruption and that increased permeability was not caused by endothelial cell growth retardation (Supplemental data 3(e)) or cytotoxicity of GM-CSF performed on HBMECs (Supplemental data 3(f)). GM-CSF treatment increased HBMEC monolayer permeability.
GM-CSF downregulates ZO-1 gene transcription
GM-CSF induced a significant decrease in ZO-1 protein expression (Figure 3(c) and (d)). To define the molecular mechanism underlying the GM-CSF-induced time-dependent decrease in ZO-1 expression, we performed real-time RT-PCR to examine the GM-CSF effect on ZO-1 gene transcription and observed a time-dependent reduction in ZO-1 mRNA levels (Figure 4(a)). To identify the GM-CSF-responsive region, the ZO-1 promoter expressing plasmid pGL3-ZO-1, containing a 2787-bp region of the ZO-1 promoter,41 was constructed. A subsequent luciferase reporter assay revealed that ZO-1 expression was most strongly inhibited by GM-CSF (Supplemental data 4). To map the GM-CSF-responsive region, different reporter constructs containing deletions in the ZO-1 promoter were constructed (Figure 4(b)). Following GM-CSF stimulation, luciferase activity decreased to 29% of that in non-GM-CSF-stimulated cells. Conversely, the activities of different constructs in which the proximal region from −2787 to −1988 of the ZO-1 promoter were deleted did not differ, regardless of the GM-CSF stimulation status. The construct containing both the region from −2787 to −1988 and −675 to 0 rescued ZO-1 expression (Figure 4(c)). Thus, the region of −2787 to −1988 and −675 to 0 are important to ZO-1 transcription. GM-CSF represses ZO-1 transcription within the proximal region from −2787 to −1988 of the ZO-1 promoter.
Figure 4.

Effect of GM-CSF on ZO-1 promoter activity and the GM-CSF-responsive region of the ZO-1 promoter. (a) GM-CSF affects ZO-1 transcription. GM-CSF was incubated with an HBMEC monolayer for the indicated time points, and mRNA expression levels were estimated by real-time RT-PCR. Values are shown as means ± standard deviations from three separate experiments. One-way ANOVA was used for repeated measurements. *p < 0.05 vs. normal HBMECs. (b) Schematic representation of different ZO-1 promoter deletion constructs. Numbers indicate the number of base pairs from the transcription start site. (c) Luciferase activities of the corresponding constructs in HBMECs. HBMECs were transfected with different ZO-1 promoter deletion mutants, and the luciferase reporter activity levels were detected 48 h after transfection. Results were normalized relative to Renilla-driven luciferase activity and expressed as means ± standard errors of the data from three separate experiments. **p < 0.01 vs. ZO-1 promoter luciferase activity in HBMECs without GM-CSF stimulation.
GM-CSF downregulates claudin-5 expression by promoting the ubiquitination pathway
Claudin-5 is expressed in nearly all blood vascular endothelial cells in the brain13 and serves as another characteristic cell adhesion molecule of brain endothelial cell tight junctions.42,43 Claudin-5 protein expression was reduced in GM-CSF-stimulated HBMECs (Figure 3(c)). However, this decrease in protein expression was not accompanied by a decrease in claudin-5 mRNA expression (Supplemental data 5), indicating a post-translational modification mediated by GM-CSF that subsequently downregulated claudin-5 protein expression.
Most proteins undergo some post-translational modification; the ubiquitin proteasome pathway (UPP) is an important protein catabolic mechanism. MG132 (carbobenzoxy-Leu-Leu-leucinal), a peptide aldehyde, effectively blocks the proteolytic activity of the 26 S proteasome complex.44 To elucidate GM-CSF stimulation mechanism leading to decreased claudin-5 expression, we evaluated the MG132 effects on GM-CSF-treated HBMECs and found that MG132 inhibited the decrease in claudin-5 protein expression (Figure 5(a)).
Figure 5.

Effect of GM-CSF on claudin-5 ubiquitination. (a) MG132 inhibits the GM-CSF-induced decrease in claudin-5 protein expression. HBMEC monolayers were incubated with MG132 and GM-CSF for 6, 12, or 24 h. (b) GM-CSF increases the claudin-5 ubiquitination level. A myc-taged-claudin-5 plasmid was transfected into HEK293T cells. GM-CSF or GM-CSF/MG132 was added to the supernatant 24 h after transfection. Immunoprecipitation with claudin-5 antibody was performed, and ubiquitin and myc expression were detected via western blotting (arrows). (c) The claudin-5 polyubiquitination levels in myc-claudin-5 and flag-ubiquitin co-transfected HEK293T cells (arrows).
To explore the ubiquitination involvement in GM-CSF-induced claudin-5 degradation, myc-tagged-claudin-5-transfected 293T cell lysates were subjected to immunoprecipitation. The results showed that GM-CSF increased the claudin-5 ubiquitination level, a process inhibited by MG132 (Figure 5(b)). Flag-tagged ubiquitin and myc-tagged claudin-5 were cotransfected into 293T cells followed by claudin-5 antibody immunoprecipitation to further clarify the claudin-5 ubiquitination effects. Western blotting with antibodies against Flag and myc indicated that claudin-5 was polyubiquitinated after GM-CSF stimulation and that MG132 attenuated this polyubiquitination (Figure 5(c)).
Intracerebral GM-CSF blockade abolished the increased in monocyte infiltration in the APP/PS1 mouse brain
Here, the APP/PS1 transgenic mouse was used as an AD model. Whole brains from APP mice exhibit a marked increase in the CD11b+CD45hi cell percentage compared with wild-type brains.4 CD11b and CD45 were used to determine the number of accumulated microglia in the brain4,18,45; in contrast to peripheral blood mononuclear cells, which can be identified via flow cytometry as a CD11b+CD45hi cell population, resident microglia comprise a distinct population of CD11b+CD45lo cells.4,18,45 To further confirm the intracerebral GM-CSF role on monocyte transmigration through BBB, a mouse GM-CSF neutralization antibody was used to block the intracerebral GM-CSF effect on transendothelial monocyte migration in APP mice. The previously observed increase in CD11b+CD45hi cells was markedly attenuated when APP mice were injected with anti-GM-CSF antibody (Figure 6(a) and (b)), indicating that GM-CSF is instrumental in the bone marrow-derived microglia accumulation in the brains of APP mice.
Figure 6.

Flow cytometric analysis of microglial accumulation in APP mice after intracerebral GM-CSF blockade. (a) Flow cytometry dot plots from an analysis of CD11b- and CD45-stained brain cells. (b) Flow cytometric quantitation of the percent of total CD11b+CD45+ brain cells (n = 4 mice for isotypic IgG group, 3 mice for anti-GM-CSF group). Two-tailed t-test was performed to compare transmigrated CD11b+CD45+ brain cells. **p < 0.01 vs. isotypic IgG intracerebral injected mouse brains.
Discussion
Microglia comprises the principal immune cell population in the brain. In AD, these brain mononuclear phagocytes are recruited from the blood and accumulate in SP. Accumulated microglia may play a protective role in the early stages of AD by promoting Aβ clearance.18 However, the mechanisms by which these cells contributed to monocyte transmigration through BBB remain unknown. A better understanding of these mechanisms will assist the development of successful cellular therapies for AD. Here, we provided the first evidence that CSF2RB on peripheral monocytes and intracerebral GM-CSF promote monocyte transmigration through the tight junctions of AD patients’ BBB.
CSF2RB, a component of receptors for GM-CSF, IL-3, and IL-5,46,47 is encoded from the linkage region 22q12.3 and expressed in most cells.48 CSF2RB expression is a common risk factor for schizophrenia and bipolar disorder.48,49 Additionally, a genetic defect in CSF2RB gives rise to pulmonary alveolar proteinosis in an autosomal recessive manner.50 However, the CSF2RB function in disease pathology has been rarely reported. We found that CSF2RB was highly expressed on peripheral monocytes in AD patients. Thus, GM-CSF bound with its receptor, CSF2RB, to promote monocyte transmigration through the tight junctions of BBB. Conversely, CSF2RA expression did not differ. The results indicated that CSF2RB, but not CSF2RA, might be used as an AD biomarker. Therefore, it is necessary to explore the mechanism underlying CSF2RB involvement in monocyte transmigration through BBB.
GM-CSF promotes inflammation in various infectious and inflammatory diseases.51 This cytokine is produced by numerous different cell types in response to cytokine or inflammatory stimuli and is involved in peripheral cell migration.52,53 Shang and Issekutz52 reported that increased GM-CSF production promoted monocyte accumulation by not only inducing monocytosis but also enhancing migration. They stimulated monocytes with GM-CSF immediately before adding the cells above human umbilical vein endothelial cells in an upper Transwell chamber, which resembled the vascular lumen. Cytokine-stimulated endothelial cells can produce sufficient GM-CSF quantities to influence the phenotypic neutrophil characteristics in an adhesion-dependent manner, leading to neutrophil transmigration.14 Endothelial cells secrete GM-CSF primarily from the luminal rather than the abluminal side.54 These studies indicated that GM-CSF could be secreted into the blood by endothelial cells; thus, GM-CSF would directly encounter its target molecules on blood cells. Although it is reported that GM-CSF levels were undetectable in AD or controls,55 recent data suggest the presence of high GM-CSF levels in the brain parenchyma and CSF of AD patients.19 Here, a mouse GM-CSF antibody was injected into the lateral ventricle to block intracerebral GM-CSF. We observed that this antibody abolished the increased monocyte infiltration into the brains of APP/PS1 mice, indicating that intracerebral GM-CSF still promotes monocyte transmigration across BBB. It is important to illuminate the molecular mechanism by which GM-CSF promotes transendothelial monocyte migration.
BBB properties are primarily determined by tight junctions between brain microvascular endothelial cells, which form a barrier not only against the passage of ions and molecules2 but also T lymphocytes5,6 and cancer cells.7,56 Generally, the passage of cells is characterized by a lack of fully continuous tight junctions between constituent cells. Tight junction disruption is generally induced by the re-distribution of tight junction proteins such as ZO-1, claudin-5, and occludin.6,56–58 However, the GM-CSF effect on BBB permeability has remained controversial. GM-CSF mediates the lipopolysaccharide-enhanced transcellular transport of HIV-1 across BBB.59 Another study reported enhanced BBB permeability in mice treated with a GM-CSF-expressing rabies virus.60 GM-CSF in blood enhances monocyte migration across in vitro BBB model.61 Here, GM-CSF downregulated ZO-1 and claudin-5 expressions and shifted the occludin distribution from the triton-insoluble to the triton-soluble fraction. Additionally, we were the first to observe that intracerebral GM-CSF directly increased brain endothelial permeability and to demonstrate that GM-CSF regulated the tight junction protein expression. These results suggest the value of further investigations of the molecular mechanism underlying GM-CSF-regulated tight junction protein expression.
The ZO-1 protein is the best characterized member of a large family of membrane-associated scaffolding and signaling molecules known as the membrane-associated guanylate kinase homologues (MAGUKs);41 these proteins are highly organized within a complicated network of interconnecting cytoskeletal and tight junction transmembrane proteins41,62 such as occludin, claudins, and the CTX (Ig) superfamily.41,61,63 The regulation of downstream molecules by ZO-1 has been well established. However, because ZO-1 is a key regulator of tight junction assembly, it is important to explore the largely unknown molecular mechanism underlying ZO-1 expression. JunD suppresses transcription through CREB binding sites within the proximal region of the ZO-1 promoter in the context of intestinal epithelial barrier function.41 Here, we observed that GM-CSF induced a time-dependent reduction in ZO-1 mRNA levels and a corresponding significant decrease in ZO-1 protein expression, indicating that GM-CSF suppresses ZO-1 transcription in the −2787 to −1988-bp region of the ZO-1 promoter, leading to an increase in epithelial permeability. Our study thus provides new insights into the mechanisms underlying the regulation of ZO-1 expression. Additionally, the −2787 to −1988-bp region of the ZO-1 promoter contains AP-1 and CREB binding sites. It is important to investigate whether GM-CSF suppresses ZO-1 expression in HBMECs by interacting with AP-1 or CREB.
Claudin-5 is another characteristic BBB component that is nearly expressed in all brain blood vessel endothelial cells13 and is involved in cerebral barrier formation.43,64,65 It is therefore important to explore the mechanism underlying the regulation of claudin-5 expression, occurring at the transcriptional,66 post-transcriptional,67 and post-translational levels.15 Here, GM-CSF does not regulate claudin-5 expression at the transcriptional or post-transcriptional levels. Claudin-5 was previously found to have a short half-life and to mainly accumulate in the intracellular compartment following proteasome inhibition, which suggested the ubiquitin/proteasome pathway involvement in its expression.68 Here, the GM-CSF-mediated downregulation of claudin-5 relied on the ubiquitin/proteasome pathway. These data are beneficial to our understanding of the regulation of claudin-5 expression. Meanwhile, we found that although claudin-5 protein expression was eventually reduced in GM-CSF-stimulated HBMECs, claudin-5 was transiently increased at the early 6 h-time point after GM-CSF treatment. We think the initial transient increase of claudin-5 in GM-CSF-stimulated HBMECs could be the initial compensatory response of claudin-5 attempting to stabilize the tight junctions.
Whether BBB disruption occurs in AD or in AD mouse models is highly controversial. Our results suggest that high levels of GM-CSF in the brain parenchyma and CSF of AD patients induced BBB opening, which is accompanied the infiltration of CSF2RB-expressing peripheral blood monocytes across BBB into the brain (Figure 7). Thus, our findings will help to understand the mechanism of monocyte infiltration in AD pathogenesis.
Figure 7.

Summary model of GM-CSF-mediated transmigration of monocytes through BBB in the AD-affected brain. Patients with AD exhibit high levels of GM-CSF in the brain parenchyma and CSF. Peripheral blood monocytes, which are CD11b+CD45lo, express the GM-CSF receptor CSF2RB. GM-CSF induces the disassembly of tight junction proteins and helps to open BBB. Consequently, peripheral blood monocytes transmigrate across BBB and infiltrate the brain, a process that is promoted by CSF2RB expression. The resident microglia exhibits a CD11b+CD45lo phenotype and can be distinguished from CD11b+CD45hi infiltrating monocytes.
Supplementary Material
Acknowledgements
The authors are grateful to Drs. Monique Stins and Kwang Sik Kim (Department of Pediatrics, John Hopkins University School of Medicine) for providing HBMECs.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by grants from the National Natural Science Foundation of China (31171291, 31571057) and Research Fund for the Doctoral Program of Higher Education of China (20132104110019).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Contributions: DSS, WDZ, BL, and YHC conceived and designed the experiments and analyzed data. DSS and YMY performed the experiments. JSJ, HZ, YBD, and KZ performed the APP/PS1 mouse experiment. D.S.S. and YHC wrote the manuscript. WGF provided technical support.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol 2008; 6: 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonzalez-Mariscal L, Betanzos A, Nava P, et al. Tight junction proteins. Prog Biophys Mol Biol 2003; 81: 1–44. [DOI] [PubMed] [Google Scholar]
- 3.Floris S, Blezer EL, Schreibelt G, et al. Blood-brain barrier permeability and monocyte infiltration in experimental allergic encephalomyelitis: a quantitative MRI study. Brain 2004; 127(Pt 3): 616–627. [DOI] [PubMed] [Google Scholar]
- 4.Zhang K, Tian L, Liu L, et al. CXCL1 contributes to beta-amyloid-induced transendothelial migration of monocytes in Alzheimer's disease. PLoS One 2013; 8: e72744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Man SM, Ma YR, Shang DS, et al. Peripheral T cells overexpress MIP-1alpha to enhance its transendothelial migration in Alzheimer's disease. Neurobiol Aging 2007; 28: 485–496. [DOI] [PubMed] [Google Scholar]
- 6.Li M, Shang DS, Zhao WD, et al. Amyloid beta interaction with receptor for advanced glycation end products up-regulates brain endothelial CCR5 expression and promotes T cells crossing the blood-brain barrier. J Immunol 2009; 182: 5778–5788. [DOI] [PubMed] [Google Scholar]
- 7.Li B, Zhao WD, Tan ZM, et al. Involvement of Rho/ROCK signalling in small cell lung cancer migration through human brain microvascular endothelial cells. FEBS Lett 2006; 580: 4252–4260. [DOI] [PubMed] [Google Scholar]
- 8.Lin MN, Shang DS, Sun W, et al. Involvement of PI3K and ROCK signaling pathways in migration of bone marrow-derived mesenchymal stem cells through human brain microvascular endothelial cell monolayers. Brain Res 2013; 1513: 1–8. [DOI] [PubMed] [Google Scholar]
- 9.Murphy MP, LeVine H., 3rd Alzheimer's disease and the amyloid-beta peptide. J Alzheimers Dis 2010; 19: 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang YM, Shang DS, Zhao WD, et al. Microglial TNF-alpha-dependent elevation of MHC class I expression on brain endothelium induced by amyloid-beta promotes T cell transendothelial migration. Neurochem Res 2013; 38: 2295–2304. [DOI] [PubMed] [Google Scholar]
- 11.Liu YJ, Guo DW, Tian L, et al. Peripheral T cells derived from Alzheimer's disease patients overexpress CXCR2 contributing to its transendothelial migration, which is microglial TNF-alpha-dependent. Neurobiol Aging 2010; 31: 175–188. [DOI] [PubMed] [Google Scholar]
- 12.Theriault P, ElAli A, Rivest S. The dynamics of monocytes and microglia in Alzheimer's disease. Alzheimers Res Ther 2015; 7: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malm TM, Koistinaho M, Pärepalo M, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis 2005; 18: 134–142. [DOI] [PubMed] [Google Scholar]
- 14.Rock RB, Gekker G, Hu S, et al. Role of microglia in central nervous system infections. Clin Microbiol Rev 2004; 17: 942–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takata K, Kitamura Y, Yanagisawa D, et al. Microglial transplantation increases amyloid-beta clearance in Alzheimer model rats. FEBS Lett 2007; 581: 475–478. [DOI] [PubMed] [Google Scholar]
- 16.Simard AR, Rivest S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J 2004; 18: 998–1000. [DOI] [PubMed] [Google Scholar]
- 17.Naert G, Rivest S. A deficiency in CCR2+ monocytes: the hidden side of Alzheimer's disease. J Mol Cell Biol 2013; 5: 284–293. [DOI] [PubMed] [Google Scholar]
- 18.El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med 2007; 13: 432–438. [DOI] [PubMed] [Google Scholar]
- 19.Tarkowski E, Wallin A, Regland B, Blennow K, Tarkowski A. Local and systemic GM-CSF increase in Alzheimer's disease and vascular dementia. Acta Neurol Scand 2001; 103(3): 166–174. [DOI] [PubMed] [Google Scholar]
- 20.Grabstein KH, Urdal DL, Tushinski RJ, Mochizuki DY, Price VL, Cantrell MA, et al. Induction of macrophage tumoricidal activity by granulocyte-macrophage colony-stimulating factor. Science 1986; 232(4749): 506–508. [DOI] [PubMed] [Google Scholar]
- 21.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med 1994; 179: 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gearing DP, King JA, Gough NM, et al. Expression cloning of a receptor for human granulocyte-macrophage colony-stimulating factor. EMBO J 1989; 8: 3667–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashida K, Kitamura T, Gorman DM, et al. Molecular cloning of a second subunit of the receptor for human granulocyte-macrophage colony-stimulating factor (GM-CSF): reconstitution of a high-affinity GM-CSF receptor. Proc Natl Acad Sci U S A 1990; 87: 9655–9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hercus TR, Thomas D, Guthridge MA, et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood 2009; 114: 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stins MF, Gilles F, Kim KS. Selective expression of adhesion molecules on human brain microvascular endothelial cells. J Neuroimmunol 1997; 76: 81–90. [DOI] [PubMed] [Google Scholar]
- 26.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984; 34: 939–944. [DOI] [PubMed] [Google Scholar]
- 27.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12: 189–198. [DOI] [PubMed] [Google Scholar]
- 28.Walker DG, Lue LF, Beach TG. Gene expression profiling of amyloid beta peptide-stimulated human post-mortem brain microglia. Neurobiol Aging 2001; 22: 957–966. [DOI] [PubMed] [Google Scholar]
- 29.Manczak M, Mao P, Nakamura K, et al. Neutralization of granulocyte macrophage colony-stimulating factor decreases amyloid beta 1-42 and suppresses microglial activity in a transgenic mouse model of Alzheimer's disease. Hum Mol Genet 2009; 18: 3876–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu W, Zhao WD, Yan JC, et al. Involvement of Src tyrosine kinase in Escherichia coli invasion of human brain microvascular endothelial cells. FEBS Lett 2010; 584: 27–32. [DOI] [PubMed] [Google Scholar]
- 31.Niu L, Heaney ML, Vera JC, et al. High-affinity binding to the GM-CSF receptor requires intact N-glycosylation sites in the extracellular domain of the beta subunit. Blood 2000; 95: 3357–3362. [PubMed] [Google Scholar]
- 32.Fleit HB, Kobasiuk CD. The human monocyte-like cell line THP-1 expresses Fc gamma RI and Fc gamma RII. J Leukoc Biol 1991; 49: 556–565. [DOI] [PubMed] [Google Scholar]
- 33.Moscicki RA, Amento EP, Krane SM, et al. Modulation of surface antigens of a human monocyte cell line, U937, during incubation with T lymphocyte-conditioned medium: detection of T4 antigen and its presence on normal blood monocytes. J Immunol 1983; 131: 743–748. [PubMed] [Google Scholar]
- 34.Park MW, Kim CH, Cheong JH, et al. Occludin expression in brain tumors and its relevance to peritumoral edema and survival. Cancer Res Treat 2006; 38: 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller F, Afonso PV, Gessain A, et al. Blood-brain barrier and retroviral infections. Virulence 2012; 3: 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 1993; 123(6 Pt 2): 1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furuse M, Fujita K, Hiiragi T, et al. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 1998; 141: 1539–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang YM, Zhou Y, Qiu LB, et al. Altered expression of matrix metalloproteinases and tight junction proteins in rats following PEMF-induced BBB permeability change. Biomed Environ Sci 2012; 25: 197–202. [DOI] [PubMed] [Google Scholar]
- 39.Yoo J, Nichols A, Mammen J, et al. Bryostatin-1 enhances barrier function in T84 epithelia through PKC-dependent regulation of tight junction proteins. Am J Physiol Cell Physiol 2003; 285: C300–C309. [DOI] [PubMed] [Google Scholar]
- 40.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 2008; 121(Pt 13): 2115–2122. [DOI] [PubMed] [Google Scholar]
- 41.Chen J, Xiao L, Rao JN, et al. JunD represses transcription and translation of the tight junction protein zona occludens-1 modulating intestinal epithelial barrier function. Mol Biol Cell 2008; 19: 3701–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ueno M. Molecular anatomy of the brain endothelial barrier: an overview of the distributional features. Curr Med Chem 2007; 14: 1199–1206. [DOI] [PubMed] [Google Scholar]
- 43.Nitta T, Hata M, Gotoh S, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 2003; 161: 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han YH, Moon HJ, You BR, et al. The effect of MG132, a proteasome inhibitor on HeLa cells in relation to cell growth, reactive oxygen species and GSH. Oncol Rep 2009; 22: 215–221. [PubMed] [Google Scholar]
- 45.Sedgwick JD, Schwender S, Imrich H, et al. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A 1991; 88: 7438–7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woodcock JM, Zacharakis B, Plaetinck G, et al. Three residues in the common beta chain of the human GM-CSF, IL-3 and IL-5 receptors are essential for GM-CSF and IL-5 but not IL-3 high affinity binding and interact with Glu21 of GM-CSF. EMBO J 1994; 13: 5176–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki T, Sakagami T, Rubin BK, Nogee LM, Wood RE, Zimmerman SL, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med 2008; 205(12): 2703–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen P, Huang K, Zhou G, et al. Common SNPs in CSF2RB are associated with major depression and schizophrenia in the Chinese Han population. World J Biol Psychiatry 2011; 12: 233–238. [DOI] [PubMed] [Google Scholar]
- 49.Moskvina V, Craddock N, Holmans P, et al. Gene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psychiatry 2009; 14: 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka T, Motoi N, Tsuchihashi Y, et al. Adult-onset hereditary pulmonary alveolar proteinosis caused by a single-base deletion in CSF2RB. J Med Genet 2011; 48: 205–209. [DOI] [PubMed] [Google Scholar]
- 51.Shim DS, Schilter HC, Knott ML, et al. Protection against Nippostrongylus brasiliensis infection in mice is independent of GM-CSF. Immunol Cell Biol 2012; 90: 553–558. [DOI] [PubMed] [Google Scholar]
- 52.Shang XZ, Issekutz AC. Enhancement of monocyte transendothelial migration by granulocyte-macrophage colony-stimulating factor: requirement for chemoattractant and CD11a/CD18 mechanisms. Eur J Immunol 1999; 29: 3571–3582. [DOI] [PubMed] [Google Scholar]
- 53.Elbjeirami WM, Donnachie EM, Burns AR, et al. Endothelium-derived GM-CSF influences expression of oncostatin M. Am J Physiol Cell Physiol 2011; 301: C947–C953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verma S, Nakaoke R, Dohgu S, et al. Release of cytokines by brain endothelial cells: a polarized response to lipopolysaccharide. Brain Behav Immun 2006; 20: 449–455. [DOI] [PubMed] [Google Scholar]
- 55.Tarkowski E, Andreasen N, Tarkowski A, et al. Intrathecal inflammation precedes development of Alzheimer's disease. J Neurol Neurosurg Psychiatry 2003; 74: 1200–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B, Wang C, Zhang Y, et al. Elevated PLGF contributes to small-cell lung cancer brain metastasis. Oncogene 2013; 32: 2952–2962. [DOI] [PubMed] [Google Scholar]
- 57.Fischer S, Wiesnet M, Renz D, et al. H2O2 induces paracellular permeability of porcine brain-derived microvascular endothelial cells by activation of the p44/42 MAP kinase pathway. Eur J Cell Biol 2005; 84: 687–697. [DOI] [PubMed] [Google Scholar]
- 58.Fischer S, Wobben M, Marti HH, et al. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res 2002; 63: 70–80. [DOI] [PubMed] [Google Scholar]
- 59.Dohgu S, Fleegal-DeMotta MA, Banks WA. Lipopolysaccharide-enhanced transcellular transport of HIV-1 across the blood-brain barrier is mediated by luminal microvessel IL-6 and GM-CSF. J Neuroinflammation 2011; 8: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H, Zhang G, Wen Y, et al. Intracerebral administration of recombinant rabies virus expressing GM-CSF prevents the development of rabies after infection with street virus. PLoS One 2011; 6: e25414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vogel DY, Kooij G, Heijnen PD, et al. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur J Immunol 2015; 45: 1808–1819. [DOI] [PubMed] [Google Scholar]
- 62.Fanning AS, Little BP, Rahner C, et al. The unique-5 and -6 motifs of ZO-1 regulate tight junction strand localization and scaffolding properties. Mol Biol Cell 2007; 18: 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol 2004; 286: C1213–C1228. [DOI] [PubMed] [Google Scholar]
- 64.Abdelilah-Seyfried S. Claudin-5a in developing zebrafish brain barriers: another brick in the wall. Bioessays 2010; 32: 768–776. [DOI] [PubMed] [Google Scholar]
- 65.Honda M, Nakagawa S, Hayashi K, et al. Adrenomedullin improves the blood-brain barrier function through the expression of claudin-5. Cell Mol Neurobiol 2006; 26: 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuan L, Le Bras A, Sacharidou A, et al. ETS-related gene (ERG) controls endothelial cell permeability via transcriptional regulation of the claudin 5 (CLDN5) gene. J Biol Chem 2012; 287: 6582–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koto T, Takubo K, Ishida S, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol 2007; 170: 1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mandel I, Paperna T, Volkowich A, et al. The ubiquitin-proteasome pathway regulates claudin 5 degradation. J Cell Biochem 2012; 113: 2415–2423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.