

Abstract
Cerebral microdialysis is a widely used clinical tool for monitoring extracellular concentrations of selected metabolites after brain injury and to guide neurocritical care. Extracellular glucose levels and lactate/pyruvate ratios have high diagnostic value because they can detect hypoglycemia and deficits in oxidative metabolism, respectively. In addition, patterns of metabolite concentrations can distinguish between ischemia and mitochondrial dysfunction, and are helpful to choose and evaluate therapy. Increased intracranial pressure can be life-threatening after brain injury, and hypertonic solutions are commonly used for pressure reduction. Recent reports have advocated use of hypertonic sodium lactate, based on claims that it is glucose sparing and provides an oxidative fuel for injured brain. However, changes in extracellular concentrations in microdialysate are not evidence that a rise in extracellular glucose level is beneficial or that lactate is metabolized and improves neuroenergetics. The increase in glucose concentration may reflect inhibition of glycolysis, glycogenolysis, and pentose phosphate shunt pathway fluxes by lactate flooding in patients with mitochondrial dysfunction. In such cases, lactate will not be metabolizable and lactate flooding may be harmful. More rigorous approaches are required to evaluate metabolic and physiological effects of administration of hypertonic sodium lactate to brain-injured patients.
Keywords: Cerebral microdialysis, brain metabolism, glucose, lactate supplementation, traumatic brain injury
Introduction
The molecular mechanisms underlying the lesions in traumatic brain injuries (TBI), subarachnoidal hemorrhage, and stroke are incompletely known. Therapy is consequently focused on efforts to support cerebral blood flow and energy metabolism based on the information obtained from multi-modality monitoring. Techniques are now available for evaluation of complex cerebral biochemical processes during critical care. The correct interpretation of the data obtained necessitates knowledge of the technical and methodological limitations of the techniques used as well as advanced knowledge of cerebral energy metabolism. The biochemical principles discussed below have been explored most extensively in patients with TBI.
Brain metabolic activities are difficult to assess comprehensively in normal human brain, and heterogeneity of patients with severe brain injury plus the temporal evolution of brain damage and recovery increase the complexity of the analysis of brain metabolism after trauma. Cerebral microdialysis is a widely used clinical tool to monitor extracellular concentrations (ECs) of selected brain metabolites after TBI, and changes in metabolite levels and lactate/pyruvate (LP) ratios provide valuable information to help evaluate the status of a patient and guide clinical care.1–3 Routine bedside monitoring relies on standard-of-care procedures (e.g. monitoring of intracranial pressure, cerebral perfusion pressure, tissue oxygen level, and cerebrovascular reactivity) along with microdialysis assays to evaluate brain status and metabolism. A limitation is that microdialysate concentration changes are not equivalent to metabolic rates or energetics, and interchangable use of these terms can lead to mis-interpretation of concentration changes, incorrect conclusions, and inadequate evaluation of hypertonic lactate supplementation as a new treatment paradigm for traumatic brain injury.
The objective of this article is to broaden the discussion of roles of lactate in energy metabolism after brain injury and emphasize the importance of rigorous analysis, conservative interpretation, and consideration of unanticipated consequences of lactate flooding of the injured brain. We describe the use of cerebral microdialysis, contrast changes in metabolite levels with metabolic rates under different conditions, evaluate effects of lactate supplementation, and provide alternative perspectives of lactate flooding. We conclude that concentration changes in the absence of metabolic assays are not sufficient to evaluate effects of lactate supplementation on brain energy metabolism after TBI.
Cerebral microdialysis in neurocritical care – A brief survey
The microdialysis technique offers the unique possibility of obtaining information regarding the chemical composition of cerebral interstitial fluid during neurocritical care. The biochemical analyses are routinely performed and displayed bedside utilizing enzymatic techniques (ISCUSflex, MDialysis, Stockholm, Sweden). The complex relations between the biochemical variables can be simplified into figures illustrating the connection between energy metabolism and glycerol indicating degradation of cellular membranes (Figure 1). The different patterns obtained during cerebral ischemia and mitochondrial dysfunction (Figure 2) may be used for choosing and evaluating the therapy given.4–6 These simplifications of the biochemical processes can be used for clinical interpretation and decision-making if the theoretical and practical possibilities and limitations of the microdialysis technique are recognized.
Figure 1.

Simplified diagram of cerebral intermediary metabolism with a focus on the glycolytic pathway and its relation to glycerol and glycerophospholipids and to the citric acid cycle. Glucose, lactate, and pyruvate are involved in major pathways of brain energy metabolism, and changes in their concentrations and lactate/pyruvate (Lac/Pyr) ratio can reflect disturbances in transport and metabolism, whereas increases in glutamate and glycerol levels reflect excitatory neurotoxicity and membrane damage, respectively. P: phosphate; α-KG: α-ketoglutarate; TCA: tricarboxylic acid. Underlined metabolites are analyzed during routine cerebral microdialysis assays and displayed at the bedside. Reference levels (means ± SD) of the various metabolites for normal human brain were obtained from Reinstrup et al.3 Modified from Nordström, Childs Nerv Syst 2010; 26:465–472, Copyright © 2009, Springer-Verlag, with permission.
Figure 2.

Schematic illustration of brain tissue oxygenation (PbtO2) and changes in the levels of lactate (Lac), pyruvate (Pyr), and the lactate/pyruvate ratio in two conditions: ischemia and mitochondrial dysfunction. Note the differences in tissue oxygenation and extracellular pyruvate levels during ischemia and mitochondrial dysfunction. Modified from Nordström et al. J Rehabil Med 2013; 45: 710–717. Copyright © 2013, The Authors, with permission.
The microdialysis technique
The microdialysis technique reflects the chemical composition of a very narrow zone surrounding the semipermeable membrane. In neurocritical care, focal cerebral lesions are common and energy metabolism is usually very different in various parts of the brain.7 Accordingly, the biochemical data obtained reveal local tissue damage/outcome but do not usually predict the clinical outcome of the patient. A correlation to clinical outcome might be expected provided the biochemical information is obtained from a focal lesion engaging a large part of the brain or if it represents global cerebral energy metabolism. Further, the levels obtained for the different chemical variables do not usually represent their true concentrations in the extracellular fluid (ECF). The relative recovery (dialysate concentration/tissue concentration) primarily depends on the permeability and the length of the dialysis membrane and the perfusion rate. These variables are standardized in clinical microdialysis and the perfusion rate is usually set at 0.3 µl/min. In addition, diffusion in the surrounding tissue affects recovery. The diffusion rate varies with variations in volume and tortuosity of the intestinal space. For routine clinical microdialysis, these variations are of little importance. However, if the microdialysis technique is utilized for scientific, quantitative measurements, it is necessary to measure continuously relative recovery.8
Biochemical analysis and interpretation
An analytical validation of the enzymatic techniques used for routine bedside analysis of glucose, lactate, and pyruvate (ISCUSflex) has recently been presented.9 Linearity ranges were 0.1–25 mmol/L, 0.2–12 mmol/L, and 19–1500 µmol/L for glucose, lactate, and pyruvate, respectively. For critical threshold, intra- and inter-assay coefficients of variation (CV) were, respectively, 3.1 and 4.5% for glucose, 3.5 and 4% for lactate, and 3.3 and 4.3% for pyruvate and inter-assay CV for LP ratio was 5.9%. The data prove that these routine analyses have the accuracy and precision required for clinical application in neurointensive care. However, extrapolation to the cellular/extracellular milieu is still indirect, and the methodology is still subject to artifacts. As a result any conflicts with previously accepted biological principles need to be carefully evaluated. For example, the unexpected observation that lactate is transported into the brain against a concentration gradient to be utilized as a source of energy in patients with brain trauma10,11 is probably erroneous, as the data are fully explained by the fact that inadequate biochemical analytical techniques were used.12
When cerebral microdialysis is used in clinical routine, it is necessary to compare with normal reference values obtained from normal human brain under physiological conditions. In accordance with clinical praxis, these data should be presented as mean normal reference value ± 2SD, with values outside this range identified as abnormal. For obvious reasons, this information has been difficult to obtain. The limited data presented for normal human brain so far (Figure 1) are probably correct and correspond to data obtained from experimental studies utilizing the same microdialysis and analytical techniques in large animals.3
Bedside evaluation of cerebral energy metabolism
Due to its high energy demand, the brain is completely dependent on aerobic energy metabolism: a continuous, sufficient supply of oxygen and substrate, as well as functioning mitochondria. Due to the shift in cytoplasmic redox state, insufficient oxidative metabolism is immediately reflected in an increase of the LP ratio. Consequently, under clinical conditions, an increase of the LP ratio is considered as the most sensitive indicator of jeopardized cerebral energy metabolism.13 During neurocritical care, an increase of the LP ratio is usually caused by insufficient tissue oxygenation (e.g. arterial hypoxia, cerebral ischemia) and/or mitochondrial dysfunction (Figure 2). The two conditions can be separated at the bedside by simultaneous monitoring of the LP ratio and brain tissue oxygenation (PbtO2). In ischemia, the increase of the LP ratio occurs simultaneously with a pronounced decrease in PbtO2, while in mitochondrial dysfunction, the increase in LP ratio occurs at a normal or increased PbtO2.5 If under clinical conditions microdialysis is used alone, a separation between the two conditions may still be obtained (Figure 2). In cerebral ischemia, the increase in LP ratio occurs at a pronounced decrease in pyruvate level, while in mitochondrial dysfunction, the increase in LP ratio is observed at a normal or increased concentration of pyruvate.5,14
An increase in LP ratio is always caused by insufficient oxidative metabolism. Decreased delivery of substrate does not, in itself, cause an increase in LP ratio. Even when blood glucose is reduced to such a low level that cerebral energy metabolism collapses, LP ratio remains normal.15 Accordingly, in a clinical situation of increased cerebral LP ratio at a low cerebral glucose concentration, the metabolic pattern is caused by insufficient oxidative metabolism. Efforts to improve cerebral energy state by delivering an alternative oxidative substrate (e.g. lactate) in this situation are futile and erroneous.16,17
Summary
Cerebral microdialysis enables real-time sampling of the concentrations of selected metabolites in ECF, and changes in metabolite levels and LP ratios provide clinicians with an indirect assessment of glucose supply-demand relationships, cytoplasmic redox state, mitochondrial function, excitotoxicity, and cellular damage (Figures 1 and 2). Most neurocritical clinicians recognize the limitations of bedside analysis of metabolite levels, but there has been a recent trend towards mis-interpretation of concentration changes in terms of metabolism.
Cerebral metabolite levels, metabolism, and energetics
Key concepts
ECF corresponds to ∼20% of the brain volume in gray matter of cerebral cortex and subcortical white matter,18,19 and concentrations of extracellular metabolites are determined by their fluxes across the blood–brain barrier (BBB) and plasma membranes of brain cells (Figure 3). These fluxes are determined by various factors, including concentration gradients, kinetic properties of transporters, and cellular rates of utilization of glucose (CMRglc), oxygen (CMRO2), and other substrates that vary with condition, blood flow rate, and fuel and oxygen availability. Metabolism involves conversion of one compound into another (Figure 3) via a series of reactions described as a pathway. Complete oxidative metabolism of glucose consumes 6O2, so the theoretical maximal ratio of CMRO2/CMRglc (i.e. the oxygen–glucose index, OGI) is 6.0. A fall in OGI below the normal range of ∼5.4–6 in the presence of normal oxygen levels is called aerobic glycolysis, i.e. upregulation of non-oxidative metabolism.20 Lactate metabolism is taken into account with the oxygen–carbohydrate index (OCI = CMRO2/(CMRglc + 0.5CMRlac), where 0.5CMRlac represents CMRglc equivalents (1 glucose = 2 lactate). Energetics or neuroenergetics involves generation and utilization of ATP for energy-dependent processes (Figure 3). Most ATP is derived from respiration even during aerobic glycolysis,21 and ATP demand governs metabolic rate but metabolic rate may or may not alter metabolite concentrations.
Figure 3.

Sampling of brain extracellular metabolite levels versus assays of metabolism. Glucose is taken up and metabolized by neurons and astrocytes via the glycolytic pathway (glucose to pyruvate), pentose phosphate shunt pathway (PPP), and tricarboxylic acid (TCA) cycle. Glutamine is synthesized in astrocytes via the pyruvate carboxylase reaction to generate a “new” molecule of oxaloacetate that condenses with acetyl CoA from a second pyruvate molecule to sequentially form α-ketoglutarate, glutamate, and glutamine. The glutamine is shuttled to neurons where it serves as precursor for the glutamate and GABA neurotransmitter pools. Glutamate released to interstitial fluid during excitatotory neurotransmission is avidly taken up into astrocytes, and converted (in part) to glutamine for transfer back to neurons. This process is called the glutamate-glutamine (Glu-Gln) cycle and the cycle rate is directly proportional to the rate of oxidative metabolism over a wide range of brain activity levels (reviewed by Rothman et al.22). Cerebral metabolic rates (CMR) can be determined by assay of arteriovenous differences (A-V) and cerebral blood flow (CBF) rates to obtain global rates, use of [18F]fluorodeoxyglucose-positron emission tomography (FDG-PET) to assay the hexokinase (HK) step and obtain total glucose utilization, or assays of oxidation of 13C-labeled substates using MRS and metabolic modeling. Glycogen is contained predominantly in astrocytes and it is a dynamic participant in brain metabolism. Microdialysis samples the ECs of compounds in ECF. After entry into cells (IC: intracellular concentration), compounds are metabolized via different pathways to generate ATP that provides energy for the cells. Note that ECs are the net balance of influxes and effluxes to and from blood and brain cells, whereas intracellular concentrations are the net balance between transport and metabolism. Glc: glucose; Lac: lactate; Pyr: pyruvate; Glu: glutamate; Gln: glutamine. Modified from Figure 1 of Dienel GA and Cruz NF. Contributions of glycogen to astrocytic energetics during brain activation. Metab Brain Dis 2015; 30: 281–298. Copyright © 2014, Springer Science+Business Media New York, with permission.
Concentration changes in the ECF are related to but are not equivalent to cellular metabolism, metabolic rate, or energetics. Metabolic rate for glucose is the flux of glucose through the extracellular and intracellular glucose pools to end products (Figure 3), and the magnitude of rate changes need not be reflected by proportionate changes in glucose level due to regulatory properties of brain. Similarly, rates of influx or efflux of lactate to or from brain need not be proportionate to its quantity in ECF. Precursor-product and concentration-rate relationships are influenced by fuel supply, “coupling” of metabolic pathways, “tightness” of linkage between glycolytic and oxidative pathways, and the level of brain activity. For example, if CMRglc increases by 2-fold during brain activation and if brain glucose transport at the BBB and cell membranes also increases by 2-fold as well as plasma glucose delivery, there will be no change in glucose concentration.
Interpretation of changes in metabolite levels requires identification of their source(s) and cause(s). For example, glucose concentration is the net result of rates of influx, efflux, and metabolism (Figure 3). Under steady-state conditions, brain glucose level in normal awake subjects is constant and approximately equal to 20% that of arterial plasma, and the ECF glucose concentration measured by microdialysis will be similar. When CMRglc is constant, changes in plasma glucose level will cause shifts in brain tissue concentration, with large decrements when glucose delivery does not satisfy demand (e.g. during hypoglycemia) and smaller increments during hyperglycemia because glucose efflux rate rises with increasing tissue concentration.23,24 On the other hand, when plasma glucose level is constant, large increases in CMRglc can reduce brain glucose concentration to a new, lower steady-state, whereas reduced CMRglc can cause glucose level to rise, as during anesthesia. Knowledge of arterial plasma metabolite levels and brain metabolic rates is necessary to interpret changes in levels of extracellular metabolites. In the absence of metabolic rate assays, brain extracellular glucose and lactate levels are more informative when compared with their arterial plasma levels.
As noted above, a limitation of microdialysis assays is correspondence of metabolite levels in a very small tissue volume to the rest of the brain. Microdialysis probes are commonly placed in white matter, gray matter, and various locations relative to the site of injury, and probe location has a high impact on values of biochemical variables.7,25 For example, sampling of tissue contralateral to a unilateral spreading depression episode will not detect changes in glucose and lactate levels caused by increased glycolysis.26,27 White matter has much lower blood flow and metabolic rates than gray matter, with white and gray CMRglc in awake humans of ∼0.18 and 0.33–0.4 µmol/g/min, respectively28; these differences are even larger in awake rats.29 Glucose levels also differ in gray and white matter in rodents,29–31 with differential gray-white changes during anesthesia, ischemia, and seizures.32–34 Direct measurement of glucose levels in human brain also shows differences in levels and transport parameters in white and gray matter.35 Thus, white-gray matter differences in metabolite level, blood flow rate, metabolic rate, and responses to injury and anesthesia require a conservative approach to interpret and extrapolate microdialysis results to other brain regions.
Summary
Metabolite concentrations, metabolic rates, and energetics are inter-related but represent different aspects of the processes required to support brain function.
Hyperosmotic agents and cerebral energy metabolism in TBI patients
Increased intracranial pressure (ICP) commonly occurs after TBI, and it is a major cause of mortality. It has been known for almost 100 years that intravenous infusion of hyperosmolar solutions may decrease cerebrospinal pressure.36 The primary mechanism for reduction of ICP is by increasing the osmotic gradient across the BBB for substances which do not freely diffuse across this barrier. The effective osmotic pressure across a membrane that is partly permeable to the solutes is, however, less than the theoretically ideal osmotic pressure. To account for this difference, the reflection coefficient (σ) was introduced. The values of σ range from 0 to 1 depending on the relative permeabilities of the membrane to water and to solute. If the membrane is impermeable to the solute but not to water σ equals 1, and if the permeability of the solute is identical to the diffusion coefficient in water the effective osmotic pressure and σ equal 0. The two hyperosmolar solutions most often used in neurocritical care are mannitol and hypertonic NaCl. The BBB reflection coefficients are for mannitol and sodium approximately 0.90 and 1.0, respectively. Some published studies have suggested that hypertonic NaCl should be considered the gold standard for treatment of increased ICP.37,38 However, a review of existing data did not support favoring boluses of hypertonic saline over mannitol in terms of ICP control.39 Sodium lactate (500 mmol/L) has been suggested as a more effective therapy of increased ICP than mannitol,40 and in a double-blind, randomized, controlled trial, this solution was more effective in reducing the number of episodes of increased ICP than was isotonic saline in patients with TBI.41 Further, it has recently been claimed that hypertonic sodium lactate (1000 mmol/L) – besides the known effects obtained by any hyperosmolar drug – will have specific beneficial effects regarding cerebral energy metabolism.17,42
If, in a patient with compromised cerebral blood flow due to high ICP, hyperosmolar therapy decreases ICP, improvement of cerebral energy metabolism would be expected. In a series of patients with cerebral hemorrhage and ICP >20 mmHg, infusion of mannitol (1 g/kg) resulted in a significant decrease in ICP and cerebral LP ratio, increased cerebral perfusion pressure, and unchanged PbtO2 and concentration of cerebral glucose.43 In a study of patients treated with hypertonic sodium lactate therapy (1000 mmol/L), the authors reported a significant decrease in ICP while LP ratio remained unaffected and PbtO2 decreased.42 The decrease in ICP is, however, fully explained by the fact that a hyperosmolar sodium solution was infused. In this, as well as in their following study,17 the authors concluded that “improvement in brain energetics” was obtained due to infusion of hypertonic lactate. This conclusion was based exclusively on the observation that tissue glucose concentration increased.
Summary
Interpretation of microdialysate metabolite concentration changes as surrogates of brain metabolic rates is seriously flawed. In the following biochemical discussion, we will clarify why this interpretation is erroneous and misleading.
Lactate supplemententation for treatment of TBI patients
Why flood the brain with lactate?
A number of publications have alluded to or emphasized the use of lactate as supplemental brain fuel after TBI by association with the notion of astrocyte-to-neuron lactate (ANL) shuttling.44 The ANL shuttle model is based on glutamate-evoked release of lactate from cultured astrocytes, with the idea that glycolysis provides the ATP needed for astrocytic uptake of neurotransmitter glutamate and its conversion to glutamine (Figure 3) and the assumption that the released lactate is oxidized by nearby neurons. Lactate is a supplemental brain fuel when its blood level is elevated during exercise or lactate infusion, but ANL shuttling has never been quantified and validated as being metabolically significant in living brain, and the cellular source(s) of lactate in brain have not been identified. In fact, there are many independent lines of strong evidence against the lactate shuttle model. For example, glutamate-evoked lactate release is not a robust phenotype of cultured astrocytes, glutamate is oxidized by cultured astrocytes to generate more ATP than glycolysis, lactate oxidation coupled with glycolysis requires a parallel rise in CMRO2 with CMRglc during brain activation that does not occur (i.e., OGI falls during brain activation), and lactate oxidation would cause trapping of labeled metabolites of glucose to the same extent as the rise in glycolytic rate, which does not occur during brain activation.20 For these reasons, lactate shuttling and utilization have been a controversial issue.45–54 Importantly, in vitro studies have shown that lactate can maintain ATP levels but not sustain neuronal signaling, underscoring unidentified but critical roles of glucose metabolism upstream of pyruvate/lactate for neuronal function.55–59 In the present context, the major issue is not lactate metabolism, but whether microdialysate metabolite levels are useful to determine if lactate flooding has beneficial metabolic and energetic effects for TBI patients. Because the metabolic efficacy of lactate supplementation depends on functional integrity of mitochondria, prior assessment of oxidative capability in each patient is required when metabolic benefits of lactate are to be evaluated.
Metabolic rates in TBI patients
TBI patients typically have below-normal CMRglc and CMRO2 (Table 1), due to sedation/anesthesia and injury. OGI is also reduced in TBI patients, indicating enhanced glycolysis, with lactate release to blood denoted by negative CMRlac (Table 1). When expressed in glucose equivalents, CMRlac is typically less than about ±5% of CMRglc in both normal and TBI brain (Table 1). However, much higher lactate release, equal to 14–22% of glucose uptake, has been reported after TBI, and prognosis of TBI patients is worst when lactate release is highest.11,60 Adverse outcome after TBI frequently derives from a “metabolic crisis” associated with “hyperglycolysis” and very high LP ratio, perhaps due to mitochondrial dysfunction with reduced CMRO261 or spreading depression that is known to increase glycolysis, brain lactate level, and lactate efflux to ∼22% of glucose influx in rat brain.26
Table 1.
Representative metabolic rates in traumatic brain-injured (TBI) and control patients.
| Ref. | Group | Blood (µmol/mL) | CMRglc (µmol/g/min) | Glc consumed in 1 h (µmol/g) | CMRO2 (µmol/g/min) | CMRlac (µmol/g/min) | OGI | OCI | 0.5CMRlac/ CMRglc (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Glucose | Lactate | |||||||||
| 1 | Control | 4.6 | 0.74 | 0.25 | 14.9 | 1.38 | −0.02 | 5.59 | 5.82 | −4.0 |
| TBI | 6.8 | 1.54 | 0.19 | 11.4 | 0.63 | 0.004 | 3.28 | 3.25 | +1.1 | |
| 2 | Control | 6.0 | 1.13 | 0.27 | 16.3 | 1.09 | −0.017 | 4.03 | 4.15 | −3.0 |
| TBI | 6.3 | 0.93 | 0.16 | 9.6 | 0.55 | −0.017 | 3.42 | 3.61 | −5.3 | |
| 3 | TBI | 0.18 | 10.8 | |||||||
| 4 | TBI | 0.16 | 9.6 | |||||||
| Mean for TBI | 0.17 ± 0.02 | 10.4 ± 0.9 | ||||||||
Note: Values are means from reference (ref.) 1, Table 2 of Glenn et al.10; ref. 2, Table 1 of Glenn et al.62; ref. 3, Table 2 of Vespa et al.61; and ref. 4, Table 2 of Hutchinson et al.63 Reported values were converted to molar units when necessary, and ratios were calculated from mean values. CMR denotes cerebral metabolic rate for the indicated substrate; Glc: glucose; Lac: lactate. The total amount of glucose consumed during a 1 h microdialysis sampling interval was calculated (CMRglc × 60 min) for comparison to net concentration changes of metabolites in microdialysate in Table 2. The mean values for CMRglc and total glucose consumed in 1 h for TBI patients for the four studies are 0.17 µmol/g/min and 10.4 µmol/g. OGI: CMRO2/CMRglc. OCI: oxygen-carbohydrate index = CMRO2/(CMRglc + 0.5 CMRlac). OGI has a theoretical maximal value of 6.0, due to the stoichiometry of 6 O2 consumed per glucose oxidized. OCI takes into account the net total carbohydrate taken up/released across the blood–brain barrier but usually does not account for any glycogen consumed. OGI and OCI values below ∼5.4–6.0 indicate that non-oxidative metabolism (mainly glycolysis) predominates. Because stress and intravenous adrenaline stimulate non-oxidative metabolism in brain and depress OGI and OCI,64 the controls in ref. 2 may have been more stressed during the procedures than those in ref. 1. Negative values for CMRlac denote release from brain. Lactate was converted to glucose equivalents by dividing by two. Note that lactate uptake and metabolism in one TBI group represents only 1% of CMRglc, whereas in all other groups ∼3–5% of the glucose metabolized was released to blood as lactate.
Assessment of metabolic effects of lactate supplementation in TBI patients
Infusion of hypertonic sodium lactate is effective in reducing intracranial pressure in TBI patients,40,41 and it is important to determine whether this procedure has additional beneficial or harmful outcomes on patients. Oddo, Magistretti, Pellerin, and colleagues are proponents of the ANL shuttle concept and they have embraced the notion of lactate as supplemental fuel for TBI patients to spare glucose and improve brain metabolism and energetics. Reports by the above group involved patients (i) with sustained decreases in microdialysate glucose levels, (ii) given 3 h of hypertonic sodium lactate infusions, or (iii) with elevated lactate levels under hypoxic and non-hypoxic conditions. The authors claimed that elevated lactate levels due to infusion into blood or generation within brain support the idea that it is fuel for injured brain, with glucose-sparing and beneficial effects on brain metabolism and neuroenergetics. However, metabolism and energetics were never measured, and their interpretation of favorable metabolic-energetic outcome is based on microdialysate metabolite concentration changes determined in visually normal subcortical white matter in conjunction with other standard measures, e.g. PbtO2 and cerebral perfusion pressure to identify ischemia/hypoxia. Their unsubstantiated conclusions have the potential to influence implementation of new treatment protocols without appropriate testing and, therefore, require careful evaluation.
How can concentration changes be interpreted when there is no direct measure of metabolism? Hutchinson et al.63 analyzed data from individual TBI patients and reported linear relationships between local CMRglc and corresponding lactate and pyruvate concentrations, but not for glucose level or LP ratio, which are critical variables for patient status diagnosis (Figures 1 and 2). However, an earlier study by Vespa et al.61 found no significant correlations among microdialysis values and CMRglc. These discordant findings suggest that changes in extracellular metabolite levels are unlikely to be predictive of metabolic rate because concentrations are influenced by many factors, including probe location and evolution of pathophysiology.
Microdialysate concentration changes vs. total glucose consumed
The conclusion that microdialysate concentration changes are not useful representations of CMRglc is further supported by comparisons to total glucose consumed during the microdialysis sampling interval and calculation of the time required to produce the observed microdialysate concentration change at a given CMRglc. In the first case, the total amount of glucose consumed during a time interval is the product of CMRglc (µmol/g/min) and time; this calculation converts glucose metabolic rate to the same units as concentration so that the total amount of glucose that traversed from blood through the extracellular glucose pool can be compared to the size of the ECF pool (see Figure 3, bottom). The reference for CMRglc calculated from means of four separate cohorts of TBI patients at different institutions was 0.17 µmol/g/min, and the mean amount of glucose consumed in 1 h was 10.4 µmol/g (Table 1). The 1 h interval corresponds to the white-matter microdialysis sampling period used by the Oddo-Magistetti group.16,17,42,65 In the second case, times to produce reported microdialysate concentration changes were calculated from the change in metabolite concentration (µmol/g) divided by the mean CMRglc in TBI patients of 0.17 µmol/g/min; the lower the metabolite level, the faster it can change.
Table 2 summarizes microdialysate metabolite levels reported in several studies of TBI patients from which changes in ECs of glucose, lactate, and pyruvate, glucose equivalents of lactate and pyruvate, and blood/brain concentration ratios were calculated. The first data set presents the mean normal reference values from Figure 1, and the second data set provides data from TBI patients (ref. 4 in Table 1) that have a CMRglc slightly below the mean TBI value and who have a high LP ratio. The third data set (ref. 3) compares a cohort of patients before and during 2 h of reduced extracellular glucose level and 4th and 5th sets are from patients before and during lactate infusions. As a first approximation, the change in extracellular glucose concentration during the 1 h microdialysis sampling interval is compared with the total amount of glucose consumed in 1 h; this is the amount of glucose that was taken up from plasma per gram brain, diffused through ECF into intracellular fluid, and metabolized (see Figure 3). In Table 2, data set 3, the extracellular glucose concentration fell by 0.6 µmol/ml during the glucose depletion episode, equivalent to 5.8% of the total glucose consumed, and this extracellular concentration change could have occurred within 3.5 min (see the bolded numbers in Table 2). Similar calculations for the lactate infusion studies 4 and 5 in Table 2 reveal that the rise in extracellular glucose concentration corresponded to about 4–8% of the total glucose consumed, and the changes could have ocurred within 2–5 min. Changes in extracellular lactate and pyruvate concentrations (after their conversion to glucose equivalents by dividing by two) are also small fractions of the total glucose consumed, <10% for lactate and <0.3% for pyruvate (bolded numbers in Table 2), and these concentration changes could take place within 5 min for lactate and within 0.1 min for pyruvate.
Table 2.
Changes in microdialysate metabolite levels in brain-injured patients with or without lactate supplementation correspond to a very small fraction of the estimated total amount of glucose metabolized during the same interval.
| Ref. | Group | Blood (µmol/mL) |
Concentration in microdialysate (µmol/mL) |
Brain/blood ratio |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Glc | Lac | Glc | Lac | Pyr | LP ratio | Glc | Lac | ||
| 1 | Normal brain of awake humans, baseline | 1.7 | 2.9 | 0.166 | 23 | ||||
| 2 | TBI baseline with CMRglc = 0.16 µmol/g/min | 1.43 | 4.55 | 0.163 | 32.1 | ||||
| 3 | TBI/SAH baseline | 7.8 | 0.9 | 1.3 | 4.0 | 0.127 | 27 | 0.17 | 4.4 |
| 2 h Glc depletion | 6.5 | 0.9 | 0.7 | 5.4 | 0.172 | 35 | 0.11 | 6.0 | |
| Difference (Glc depleted-baseline) | −1.3 | 0 | −0.6 | 1.4 | 0.045 | ||||
| Glc equivalents of difference | −0.6 | 0.7 | 0.023 | ||||||
| % of Glc metabolized in 1 h | 5.8% | 6.8% | 0.2% | ||||||
| Time to cause concentration change (min) | 3.5 | 4.1 | 0.1 | ||||||
| 4 | TBI baseline | 1.0 | 1.4 | 3.1 | 0.116 | 27.0 | |||
| TBI + 3 h Lac infusion | 6.1 | 2.2 | 5.1 | 0.180 | 28.0 | ||||
| Difference (infused – baseline) | 5.1 | 0.8 | 2.0 | 0.065 | |||||
| Glc equivalents of difference | 0.8 | 1.0 | 0.03 | ||||||
| % of Glc metabolized in 1 h | 7.7% | 9.7% | 0.3% | ||||||
| Time to cause concentration change (min) | 4.6 | 2.9 | 0.1 | ||||||
| 5 | TBI baseline | 7.4 | 1.0 | 1.5 | 2.8 | 24 | 0.20 | 2.8 | |
| TBI + 3 h Lac infusion | ∼7.5 | ∼5 | ∼1.9 | ∼4.4 | ∼0.25 | 0.8 | |||
| Difference (infused – baseline) | 0.1 | 4 | ∼0.4 | ∼1.6 | |||||
| Glc equivalents of difference | 0.4 | 0.8 | |||||||
| % of Glc metabolized in 1 h | 3.9% | 7.7% | |||||||
| Time to cause concentration change (min) | 2.3 | 4.6 | |||||||
Note: Values are means from reference (ref.) 1: Table 2 of Reinstrup et al.3; ref. 2: Table 2 of Hutchinson et al.63; ref. 3: Table 2 of Patet et al.16; ref. 4: text and Figure 3 of Bouzat et al.42; ref. 5: Table 2 and maximal values that were emphasized by the authors that were from their text and estimated from their Figure 1 (note: these values are not as accurate as desired due to lack of ordinate scale marks in their Figures 1 and 2 and small differences in baseline values reported in their Table 1 and Figure 1) of Quintard et al.17 TBI: traumatic brain injury; SAH: subarachnoid hemorrhage; Glc: glucose; Lac: lactate; Pyr: pyruvate; LP: lactate/pyruvate ratio. Brain/blood ratios were calculated from reported mean values. The estimated total amount of glucose metabolized in a 1 h microdialysis sampling interval (10.4 µmol/g) was calculated using the mean CMRglc in four cohorts of TBI patients in Table 1 (0.17 µmol/g/min × 60 min). For simplicity, 1 g tissue was equated with 1 mL (=1 g) microdialysate and the small volume fraction of extracellular fluid (∼20%,18,19) compared with brain tissue was disregarded. Glucose equivalents for pyruvate and lactate were calculated by dividing their concentrations by two; these values were used to calculate % of total glucose metabolized in 1 h (i.e., concentration difference/glc metabolized) and calculate the time to cause the concentration change (i.e., concentration difference/mean CMRglc).
Impact of small ECF volume
When the ECF volume fraction and brain water content are taken into account, the extracellular concentration changes as percent of total glucose metabolized are actually much smaller, only about 15% (i.e. 0.2 × 0.77) of the bolded values shown in Table 2. For example, for ref. 3, Table 2, the change in glucose concentration is 0.6 µmol per ml of ECF. Multiply 0.6 µmol/ml by 0.2 ml ECF/ml brain water and by 0.77 ml brain water/g brain tissue to obtain 0.09 µmol/g as the concentration change in brain tissue represented by the change in ECF. This amount corresponds to ∼0.9% of the 10.4 µmol/g glucose taken up from plasma and metabolized. In other words, for each molecule of glucose depleted from ECF, 116 molecules of glucose passed through the same pool from plasma to brain cells where they were metabolized. Similar calculations can be made for the changes in all metabolite levels during lactate supplementation. All changes in ECF concentrations are tiny fractions of the fluxes through the metabolite pools, demonstrating that any relationship of extracellular concentration change to metabolic rate is weak. Furthermore, even if the reference CMRglc (Table 1) differed by ± 100% from the true rate in the TBI patients in Table 2 (e.g. due to regional or gray-white matter rate differences), the percentages of glucose metabolized and the times to generate the concentration changes would still be small. Finally, the observed concentration changes can occur within a small fraction of the 1 h duration of the microdialysate sampling interval, indicating that they must be either very large or prolonged to change by >10–15% in 1 h samples.
Effects of lactate supplied to brain vs. lactate generated within brain
A striking outcome of the analysis in Table 2 is that the data derived during 3 h hypertonic lactate infusion (calculated for references 4 and 5) were similar to those in “2 h glucose-depleted” subjects (calculated for reference 3). The glucose-depleted cohort probably had high glycolytic rates, based on near-normal blood glucose level, elevated pyruvate level, high LP ratio, and low brain/blood glucose ratio, characteristics similar to the profile of mitochondrial dysfunction in Figure 2. The lactate dialysate concentration changes as a percent of glucose consumed and short times to achieve the changes do not distinguish between effects of lactate supplementation and glycolytic lactate production in brain and the two situations could not be differentiated in a blinded study.
Peripheral effects of lactate flooding
Lactate supplementation probably increases pyruvate levels in blood due to conversion from lactate in peripheral tissues and red blood cells, causing greater pyruvate uptake into brain. Interpretation of brain metabolite levels and LP ratio after hypertonic sodium lactate infusion, therefore, requires knowledge of glucose, lactate, and pyuvate levels and LP ratio in arterial plasma.46 Because brain pyruvate concentrations are very low, their levels can change by the reported amount within 0.1 min (Table 2). This six-second response time for pyruvate compared with longer times for lactate (2.9–4.6 min, Table 2) indicates that rapid changes in pyruvate level can have a high impact on LP ratio.
Summary
Extracellular metabolite concentration changes are influenced by the actual blood and brain concentrations relative to their transport and metabolic rates, and they are not sensitive indicators of metabolic rate, metabolic improvement, or metabolic decrement. Significantly, analysis of concentration changes does not distinguish between effects of lactate supplementation and “hyperglycolysis.” Therefore, in the absence of metabolic studies, it is not appropriate to interpret extracellular concentration changes in terms of metabolism or neuroenergetics, and such conclusions are not useful for evaluation of patient outcome.
Concentration-metabolic rate discordance under conditions relevant to TBI
Sensory stimulation, physiological stress, adrenaline release to blood, spreading depression, and seizures are all pertinent to patients with severe brain injury. Each of these situations could independently contribute to changes in brain metabolite levels and metabolic rates after TBI and increase the complexity of interpretation of microdialysate lactate concentration changes. To extend the number of situations in which predictors of cerebral metabolic rates would be useful for analysis of TBI patients, examples from studies in experimental animals (that cannot be carried out in human patients) are tabulated in Table 3. The calculations are similar to those described for Table 2, except that all but one of the data sets in Table 3 are brain tissue concentrations, i.e. intracellular plus ECs.
Table 3.
The magnitude of change of brain metabolite levels in experimental animals does not provide useful information about the corresponding change in metabolic rate under different conditions.
| Ref. | Group | Blood or plasma |
Brain tissue |
Extracellular |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glc | Lac | Glc | Lac | Glyc. | Pyr | LP | CMRglc | CMRlac | CMRO2 | Glc | Lac | ||
| 1 | Control | 6.8 | 0.5 | 2.8 | 1.0 | 6.1 | 0.75 | 4.1 | |||||
| Sensory stimulation, 5 min | 7.8 | 2.0 | 3.1 | 1.9 | 5.4 | 1.08 | 5.4 | ||||||
| Difference | 1 | 1.5 | 0.3 | 0.9 | −0.7 | 0.33 | 1.3 | ||||||
| % of Glc metabolized | 4.9% | 7.4% | 11.5% | 144%C | 132%C | ||||||||
| Time to cause conc. change (min) | 0.3 | 0.4 | |||||||||||
| 2 | Control | 10.6 | 0.92 | 2.38 | 0.34 | 4.6 | 1.07 | ||||||
| Sensory stimulation , 10 min | 12.9 | 1.31 | 2.82 | 0.85 | 4.0 | 1.36 | |||||||
| Difference | 2.3 | 0.39 | 0.44 | 0.51 | −0.6 | 0.29 | |||||||
| % of Glc metabolized | 3.1% | 1.8% | 4.2% | 127%C | |||||||||
| Time to cause conc. change (min) | 0.3 | 0.2 | |||||||||||
| 3 | Control | Est. 0.55 | 0.8 | 0.3 | |||||||||
| Visual stimulation, 0.5 min | Est. 0.63 | 0.3 | 0.5 | ||||||||||
| Difference | 0.08 | −0.5 | 0.2 | ||||||||||
| % of Glc metabolized | 115%C | 61% | 12.2% | ||||||||||
| 4 | Control | 9 | 2.99 | 1.55 | 2.31 | 0.11 | 14.1 | (Est. 0.68) | 4.09 | ||||
| Adrenaline, 10 min | 14.4 | 5.02 | 1.59 | 2.42 | 0.11 | 14.5 | (Est. 1.37) | 8.19 | |||||
| Difference | 5.4 | 2.03 | 0.04 | 0.11 | 0 | 0.68 | 4.1 | ||||||
| % of Glc metabolized | 15% | 0.1% | 0.8% | 0 | 200%C | 200%C | |||||||
| Time to cause conc. change (min) | 1.5 | 0.01 | |||||||||||
| 5 | Control | 1.98 | 2.47 | 0.76 | |||||||||
| Spreading depression, 25 min | 1.38 | 8.48 | 1.10 | −0.98 | |||||||||
| Difference | −0.6 | 6.01 | 0.34 | ||||||||||
| % of Glc metabolized | −2.1% | 10.7% | 145%C | ||||||||||
| Time to cause conc. change (min) | 0.5 | 2.7 | |||||||||||
| 6 | Control | 10.4 | 4.16 | 1.53 | 1.84 | 0.141 | 11.1 | 0.77 | 4.4 | ||||
| Seizures, 1 min | 11.1 | 1.37 | 6.21 | 1.38 | 0.289 | 21.6 | 3.17 | 12.2 | |||||
| Difference | 0.7 | −2.79 | 4.68 | −0.48 | 0.148 | 2.4 | 7.8 | ||||||
| % of Glc metabolized | −43% | 36% | −7% | 1% | 412%C | 277%C | |||||||
| Time to cause conc. change (min) | 0.9 | 0.7 | |||||||||||
Note: Mean values are from references (ref.) 166,67; 268; 369; 470; 526,27; 6.71,72 Units are as follows: blood or arterial plasma concentration, µmol/mL; brain concentration: µmol/g; metabolic rate: µmol/g/min; brain extracellular concentration, µmol/mL. Glycogen concentration is expressed as glucosyl units. Abbreviations: Glc: glucose; Lac: lactate; Glyc: glycogen; Pyr: pyruvate; LP: lactate/pyruvate ratio; CMR: cerebral metabolic rate for the indicated substrate. The negative value for CMRlac denotes lactate release from brain, calculated as the twice the difference in metabolic rates based on total glucose utilization measured with [14C]deoxyglucose and trapping of metabolites of [6-14C]glucose. This difference reflects the rate of release of labeled metabolites from brain; half of this rate corresponded to lactate efflux to blood, directly measured relative to glucose influx by arteriovenous difference assays in awake rats,26,27 and the remainder is assumed to be released by other routes including perivascular flow to the lymphatic drainage.73,74 Lactate and pyruvate were converted to glucose equivalents by dividing by two prior to calculation of % of glucose metabolized. Total glucose metabolized was calculated as the product of CMRglc and experimental duration plus the decreases in tissue glucose and glycogen concentrations, if any. Changes in metabolic rate are expressed as percent of control (c). In the visual stimulation assays in anesthetized cats of Li and Freeman,69 the example is taken from their Figure 5, using the values presented and discussed by Leloup75; similar changes were obtained in the other experimental conditions used by Li and Freeman. The rise in CMRglc during visual stimulation was estimated (Est.) as 15%, based on the reported 15% increase in cerebral blood flow (CBF), and parallel changes in CBF and CMRglc under many conditions. The changes in extracellular glucose and lactate were coincident with neuronal spiking activity, so time to cause the change was not tabulated. Total glucose metabolized was estimated by using CMRglc for visual cortex of awake adult cats of 1.1 µmol/g/min,76 multiplied by 0.5 (assuming a ∼50% decrease due to use of fentanyl and propofol77,78) to obtain baseline rate of blood glucose consumption of 0.55 µmol/g/min and an activated rate of 0.63 µmol/g/min. This rate would consume 0.32 µmol/g blood glucose in 0.5 min to which the 0.5 µmol/mL decrement in extracellular glucose was added (assume 1 mL = 1 g, ignore the volume fraction of extracellular space, assume intracellular change was similar) to give a total of 0.82 glucose µmol/g consumed for calculation of % of glucose metabolized. CMRglc was not measured in the adrenaline study, and its minimal value was estimated by dividing CMRO2 by 6. The estimated CMRglc would be higher if the oxygen–glucose index were less than 6.
Activation of normal brain causes aerobic glycolysis
Neurointensive-care patients receive various types of physiological stimuli that can influence brain blood flow and metabolic activity when awake or sedated. Generalized sensory stimulation of awake rats for 5 or 10 min increased CMRglc more than CMRO2 thereby causing OGI to fall, small amounts of lactate accumulated in brain, and glycogen was consumed (Table 3,references (ref.) 1 and 2). CMRglc rose by ∼25–45%, and total tissue glucose and lactate levels changed but the concentration differences corresponded to only ∼2–8% of glucose consumed; glycogen consumption corresponded to 4–12% of glucose metabolized (Table 3). These animals were not fasted and they moved in response to stimulation, causing blood (ref. 1) and plasma (ref. 2) glucose and lactate levels to increase and contribute to increased brain levels. Because the brain/plasma or brain/blood ratios for glucose were similar during rest and stimulation in both studies, the rise in brain level was due to increases in blood. Thus, the higher metabolic rate during stimulation was maintained by increased flux of glucose from blood to brain cells without reducing brain tissue glucose level. In the absence of knowledge of OGI and CMRglc, the 10% rise in brain glucose level might be mis-interpreted as a consequence of reduced CMRglc or utilization of endogenously generated lactate as supplemental fuel.
Brief visual stimulation of anesthetized cats and use of high-speed electrochemical sensors and microelectrodes revealed immediate decreases in extracellular glucose level and concomitant increases in extracellular lactate level that coincided with and were proportional to neuronal spiking rate.69 One representative example is provided in Table 3 (ref. 3), and calculation of these extracellular changes as percent of total glucose consumed in Table 3 took into account the estimated metabolic flux of blood glucose through the extracellular pool as explained in the legend. This calculation had the effect of reducing the apparent relationship between changes in lactate and glucose levels. The + 0.1 µmol/L glucose equivalents for lactate vs. −0.5 µmol/L glucose correspond to a rise in extracellular lactate equivalent to 20% of the decrease in glucose level. However, when compared with the estimated amount of total glucose consumed (0.32 µmol/g delivered from blood plus 0.5 µmol/g extracellular glucose consumed =0.82 µmol/g), the lactate released to ECF actually corresponds to 12% of total glucose metabolized, not 20%. The results were appropriately interpreted by the authors who took into account factors that influence extracellular metabolite concentrations. On the other hand, the editorial highlight of this study by Leloup75 went beyond the data and authors' interpretation to associate their work with ANL shuttling. LeLoup's discussion included studies by Hu and Wilson79,80 that measured extracellular glucose and lactate concentration changes in response to strong electrical stimuli that may have caused seizures. These concentration changes were ascribed to lactate shuttling,79,80 but data were mis-interpreted because a change in lactate level does not provide any information about lactate shuttling. Furthermore, the contributions of changes in extracellular glucose and lactate levels to total metabolism were overestimated by Hu and Wilson because they did not take into account the metabolic through-flux of blood-borne glucose (see Table 7 of Dienel47).
Similar discordance between rate and concentration changes has been found in studies of human visual cortex. Positron-emission tomographic (PET) and 1H magnetic resonance spectroscopy (MRS) studies of visual stimulation paradigms revealed increased glucose consumption by over 30% with transient lactate concentration increases of only 0.2–0.3 µmol/g and even smaller drops in glucose level.81–86 Although the MRS measures total brain glucose (mostly cellular), rapid diffusion of both glucose and lactate across neuronal and glial membranes suggests that these changes are reflected in the ECF and they are consistent with the findings described above. MRS assays of labeled metabolites recovered in the microdialysate after insertion of labeled precursors into brain87,88 are anticipated to be much less useful than in vivo MRS assays after intravenous infusion of labeled precursors because the recovery of labeled compounds in microdialysate is low, long collection times must be used (24 h-pooled samples), and samples only report ECF, excluding predominantly labeled intracellular compounds (e.g. glutamate) relevant to glutamate-glutamine cycling and oxidative metabolism rates. Demonstration of lactate oxidation88 provides no evidence to confirm the ANL model, as erroneously claimed.89
Stress
TBI patients are subjected to various types of stressful events that can cause adrenal release of adrenaline into blood and influence brain metabolism via vagus nerve signaling and norepinephrine release.20,90 Adrenaline treatment increased CMRO2 2-fold (Table 3, ref. 4). CMRglc was not measured in this study, but, when calculated from CMRO2 and OGI, CMRglc would double if OGI = 6, and it would increase further if OGI were <6. Adrenaline increased plasma glucose level by 60%, with a proportionate increase in brain glucose level; brain/blood ratios were similar to control. The net change in brain glucose level corresponded to 15% of the total glucose consumed, whereas brain lactate and glycogen levels were stable. This study is notable because increased respiration rate was associated with elevated blood and brain glucose levels, presumably due to stimulation of liver and muscle glycogenolysis. In this case, increased glucose supply from blood matched the elevated oxidative rate, and the rise in brain glucose level reflected that in blood, not glucose sparing or a reduction in CMRglc.
Spreading depression
Spreading depression is common after brain injury and subarachnoid hemorrhage, and repeated spreading depolarizations in rat brain increased CMRglc by 45% (Table 3 , ref. 5), with a small decrease in tissue glucose level indicative of the inability of glucose supply to fully match increased demand. There was also a 3.4-fold rise in tissue lactate level. Both glucose and lactate concentration changes corresponded to small fractions of total glucose consumed and could have been attained within minutes. On the basis of parallel assays27 with [14C]deoxyglucose to measure total CMRglc at the hexokinase step and with [6-14C]glucose to estimate oxidative metabolic rate based on trapping of labeled metabolites in tissue via dilution into the large amino acid pools (see Figure 3), CMRlac was calculated to be −0.98 µmol/g/min (rate of net release from brain). Arteriovenous assays of lactate release gave CMRlac about half of this rate,26 and the rest of the lactate is assumed to be released from brain via perivascular-lymphatic drainage.73,74 To sum up, levels of lactate retained in brain do not fully represent the extent of glycolytic upregulation. Brain lactate generated by hyperglycolysis is rapidly released, little lactate, if any, is oxidized, and assays of brain metabolite concentrations, lactate release to blood, and CMRglc at the hexokinase step are not sufficient to fully evaluate lactate production and release from brain.
Seizures
Seizures may also occur after brain injury, and a brief seizure that increased CMRO2 and CMRglc by about 3- and 4-fold, respectively, reduced respective glucose and glycogen levels by 60% and 26%, raised lactate level 4-fold, and doubled pyruvate level, all within one minute (Table 3, ref. 6). OGI fell from 5.7 to 3.8 and the LP ratio doubled, indicative of hyperglycolysis. The large increase in respiration implies adequate oxygen levels and functioning mitochondria. In conjunction with increases in lactate and pyruvate levels, these metabolic changes resemble the profile exhibited by mitochondrial dysfunction (Figure 2), but it is more likely that fast, massive upregulation of glyolytic rate during the seizure greatly exceeded oxidative capacity (and probably BBB lactate transport capacity) to resemble effects of mitochondrial dysfunction. Changes in glucose and lactate levels corresponded to large fractions of total glucose consumed (Table 3) and demonstrated that glucose supply did not match the high metabolic demand. Thus, spreading depression and seizures increase CMRglc and brain lactate accumulation, with depletion of brain glucose. If lactate were a preferential fuel, it should be quickly oxidized. Instead, much lactate is released to blood, presumably due to limitations of its oxidation in the absence of prior injury.
Summary
Sensory stimulation, stress responses, spreading depression, and seizures can have strong, independent influences on brain metabolism, contributing to the effects of the injury and recovery processes. These situations increase the complexity of interaction of plasma and brain glucose levels, metabolic rate, and transport, as illustrated in Table 3. All concentration changes, except during seizures, corresponded to a small fraction (0.1–12%) of the total glucose consumed during the assay interval, and they could have occurred within 3 min. The magnitude and direction of changes in tissue glucose level do not reflect the magnitude of change in CMRglc. Changes in extracellular metabolite levels are informative but insufficient to evaluate changes in lactate or glucose metabolism or metabolic benefit from lactate flooding.
Contributions of lactate supplementation to brain oxidative metabolism
In the absence of metabolic studies to validate cerebral metabolism of lactate supplements given to TBI patients, potential contributions of lactate to brain oxidative metabolism can be estimated from studies in normal humans during lactate infusions or graded-intensity exercise. Studies using MRS and arteriovenous (A-V) differences have looked at the relationship between plasma lactate concentration, brain levels of lactate, and the rate of lactate oxidation,52–54 and at physiological plasma lactate levels of <1 mmol/L, net lactate oxidation by brain is negligible. Even at the relatively high resting physiological level of ∼3 mmol/L lactate in plasma and 2 µmol/g lactate in brain achieved by modest lactate infusions into sedentary human subjects, <10% of brain energy metabolism is supplied by lactate (Figure 4). Resting brain lactate levels in carefully handled sedentary animals and humans are generally <1 µmol/g and rise to ∼2 µmol/g during brain activation, and any utilization of this lactate will contribute <10% to total oxidation, with glucose providing >90% (green arrows and text, Figure 4). The higher microdialysate lactate levels in TBI patients (∼3–5 mmol/L, Table 2), if oxidized, may contribute ∼10–20% to total oxidation. The above studies showed that lactate has the potential of supplying up to ∼40% of total brain oxidative metabolism, but this occurs at extremely high lactate levels (>15 mmol/L) that are not viable from infusions and only occur physiologically during prolonged, intense exercise.
Figure 4.
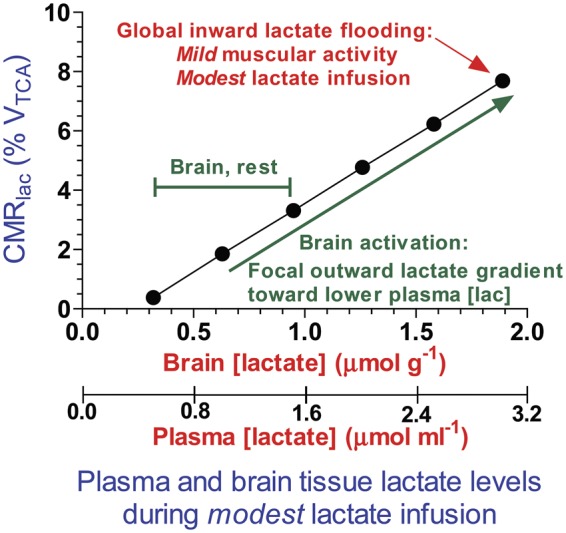
Fractional contribution of brain lactate to total oxidation as function of brain lactate concentration. Boumezbeur et al.54 infused [13C]lactate into human subjects and measured its oxidation rate in brain. The linear regressions from their Figure 6 were used to calculate brain lactate levels from arterial plasma lactate levels at 0.5 µmol/mL intervals using their equation [lactate]brain = 0.63 [lactate]plasma. These data were plotted against the corresponding calculated values for CMRlactate, calculated with their equation, CMRlac = 0.019[lac]plasma – 0.007; CMRlac is expressed as % VTCA, where VTCA = 0.65 µmol/g min and represents the total oxidation rate in neurons plus glia.54 The plotted points illustrate the approximate fractional rates of lactate oxidation at plasma and brain lactate levels arising from the modest lactate intravenous infusion schedule. Actual values measured by Boumezbeur et al.54 and plotted in their Figure 6(c) would be distributed around this regression line. These results demonstrate that within the range of brain lactate levels in normal resting and activated brain in sedentary subjects, lactate contributes ∼2–8% to total oxidation and glucose accounts for the remaining 92–98%. The green text indicates the range of brain lactate levels in normal activated brain in sedentary rodents that have lower arterial plasma lactate levels and an outward, brain-to-blood lactate gradient. The red text denotes the point above which arterial plasma levels rise during progressively increasing intensity of exercise or of higher lactate infusion schedules causing an inward, blood-to-brain lactate gradient. CMRlac: cerebral metabolic rate for lactate; TCA: tricarboxylic acid; VTCA: total oxidation rate in neurons and astrocytes. Modified from Figure 10(c) of Dienel GA, Fueling and imaging brain activation. ASN Neuro 2012; 4:267–321.Copyright © 2012, The Author, with permission.
Summary
Plasma lactate is not a major fuel even under several fold elevated conditions. Based on 13C MRS studies under physiological and several fold elevated plasma lactate levels, the proportion of neuronal and glial lactate oxidation is the same as that for glucose, which argues against preferential neuronal uptake. In addition, lactate did not increase total brain metabolism as measured by MRS.54
Alternative perspectives of changes in microdialysate glucose and lactate levels
The studies in Table 2 provide a number of interesting observations that the authors interpreted with a focus on lactate as supplemental fuel. The goal of the following discussion is to expand the conversation on lactate metabolism by providing additional considerations and alternative data interpretations related to use of hyperosmotic sodium lactate supplementation.
Glucose depletion
In the Patet et al.16 study (Table 2, ref. 3), the mean glucose level in the “glucose-depleted” group was 0.7 mmol/L (1 mmol/L when corrected for 70% recovery in microdialysate), and 80% of the glucose depletion episodes were associated with LP ratios >25. These findings, along with increased levels of lactate and pyruvate, are consistent with increased glycolysis caused by unidentified circumstances, as concluded by the authors. Hyperglycolysis may have arisen from spreading depression, seizures, or mitochondrial dysfunction (Figure 2, Table 3). Lactate was probably not an oxidative fuel in the glucose-depleted patients because the LP ratio is substantially elevated. Lactate efflux from brain to blood down its 6:1 concentration gradient (Table 2) was not measured by Patet et al., but was probably substantial, based on studies in TBI patients (Table 1) and animal studies of spreading depression (ref. 5, Table 3). The Km (substrate concentration at which the reaction velocity is half-maximal) of brain hexokinase for glucose is ∼0.05 mmol/L,91 and, based on Michaelis–Menten kinetics (v = VmaxS/(Km + S), where v = reaction velocity, Vmax is maximal velocity, and S is substrate level), hexokinase would operate at 93% of Vmax at 0.7 mmol/L glucose. In other words, this glucose level should be able to support CMRglc in white matter where the probe was implanted. Based on analysis of the results in Tables 2 and 3, the percent changes in microdialysate lactate and glucose concentrations presented in Figure 5 of the Patet et al.16 report are not useful for analysis of CMRglc and CMRlac. Furthermore, there is no evidence for their assumptions that astrocytes are the source of the lactate, that lactate is supplemental fuel, for the causes of increased glycolysis, or justification for extrapolation of values in “normal” white matter to other brain regions. Spreading depolarizations can cause complex metabolic changes,26,27,92 and monitoring of patients for spreading depression may help establish the basis of hypgerglycolysis. Conclusions regarding lactate metabolism65 cannot be drawn from its concentration, as emphasized by Veech.93
Figure 5.

Metabolic pathways that may be affected by lactate flooding of brain after traumatic brain injury. Entry of large amounts of exogenous lactate into a brain cell drives the lactate dehydrogenase reaction (LDH) towards pyruvate and NADH production, and at the same time co-transports H+ and generates a second H+ that can acidify the cytoplasm. Phosphofructokinase (PFK), one of the major regulatory enzymes of the glycolytic pathway, is very sensitive to small decreases in pH that inhibit its activity. Increased levels of glucose-6-phosphate will enhance its regulation of hexokinase activity by feedback inhibition. (Brown lines with a ball at one end denote inhibition.) Reduction of the NAD+ availablity will also impair glycolytic flux. Lactate flooding-mediated lowering of pH and NADH generation has the potential to impair glycolytic, glycogenolytic, and pentose-phosphate (P) shunt pathway fluxes. Cytoplasmic NAD+ is regenerated from NADH by the action of LDH or the malate-aspartate shuttle (MAS). Note that lactate cannot be oxidized if the MAS is impaired or if there is mitochondrial dysfunction. HK: hexokinase; Glc: glucose; Glc-6-P: glucose-6-phosphate (P); Fru-6-P: fructose-6-P; Fru-1,6-P2: fructose-1,6-P2; Lac: lactate; PDH: pyruvate dehydrogenase; PC: pyruvate carboxylase; OAA: oxaloacetate; cit: citrate; αKG: α-ketoglutarate; MCT: monocarboxylic acid transporter; GLUT: glucose transporter; NE: norepinephrine; LC: locus coeruleus. Modified from Figure 1 of Dienel GA. Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab 2012; 32: 1107–1138. Copyright © 2012, International Society for Cerebral Blood Flow and Metabolism, with permission.
“Glucose sparing” and mitochondrial dysfunction
Lactate infusion into TBI patients by Bouzat et al. (Table 2, ref. 4) increased extracellular lactate, pyruvate, and glucose levels, and these findings were interpreted as beneficial, based on “glucose sparing” and provision of supplemental oxidative fuel. Limitations of this study have been previously identified,46,94 including lack of assays of brain metabolism and blood pyruvate level; changes in extracellular glucose or lactate concentration do not provide sufficient information to evaluate metabolism and energetics. Also, it is likely that increased brain pyruvate was due, in part, to uptake of blood pyruvate after peripheral lactate metabolism, so the brain LP ratios and percent changes in brain pyruvate level presented in Table 2 and Figure 5 of Bouzat et al.42 have no interpretive value. Another critical issue is that 67% of TBI subjects had a baseline LP ratio >25, raising the likelihood of mitochondrial dysfunction or “hyperglycolysis” prior to lactate infusion. Increased brain glucose level secondary to lactate flooding in TBI patients that may have oxidative metabolism impairment is also likely in the Quintard et al.17 report (Table 2, ref. 5). In this study, microdialysate glucose levels increased only in patients with LP ratio >25, not in those with an LP ratio <25. The high LP ratio suggests the inability to metabolize the lactate, and raises the concern that capacity for oxidative metabolism was not verified before lactate flooding. In fact, when oxidative metabolism and PbtO2 are adequate (LP ratio < 25), glucose oxidation is probably sufficient to support brain's energy demands and exogenous lactate is not necessary.
It is also noteworthy that publication titles and some topic headings in the lactate-related reports in Table 2 are not appropriate because neuroenergetics, neuroenergetic response, and metabolism of glucose and lactate were not measured. Accurate descriptive language and rigorous interpretation of data are necessary for development and evaluation of new experimental protocols for use in TBI patients. To summarize, neurocritical intensivists should ensure that cerebral oxidative metabolism is not impaired when hypertonic sodium lactate is used to reduce intracerebral pressure or modify brain energy metabolism. If oxidative metabolism is abnormal, there is no point in increasing brain lactate level by lactate supplementation because lactate will be poorly metabolized, if at all, and high lactate levels are likely to worsen outcome.
Potential consequences of lactate flooding
Lactate flooding causes intracellular acidification95 that may reduce the activity of a major glycolytic regulatory enzyme, phosphofructokinase (Figure 5). This enzyme has a steep activity-pH profile and is inhibited by small decreases in pH.96,97 Also, lactate flooding may raise NADH levels, thereby reducing the availability of NAD+ for glycolysis (Figure 5). High NADH levels and phosphofructokinase inhibition will also impair utilization of glycogen in astrocytes (Figures 3 and 4), potentially disrupting roles of glycogen in K+ and Ca2+ homeostasis.98,99 The pentose shunt pathway is important for management of oxidative stress and provision of precursors for biosynthesis (Figure 5). Pentose shunt flux is high shortly after brain injury, and lactate produced after passage through this pathway was released to blood where it was assayed in TBI and control patients.100 Less lactate was subsequently found to be generated from the pentose shunt compared with glycolysis in TBI patients,101 but these pathway fluxes may vary with severity and progression of injury. Inhibition of glycolysis by lactate will impair pentose shunt flux because glucose-derived carbon re-enters the glycolytic pathway upstream of the metabolic steps requiring action of phosphofructokinase and NAD+ availability (Figure 5). To sum up, lactate flooding may inhibit glycolytic and pentose shunt fluxes in neurons and astrocytes and impair glycogenolysis in astrocytes. Lactate can “spare glucose” in normal subjects during high demand for glucose (e.g., exercise), but in brain-injured patients, apparent glucose sparing may actually reflect reduced glycolytic flux required for neuronal signaling, degraded capacity for oxidative stress management, and impaired astrocytic regulation of ionic homeostasis.
Lactate signaling evoked by lactate flooding is another important consideration when evaluating its effects on injured brain. For example, lactate acts via the HCA-1 receptor (Figure 5) to reduce cAMP levels102 and also inhibit excitatory and inhibitory firing of cultured neurons by 50% at 4.2 mmol/L.103 This level of lactate is approximated or exceeded in the TBI studies (Table 2) and during spreading depression and seizures in rat brain (Table 3). In addition, 12.5 mmol/L lactate attenuates hippocampal pyramidal cell firing in vivo by 85%,104 and 10 mmol/L lactate suppresses field potentials, population spikes, and population excitatory postsynaptic potentials in hippocampal slices.55 Thus, lactate supplementation should enhance neuronal shutdown, thereby reducing neuronal energy demand and contributing to glucose sparing independent of any lactate metabolism.
Another interesting consequence of lactate signaling is activation of an unidentified receptor in the locus coeruleus (Figure 5) by low lactate concentrations (∼0.5 mmol/L); receptor activation increases norepinephrine release in the locus coeruleus and in the brain, thereby modulating neural activity105 with a strong influence astrocytic metabolism via the actions of norepinephrine on their adrenergic receptors.20 It is not clear how suppression of neuronal activity over and above that from anesthesia would influence recovery from brain injury, but signaling and metabolic properties of lactate flooding must be taken into account with evaluating outcome.
Summary
Lactate flooding has the potential to inhibit glycolytic, glycogenolytic, and pentose shunt fluxes, further compromising the brain's ability to upregulate cytosolic ATP production, control ionic gradients and extracellular neurotransmitter glutamate levels, and manage oxidative stress, particularly during hyperglycolysis or mitochondrial dysfunction (Figures 5 and 6). These complications, along with previous concerns,12,46,94 need to be addressed before lactate supplementation is used clinically.
Figure 6.

Influence of lactate flooding after traumatic brain injury on aspects of brain metabolism and signaling. Summary of potential consequences of elevated lactate on brain functions. Superscripts refer to the following references: 1,98,99; 2,106–108; 3,105; 4,102; 5,103; 6,104; 7,109.
Concluding comments
Extracellular metabolite concentrations and their temporal changes are extremely important to help distinguish among major types of metabolic changes after brain injury and guide therapy,1,2 but additional assays are required to evaluate metabolic changes. “Surrogate endpoints,”87 i.e. lactate, glucose, and pyruvate levels and LP ratios in microdialysates, provide therapeutic guidelines, but are not sufficient to evaluate adequately the metabolic and neuroenergetic effects of lactate flooding. Metabolic assays are necessary to verify oxidative capability of mitochondria and to identify beneficial and undesirable effects of lactate in brain-injured patients (Figure 6). Reduction of intracranial pressure by hypertonic sodium lactate is extremely important and its benefits may outweigh any potential effects on glycolysis, but the procedure needs to be properly evaluated. Glycolytic upregulation during brain activation and under abnormal conditions serves unidentified functions, and suppression of glycolysis by lactate supplementation during “hyperglycolysis” or mitochondrial dysfunction may be harmful. The major issue is not whether lactate can be metabolized, but how much does it contribute relative to glucose and is it metabolically and physiologically beneficial? Lactate contributes < 10% to total oxidation when plasma lactate levels are ≤3 mmol/L (Figure 454). It is necessary for proponents of lactate supplementation to demonstrate that a 10–20% contribution by cerebral oxidation of lactate in brain is advantageous to TBI patients with normal mitochondrial function and that lactate flooding is not harmful to patients with mitochondrial dysfunction or hyperglycolysis. Peripheral effects of lactate metabolism during its supplementation (e.g. to generate glucose via gluconeogenesis62,110) may also be important, and these types of contributions to brain metabolism must be distinguished from uptake and oxidation of lactate in brain.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: GD and C-HN received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. DLR was supported by the National Institutes of Health (grant number R01NS087568A).
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: GD and DLR declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. C-HN is consulting for MDialysis, Stockholm, Sweden.
References
- 1.Nordström CH, Nielsen TH, Jacobsen A. Techniques and strategies in neurocritical care originating from southern Scandinavia. J Rehabil Med 2013; 45: 710–717. [DOI] [PubMed] [Google Scholar]
- 2.Nordström CH. Cerebral energy metabolism and microdialysis in neurocritical care. Childs Nerv Syst 2010; 26: 465–472. [DOI] [PubMed] [Google Scholar]
- 3.Reinstrup P, Ståhl N, Mellergård P, et al. Intracerebral microdialysis in clinical practice: baseline values for chemical markers during wakefulness, anesthesia, and neurosurgery. Neurosurgery 2000; 47: 701–710. [DOI] [PubMed] [Google Scholar]
- 4.Amer-Wahlin I, Nord A, Bottalico B, et al. Fetal cerebral energy metabolism and electrocardiogram during experimental umbilical cord occlusion and resuscitation. J Matern Fetal Neonatal Med 2010; 23: 158–166. [DOI] [PubMed] [Google Scholar]
- 5.Nielsen TH, Olsen NV, Toft P, et al. Cerebral energy metabolism during mitochondrial dysfunction induced by cyanide in piglets. Acta Anaesthesiol Scand 2013; 57: 793–801. [DOI] [PubMed] [Google Scholar]
- 6.Reinstrup P, Nordström CH. Prostacyclin infusion may prevent secondary damage in pericontusional brain tissue. Neurocrit Care 2011; 14: 441–446. [DOI] [PubMed] [Google Scholar]
- 7.Engström M, Polito A, Reinstrup P, et al. Intracerebral microdialysis in severe brain trauma: the importance of catheter location. J Neurosurg 2005; 102: 460–469. [DOI] [PubMed] [Google Scholar]
- 8.Ederoth P, Tunblad K, Bouw R, et al. Blood-brain barrier transport of morphine in patients with severe brain trauma. Br J Clin Pharmacol 2004; 57: 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tholance Y, Barcelos G, Quadrio I, et al. Analytical validation of microdialysis analyzer for monitoring glucose, lactate and pyruvate in cerebral microdialysates. Clin Chim Acta 2011; 412: 647–654. [DOI] [PubMed] [Google Scholar]
- 10.Glenn TC, Kelly DF, Boscardin WJ, et al. Energy Dysfunction as a Predictor of Outcome After Moderate or Severe Head Injury: Indices of Oxygen, Glucose, and Lactate Metabolism. J Cereb Blood Flow Metab 2003; 23: 1239–1250. [DOI] [PubMed] [Google Scholar]
- 11.Jalloh I, Helmy A, Shannon RJ, et al. Lactate uptake by the injured human brain: evidence from an arteriovenous gradient and cerebral microdialysis study. J Neurotrauma 2013; 30: 2031–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nordström CH, Nielsen T, Nielsen H. Lactate uptake against a concentration gradient: misinterpretation of analytical imprecision. J Neurotrauma 2014; 31: 1528. [DOI] [PubMed] [Google Scholar]
- 13.Hutchinson P, Jalloh I, Helmy A, et al. Consensus statement from the 2014 international microdialysis forum. Intensive Care Med 2015; 41: 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nielsen TH, Schalen W, Stahl N, et al. Bedside diagnosis of mitochondrial dysfunction after malignant middle cerebral artery infarction. Neurocrit Care 2014; 21: 35–42. [DOI] [PubMed] [Google Scholar]
- 15.Agardh CD, Folbergrova J, Siesjo BK. Cerebral metabolic changes in profound, insulin-induced hypoglycemia, and in the recovery period following glucose administration. J Neurochem 1978; 31: 1135–1142. [DOI] [PubMed] [Google Scholar]
- 16.Patet C, Quintard H, Suys T, et al. Neuroenergetic response to prolonged cerebral glucose depletion after severe brain injury and the role of lactate. J Neurotrauma 2015; 32: 1560–1566. [DOI] [PubMed] [Google Scholar]
- 17.Quintard H, Patet C, Zerlauth JB, et al. Improvement of neuroenergetics by hypertonic lactate therapy in patients with traumatic brain injury is dependent on baseline cerebral lactate/pyruvate ratio. J Neurotrauma 2016; 33: 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Syková E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev 2008; 88: 1277–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehmenkühler A, Syková E, Svoboda J, et al. Extracellular space parameters in the rat neocortex and subcortical white matter during postnatal development determined by diffusion analysis. Neuroscience 1993; 55: 339–351. [DOI] [PubMed] [Google Scholar]
- 20.Dienel GA, Cruz NF. Aerobic glycolysis during brain activation: adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J Neurochem 2016; 138: 14–52. [DOI] [PubMed] [Google Scholar]
- 21.Hyder F, Herman P, Bailey CJ, et al. Uniform distributions of glucose oxidation and oxygen extraction in gray matter of normal human brain: no evidence of regional differences of aerobic glycolysis. J Cereb Blood Flow Metab 2016; 36: 903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothman DL, De Feyter HM, de Graaf RA, et al. 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed 2011; 24: 943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holden JE, Mori K, Dienel GA, et al. Modeling the dependence of hexose distribution volumes in brain on plasma glucose concentration: implications for estimation of the local 2-deoxyglucose lumped constant. J Cereb Blood Flow Metab 1991; 11: 171–182. [DOI] [PubMed] [Google Scholar]
- 24.Dienel GA, Cruz NF, Mori K, et al. Direct measurement of the lambda of the lumped constant of the deoxyglucose method in rat brain: determination of lambda and lumped constant from tissue glucose concentration or equilibrium brain/plasma distribution ratio for methylglucose. J Cereb Blood Flow Metab 1991; 11: 25–34. [DOI] [PubMed] [Google Scholar]
- 25.Timofeev I, Czosnyka M, Carpenter KL, et al. Interaction between brain chemistry and physiology after traumatic brain injury: impact of autoregulation and microdialysis catheter location. J Neurotrauma 2011; 28: 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cruz NF, Adachi K, Dienel GA. Rapid efflux of lactate from cerebral cortex during K+ -induced spreading cortical depression. J Cereb Blood Flow Metab 1999; 19: 380–392. [DOI] [PubMed] [Google Scholar]
- 27.Adachi K, Cruz NF, Sokoloff L, et al. Labeling of metabolic pools by [6-14C]glucose during K(+)-induced stimulation of glucose utilization in rat brain. J Cereb Blood Flow Metab 1995; 15: 97–110. [DOI] [PubMed] [Google Scholar]
- 28.Heiss WD, Pawlik G, Herholz K, et al. Regional kinetic constants and cerebral metabolic rate for glucose in normal human volunteers determined by dynamic positron emission tomography of [18F]-2-fluoro-2-deoxy-D-glucose. J Cereb Blood Flow Metab 1984; 4: 212–223. [DOI] [PubMed] [Google Scholar]
- 29.Sokoloff L, Reivich M, Kennedy C, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 1977; 28: 897–916. [DOI] [PubMed] [Google Scholar]
- 30.Gjedde A, Diemer NH. Autoradiographic determination of regional brain glucose content. J Cereb Blood Flow Metab 1983; 3: 303–310. [DOI] [PubMed] [Google Scholar]
- 31.Nakanishi H, Cruz NF, Adachi K, et al. Influence of glucose supply and demand on determination of brain glucose content with labeled methylglucose. J Cereb Blood Flow Metab 1996; 16: 439–449. [DOI] [PubMed] [Google Scholar]
- 32.Folbergrová J, Lowry OH, Passonneau JV. Changes in metabolites of the energy reserves in individual layers of mouse cerebral cortex and subjacent white matter during ischaemia and anaesthesia. J Neurochem 1970; 17: 1155–1162. [DOI] [PubMed] [Google Scholar]
- 33.Folbergrová J, Passonneau JV, Lowry OH, Schulz DW. Glycogen, ammonia, and related metabolites in the brain during seizures evoked by methionine sulphoximine. J Neurochem 1969; 16: 191–203. [DOI] [PubMed] [Google Scholar]
- 34.Gatfield PD, Lowry OH, Schulz DW, et al. Regional energy reserves in mouse brain and changes with ischaemia and anaesthesia. J Neurochem 1966; 13: 185–195. [DOI] [PubMed] [Google Scholar]
- 35.de Graaf RA, Pan JW, Telang F, et al. Differentiation of glucose transport in human brain gray and white matter. J Cereb Blood Flow Metab 2001; 21: 483–492. [DOI] [PubMed] [Google Scholar]
- 36.Wise BL, Chater N. The value of hypertonic mannitol solution in decreasing brain mass and lowering cerebro-spinal-fluid pressure. J Neurosurg 1962; 19: 1038–1043. [DOI] [PubMed] [Google Scholar]
- 37.Marko NF. Hypertonic saline, not mannitol, should be considered gold-standard medical therapy for intracranial hypertension. Crit Care 2012; 16: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mortazavi MM, Romeo AK, Deep A, et al. Hypertonic saline for treating raised intracranial pressure: literature review with meta-analysis. J Neurosurg 2012; 116: 210–221. [DOI] [PubMed] [Google Scholar]
- 39.Diringer MN. New trends in hyperosmolar therapy? Curr Opin Crit Care 2013; 19: 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ichai C, Armando G, Orban JC, et al. Sodium lactate versus mannitol in the treatment of intracranial hypertensive episodes in severe traumatic brain-injured patients. Intensive Care Med 2009; 35: 471–479. [DOI] [PubMed] [Google Scholar]
- 41.Ichai C, Payen JF, Orban JC, et al. Half-molar sodium lactate infusion to prevent intracranial hypertensive episodes in severe traumatic brain injured patients: a randomized controlled trial. Intensive Care Med 2013; 39: 1413–1422. [DOI] [PubMed] [Google Scholar]
- 42.Bouzat P, Sala N, Suys T, et al. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med 2014; 40: 412–421. [DOI] [PubMed] [Google Scholar]
- 43.Helbok R, Kurtz P, Schmidt JM, et al. Effect of mannitol on brain metabolism and tissue oxygenation in severe haemorrhagic stroke. J Neurol Neurosurg Psychiatry 2011; 82: 378–383. [DOI] [PubMed] [Google Scholar]
- 44.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 1994; 91: 10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chih CP, Roberts EL., Jr Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J Cereb Blood Flow Metab 2003; 23: 1263–1281. [DOI] [PubMed] [Google Scholar]
- 46.Dienel GA. Lactate shuttling and lactate use as fuel after traumatic brain injury: metabolic considerations. J Cereb Blood Flow Metab 2014; 34: 1736–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dienel GA. Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab 2012; 32: 1107–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel AB, Lai JC, Chowdhury GM, et al. Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc Natl Acad Sci U S A 2014; 111: 5385–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mangia S, DiNuzzo M, Giove F, et al. Response to ‘comment on recent modeling studies of astrocyte-neuron metabolic interactions’: much ado about nothing. J Cereb Blood Flow Metab 2011; 31: 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DiNuzzo M, Mangia S, Maraviglia B, et al. Changes in glucose uptake rather than lactate shuttle take center stage in subserving neuroenergetics: evidence from mathematical modeling. J Cereb Blood Flow Metab 2010; 30: 586–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mangia S, Simpson IA, Vannucci SJ, et al. The in vivo neuron-to-astrocyte lactate shuttle in human brain: evidence from modeling of measured lactate levels during visual stimulation. J Neurochem 2009; 109(Suppl 1): 55–62. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Hall G, Stromstad M, Rasmussen P, et al. Blood lactate is an important energy source for the human brain. J Cereb Blood Flow Metab 2009; 29: 1121–1129. [DOI] [PubMed] [Google Scholar]
- 53.Quistorff B, Secher NH, Van Lieshout JJ. Lactate fuels the human brain during exercise. FASEB J 2008; 22: 3443–3449. [DOI] [PubMed] [Google Scholar]
- 54.Boumezbeur F, Petersen KF, Cline GW, et al. The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J Neurosci 2010; 30: 13983–13991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cox DW, Bachelard HS. Partial attenuation of dentate granule cell evoked activity by the alternative substrates, lactate and pyruvate: evidence for a postsynaptic action. Exp Brain Res 1988; 69: 368–372. [DOI] [PubMed] [Google Scholar]
- 56.Bak LK, Obel LF, Walls AB, et al. Novel model of neuronal bioenergetics: post-synaptic utilization of glucose but not lactate correlates positively with Ca2+ signaling in cultured mouse glutamatergic neurons. ASN Neuro 2012; 4: 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ivanov AI, Malkov AE, Waseem T, et al. Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J Cereb Blood Flow Metab 2014; 34: 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galow LV, Schneider J, Lewen A, Ta TT, Papageorgiou IE, Kann O. Energy substrates that fuel fast neuronal network oscillations. Front Neurosci 2014; 8: 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okada Y, Lipton P. Glucose, oxidative energy metabolism, and neural function in brain slices – glycolysis plays a key role in neural activity. In: Gibson GE, Dienel GA. (eds). Brain energetics integration of molecular and cellular processes, 3 ed Berlin: Springer-Verlag, 2007, pp. 17–39. [Google Scholar]
- 60.Soustiel JF, Glenn TC, Shik V, et al. Monitoring of cerebral blood flow and metabolism in traumatic brain injury. J Neurotrauma 2005; 22: 955–965. [DOI] [PubMed] [Google Scholar]
- 61.Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab 2005; 25: 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glenn TC, Martin NA, Horning MA, et al. Lactate: Brain fuel in human traumatic brain injury: a comparison with normal healthy control subjects. J Neurotrauma 2015; 32: 820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hutchinson PJ, O'Connell MT, Seal A, et al. A combined microdialysis and FDG-PET study of glucose metabolism in head injury. Acta Neurochir (Wien) 2009; 151: 51–61. discussion 61. [DOI] [PubMed] [Google Scholar]
- 64.Seifert T, Secher NH. Sympathetic influence on cerebral blood flow and metabolism during exercise in humans. Prog Neurobiol 2011; 95: 406–426. [DOI] [PubMed] [Google Scholar]
- 65.Oddo M, Levine JM, Frangos S, et al. Brain lactate metabolism in humans with subarachnoid hemorrhage. Stroke 2012; 43: 1418–1421. [DOI] [PubMed] [Google Scholar]
- 66.Madsen PL, Linde R, Hasselbalch SG, et al. Activation-induced resetting of cerebral oxygen and glucose uptake in the rat. J Cereb Blood Flow Metab 1998; 18: 742–748. [DOI] [PubMed] [Google Scholar]
- 67.Madsen PL, Cruz NF, Sokoloff L, et al. Cerebral oxygen/glucose ratio is low during sensory stimulation and rises above normal during recovery: excess glucose consumption during stimulation is not accounted for by lactate efflux from or accumulation in brain tissue. J Cereb Blood Flow Metab 1999; 19: 393–400. [DOI] [PubMed] [Google Scholar]
- 68.Dienel GA, Ball KK, Cruz NF. A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: implications for glycogen turnover. J Neurochem 2007; 102: 466–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li B, Freeman RD. Neurometabolic coupling between neural activity, glucose, and lactate in activated visual cortex. J Neurochem 2015; 135: 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dahlgren N, Rosén I, Sakabe T, et al. Cerebral functional, metabolic and circulatory effects of intravenous infusion of adrenaline in the rat. Brain Res 1980; 184: 143–152. [DOI] [PubMed] [Google Scholar]
- 71.Börgstrom L, Chapman AG, Siesjö BK. Glucose consumption in the cerebral cortex of rat during bicuculline-induced status epilipticus. J Neurochem 1976; 27: 971–973. [DOI] [PubMed] [Google Scholar]
- 72.Chapman AG, Meldrum BS, Siesjö BK. Cerebral metabolic changes during prolonged epileptic seizures in rats. J Neurochem 1977; 28: 1025–1035. [DOI] [PubMed] [Google Scholar]
- 73.Dienel GA. Fueling and imaging brain activation. ASN Neuro 2012; 4: 267–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ball KK, Cruz NF, Mrak RE, et al. Trafficking of glucose, lactate, and amyloid-beta from the inferior colliculus through perivascular routes. J Cereb Blood Flow Metab 2010; 30: 162–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leloup C. An integrated energy plan for activated neurons. J Neurochem 2015; 135: 639–642. [DOI] [PubMed] [Google Scholar]
- 76.Chugani HT, Hovda DA, Villablanca JR, et al. Metabolic maturation of the brain: a study of local cerebral glucose utilization in the developing cat. J Cereb Blood Flow Metab 1991; 11: 35–47. [DOI] [PubMed] [Google Scholar]
- 77.Cavazzuti M, Porro CA, Barbieri A, et al. Brain and spinal cord metabolic activity during propofol anaesthesia. Br J Anaesth 1991; 66: 490–495. [DOI] [PubMed] [Google Scholar]
- 78.Dam MDM, Ori MDC, Pizzolato MDG, et al. The effects of propofol anesthesia on local cerebral glucose utilization in the rat. Anesthesiol 1990; 73: 499–505. [DOI] [PubMed] [Google Scholar]
- 79.Hu Y, Wilson GS. A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J Neurochem 1997; 69: 1484–1490. [DOI] [PubMed] [Google Scholar]
- 80.Hu Y, Wilson GS. Rapid changes in local extracellular rat brain glucose observed with an in vivo glucose sensor. J Neurochem 1997; 68: 1745–1752. [DOI] [PubMed] [Google Scholar]
- 81.Mintun MA, Vlassenko AG, Shulman GL, et al. Time-related increase of oxygen utilization in continuously activated human visual cortex. Neuroimage 2002; 16: 531–537. [DOI] [PubMed] [Google Scholar]
- 82.Prichard J, Rothman D, Novotny E, et al. Lactate rise detected by 1H NMR in human visual cortex during physiologic stimulation. Proc Natl Acad Sci U S A 1991; 88: 5829–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen W, Novotny EJ, Zhu XH, et al. Localized 1H NMR measurement of glucose consumption in the human brain during visual stimulation. Proc Natl Acad Sci U S A 1993; 90: 9896–9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mangia S, Tkac I, Gruetter R, et al. Sustained neuronal activation raises oxidative metabolism to a new steady-state level: evidence from 1H NMR spectroscopy in the human visual cortex. J Cereb Blood Flow Metab 2007; 27: 1055–1063. [DOI] [PubMed] [Google Scholar]
- 85.Frahm J, Kruger G, Merboldt KD, et al. Dynamic uncoupling and recoupling of perfusion and oxidative metabolism during focal brain activation in man. Magn Reson Med 1996; 35: 143–148. [DOI] [PubMed] [Google Scholar]
- 86.Schaller B, Xin L, O'Brien K, et al. Are glutamate and lactate increases ubiquitous to physiological activation? A (1)H functional MR spectroscopy study during motor activation in human brain at 7Tesla. Neuroimage 2014; 93(Pt 1): 138–145. [DOI] [PubMed] [Google Scholar]
- 87.Carpenter KL, Jalloh I, Hutchinson PJ. Glycolysis and the significance of lactate in traumatic brain injury. Front Neurosci 2015; 9: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gallagher CN, Carpenter KL, Grice P, et al. The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain 2009; 132: 2839–2849. [DOI] [PubMed] [Google Scholar]
- 89.Patet C, Suys T, Carteron L, et al. Cerebral lactate metabolism after traumatic brain injury. Curr Neurol Neurosci Rep 2016; 16: 1–7. [DOI] [PubMed] [Google Scholar]
- 90.Osborne D, Pearson-Leary J, McNay E. The neuroenergetics of stress hormones in the hippocampus and implications for memory. Front Neurosci 2015; 9: 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol 2003; 206: 2049–2057. [DOI] [PubMed] [Google Scholar]
- 92.Feuerstein D, Backes H, Gramer M, et al. Regulation of cerebral metabolism during cortical spreading depression. J Cereb Blood Flow Metab. 2016; 36: 1965–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Veech RL. The metabolism of lactate. NMR Biomed 1991; 4: 53–58. [DOI] [PubMed] [Google Scholar]
- 94.Nordström CH, Nielsen TH. Exogenous lactate supplementation to the injured brain: misleading conclusions with clinical implications. Intensive Care Med 2014; 40: 919. [DOI] [PubMed] [Google Scholar]
- 95.Nedergaard M, Goldman SA. Carrier-mediated transport of lactic acid in cultured neurons and astrocytes. Am J Physiol 1993; 265: R282–R289. [DOI] [PubMed] [Google Scholar]
- 96.Trivedi B, Danforth WH. Effect of pH on the kinetics of frog muscle phosphofructokinase. J Biol Chem 1966; 241: 4110–4112. [PubMed] [Google Scholar]
- 97.Halperin ML, Connors HP, Relman AS, et al. Factors that control the effect of pH on glycolysis in leukocytes. J Biol Chem 1969; 244: 384–390. [PubMed] [Google Scholar]
- 98.Müller MS. Functional impact of glycogen degradation on astrocytic signalling. Biochem Soc Trans 2014; 42: 1311–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hertz L, Xu J, Song D, et al. Astrocytic glycogenolysis: mechanisms and functions. Metab Brain Dis 2015; 30: 317–333. [DOI] [PubMed] [Google Scholar]
- 100.Dusick JR, Glenn TC, Lee WN, et al. Increased pentose phosphate pathway flux after clinical traumatic brain injury: a [1,2-13C2]glucose labeling study in humans. J Cereb Blood Flow Metab 2007; 27: 1593–1602. [DOI] [PubMed] [Google Scholar]
- 101.Jalloh I, Carpenter KLH, Grice P, et al. Glycolysis and the pentose phosphate pathway after human traumatic brain injury: microdialysis studies using 1,2-13C2 glucose. J Cereb Blood Flow Metab 2015; 35: 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lauritzen KH, Morland C, Puchades M, et al. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb Cortex 2014; 24: 2784–2795. [DOI] [PubMed] [Google Scholar]
- 103.Bozzo L, Puyal J, Chatton JY. Lactate modulates the activity of primary cortical neurons through a receptor-mediated pathway. PloS One 2013; 8: e71721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gilbert E, Tang JM, Ludvig N, et al. Elevated lactate suppresses neuronal firing in vivo and inhibits glucose metabolism in hippocampal slice cultures. Brain Res 2006; 1117: 213–223. [DOI] [PubMed] [Google Scholar]
- 105.Tang F, Lane S, Korsak A, et al. Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat Commun 2014; 5: 3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hein TW, Xu W, Kuo L. Dilation of retinal arterioles in response to lactate: role of nitric oxide, guanylyl cyclase, and ATP-sensitive potassium channels. Invest Ophthalmol Vis Sci 2006; 47: 693–699. [DOI] [PubMed] [Google Scholar]
- 107.Yamanishi S, Katsumura K, Kobayashi T, et al. Extracellular lactate as a dynamic vasoactive signal in the rat retinal microvasculature. Am J Physiol Heart Circ Physiol 2006; 290: H925–H934. [DOI] [PubMed] [Google Scholar]
- 108.Gordon GR, Choi HB, Rungta RL, et al. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 2008; 456: 745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Winkler U, Hirrlinger J. Crosstalk of signaling and metabolism mediated by the NAD(+)/NADH redox state in brain cells. Neurochem Res 2015; 40: 2394–2401. [DOI] [PubMed] [Google Scholar]
- 110.Glenn TC, Martin NA, McArthur DL, et al. Endogenous Nutritive Support after Traumatic Brain Injury: Peripheral Lactate Production for Glucose Supply via Gluconeogenesis. J Neurotrauma 2015; 32: 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]