

Abstract
We examined the brains of 266 patients with prion diseases (PrionD) and found that 46 (17%) had Alzheimer disease (AD)-like changes. To explore potential mechanistic links between PrionD and AD, we exposed human brain aggregates (Hu BrnAggs) to brain homogenate from a patient with sporadic Creutzfeldt-Jakob disease (CJD) and found that the neurons in the Hu BrnAggs produced many β-amyloid (β42) inclusions, whereas uninfected, control-exposed Hu BrnAggs did not. Western blots of 20-pooled CJD-infected BrnAggs verified higher Aβ42 levels than controls. We next examined the CA1 region of the hippocampus from 14 patients with PrionD and found that 5 patients had low levels of scrapie-associated prion protein (PrPSc), many Aβ42 intraneuronal inclusions, low APOE-4, and no significant nerve cell loss. Seven patients had high levels of PrPSc, low Aβ42, high APOE-4 and 40% nerve cell loss, suggesting that APOE-4 and PrPSc together cause neuron loss in PrionD. There were also increased levels of hyperphosphorylated tau protein (Hτ) and Hτ-positive neuropil threads and neuron bodies in both PrionD and AD groups. The brains of 6 age-matched control patients without dementia did not contain Aβ42 deposits; however, there were rare Hτ-positive threads in 5 controls and 2 controls had a few Hτ-positive nerve cell bodies. We conclude that PrionD may trigger biochemical changes similar to AD and suggest that PrionD are diseases of PrPSc, Aβ42, APOE-4 and abnormal tau.
Keywords: A-β42, Alzheimer disease, Codon 129, Creutzfeldt-Jakob disease, Male:female ratio, PrPSc, Tau hyperphosprylation
INTRODUCTION
As part of collaborative studies with Dr. Stanley Prusiner on the biology and treatment of prion and other neurodegenerative diseases, we have collected and analyzed brains from 266 patients with prion diseases (PrionD), including 240 with sporadic Creutzfeldt-Jakob disease (sCJD), 19 with familial CJD, and 7 with Gerstmann-Sträussler-Scheinker disease (GSS). PrionD and Alzheimer disease (AD) may share certain pathogenic mechanisms. In 1998, Hainfellner et al reported 10.9% overlap of AD in 110 cases of CJD (1). They concluded that the coexistence of AD-type pathology with CJD most probably represented coincidental events. On the other hand, Lauren et al reported that the cellular prion protein PrPC is a high-affinity receptor for β-amyloid (Aβ42) oligomers in AD and that binding to PrPC mediates the deleterious effects of Aβ42 (2). Parkin et al showed that PrPC inhibits the β-secretase (BACE-1) cleavage of the amyloid precursor protein (βAPP), which is the first step in Aβ40 and Aβ42 formation (3). Kovacs et al reported that familial CJD associated with the E200K PRNP mutation forms a complex proteinopathy consisting of scrapie-associated prion protein (PrPSc), tau, α-synuclein, and deposition of Aβ (4). They also reported that Aβ deposits may be detected in sCJD.
Here, we investigated how the replacement of PrPC with PrPSc may promote Aβ42 production and how PrionD might induce AD-like pathological changes in some patients. First, we determined how many definite AD or early (incipient) AD cases, defined by the presence of extracellular Aβ42 neuritic plaques, occurred in our 266 cases of PrionD. Next, we wanted to know whether intracellular Aβ42 peptide is formed in neurons and glial cells in the absence of Aβ42 plaque formation in PrionD. Because abnormal hyperphosphorylated tau protein (Hτ) and Aβ42 neuritic plaques define AD, we wanted to know whether Hτ is formed in the brains of patients with PrionD. We report the concomitant occurrence of neuropathologic findings consistent with PrionD and AD, which suggest that Aβ peptides and abnormal tau are induced in PrionD. For controls we used 6 age-matched patients with no neurological diseases. Other control data were obtained from the literature and included reports of examinations of the brains of a large number of non-demented patients (5–9).
To test the interaction of PrPC, PrPSc, Aβ42 and abnormal tau we used an in vitro cell model of brain aggregates (BrnAggs). After 20 days in culture, these BrnAggs contain mature neurons with dendrites, axons and synapses, as well as other cellular CNS elements including astrocytes, oligodendrocytes and microglia. In a previous study, we showed that the susceptibility of BrnAggs to prions was similar to in vivo disease in mice and that BrnAgg provided a clear view of the distribution of PrPSc in the plasma membrane and subcellular structures (10).
MATERIALS AND METHODS
Patients
We examined 266 consecutive cases of PrionD in the Neuropathology Research Laboratory of the Department of Pathology at UCSF. We also studied 127 AD cases from the files of the Neuropathology Unit. As controls, we examined the brains from 6 (3 males, 3 females) age-matched (age range of 54 to 69 years), non-demented individuals.
Estimates of Gross Brain Abnormalities
Brains were characterized by the degree of atrophy (i.e. none, mild, moderate, or severe), based on brain weight and visual inspection of the cerebral cortex. Atrophy of the hippocampus was scored as positive if the inferior horn was dilated and the size of hippocampus was smaller than normal by visual inspection. For hydrocephalus, protrusion of the lateral ventricles 0.2 to 1.5 cm into the frontal lobe in a coronal section taken immediately rostral to the temporal poles qualified as mild hydrocephalus; extensive rounding and dilatation of the anterior horn, body, and inferior horn of the lateral ventricle and the third ventricle were considered severe. Depigmentation of the locus coeruleus and substantia nigra was also evaluated.
Histoblot Technique
Cryostat sections of brain samples were prepared from fresh frozen tissue. Coronal (full and half) sections of fresh frozen human brains were cut to 25 μm thickness with a Leica Cryopolycut microtome (now obtained from International Medical Equipment, Inc., San Marcos, CA). Routine frozen sections of brain (1–2 × 3 inches in size) were cut (10-μm thickness) with a Microm HM505 cryostat (Carl Zeiss, Thornwood, NY) and pressed onto nitrocellulose membrane. The resulting histoblots were treated with proteinase K to eliminate PrPC, and then with 3 M guanidinium to denature PrPSc for immunohistochemical staining of protease-resistant PrPSc, which is diagnostic of PrionD (11, 12). The same technique, without the denaturation step, was used to stain for protease-resistant Aβ peptides that are characteristic of AD (13). The histological diagnoses of incipient and definite AD were established by semiquantitative estimate of the number of neocortical Aβ42 neuritic plaques using The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria (14).
Neurohistology and Molecular Biology
Brain samples were fixed in 10% buffered formalin and embedded in paraffin using standard procedures. The degree of vacuolation as a percentage of the gray matter area, pattern, and laminar distribution of vacuolation (assessed with hematoxylin and eosin stain) were estimated. Hematoxylin and eosin-stained tissue was also evaluated for the presence of other lesions such as infarcts and Lewy bodies. Fresh frozen and formalin-fixed tissues were sent to the National Prion Disease Surveillance Center for genetic testing of PRNP gene mutations and the codon-129 polymorphism, as well as for Western blot determination of the PrPSc phenotypes.
Immunohistochemistry of Human Brains
Brain sections were cut (8-μm thickness), deparaffinized, and processed for immunohistochemistry. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol. Non-specific antibody binding sites were blocked with 10% normal horse serum. For the detection of human PrPSc, we used mouse 3F4 Ab (1:1000) and HuM-P Ab conjugated to horseradish peroxidase (HRP) (1:10,000) (gifts from Dr. Stanley Prusiner, UCSF, San Francisco, CA). The intensity of PrPSc immunostaining was scored semiquantitatively as mild (1+), moderate (2+), moderately severe (3+), or severe (4+).
Abnormal Hτ tau was detected with the mouse AT8 antibody (1:250), which is specific for phosphorylated, paired-helical filamentous tau Ser202/Thr205 (Life Technologies, Cat # MN1020B, Carlsbad, CA). Aβ42 was stained with the 4G8 monoclonal antibody (1:500) specific for the Aβ42 at epitope 12–24 (BioLegend Co., formerly Covance antibody products, Cat #SIG-39220, San Diego, CA). Cortical Lewy bodies were detected with the mouse monoclonal α-synuclein antibody (1:250) (LB509, Cat # ab 27766, Abcam, Cambridge, MA). Sections to be stained with the 3F4 and AT8 antibodies were subjected to hydrolytic autoclaving for 10 minutes at 121°C in citrate buffer. Sections to be stained with 4G8 were immersed in formic acid for 5 minutes. Tissue stained with α-synuclein antibodies were not pretreated. All sections were incubated with primary antibodies overnight at room temperature and with biotinylated horse anti-mouse secondary antibody (1:200) (Cat # BA-2000, Vector Laboratories, Inc., Burlingame, CA), for 30 minutes at room temperature. Antibody binding was detected using a Vectastain ABC kit (PK-4000) and visualized with 3,3′-diaminobenzidine (Vector Laboratories, DAB peroxidase substrate kit SK-4100).
Scoring Cortical Hτ
Hτ immunopositivity in the locus coeruleus/raphe nuclei (LC/RN) was scored from 0 to 4+ in nerve cell bodies, neuropil threads, dystrophic neurites, and then multiplied by the total area of the LC/RN involved. The total Hτ load in the mesial temporal lobe (MTL) was estimated by multiplying the Hτ score by the extent of the affected layers in cortex (×1 if the Hτ was found in 2–3 layers; ×2 if Hτ was found in all 6 layers). The final scores were multiplied by the number of regions of the MTL in which Hτ was found, which varied from 1 to 6 depending on the number of affected regions (the hippocampus, subiculum, presubiculum, entorhinal cortex, transentorhinal cortex, and the inferior temporal cortex).
Brain Aggregates
Mouse (Mo) brain aggregates (BrnAggs) were prepared from E15 day gestation embryos obtained from pregnant Tg(tau-P301L)4510 mice (gift from Dr. George Carlson, McLaughlin Research Institute, Great Falls, MT). The fetal brains were genotyped by PCR for the presence of tau(P301L) and calmodulin kinase II promoter system used to construct the transgenic mice (15). Human fetal brain tissues at 15 to 17 weeks gestation were obtained from Advanced Bioscience Resources, Inc. (Alameda, CA) and used to prepare the Hu BrnAggs cell cultures. All experiments involving human brain tissue were done in Biohazard Safety Level 3 in accordance to our Biological Use Authorization.
BrnAggs were prepared as previously described (10). Briefly, Mo and Hu brain cells were dissociated through two nylon meshes. Following 2 washes with DMEM H21 containing glucose (12 g/L), fungizone (2.5 mg/L) and gentamicin (50 mg/L), the dissociated neural cells were resuspended in a modified growth medium DMEM H21 containing glucose (6 g/L), gentamicin (50 mg/L), fungizone (2.5 mg/L), and 10% fetal bovine serum at a density of 1 × 107 cells per ml. Four ml of cells were placed in 25 ml Delong flasks and kept at constant rotation (37°C, 10 % CO2). The next day, 1 ml of exchange medium consisting of DMEM H21 supplemented with 6 g/L glucose, 50 mg/L gentamicin, and 15% fetal bovine serum was added to the flask. After 2 to 3 days, the rotated BrnAggs were transferred to 50 ml Delong flasks to which 5 ml of exchange medium was added for a total of 10 ml. Exchange medium was refreshed every 2 to 3 days by removing 5 ml of conditioned medium and replacing it with 5 ml of fresh exchange medium. For Mo BrnAggs, after 6 to 8 days in culture, the growing and fusing BrnAggs were transferred to a 24-well culture plate, which was also maintained in constant rotation. For Hu BrnAggs, this process was done at approximately 12 to 14 days. The medium was changed every 2 to 3 days by discarding 500 μl of conditioned medium and replacing it with an equal volume of exchange medium.
Exposure of BrnAggs to Prions
After 15 days in culture, Mo BrnAggs were exposed for 10 days to a 1:50 dilution of RML prions derived from scrapie-infected CD1 mouse brains. After 41 days in culture, Mo BrnAggs were harvested and analyzed by immunohistochemistry. The paraffin-embedded sections were stained for phosphorylated tau with either mouse anti-tau13 antibody, 1:500 dilution (Abcam, Cat# ab24634), or mouse anti-tau CP13 antibody diluted 1:250 (gift from Dr. Peter Davies, Albert Einstein College of Medicine of Yeshiva University, New York, NY). The secondary antibody, goat anti-mouse 488 Alexa Fluor, 1:200 dilution (Cat# A11029, Life Technologies, Carlsbad, CA). Three to 4 stacks of approximately 40 1-μm-thick serial sections were captured by confocal microscopy (Zeiss, LSM 510, Jena, Germany) or whole BrnAggs were examined throughout with a 40× lens by a fluorescence microscope (Leica, DM IRB, Wetzlar, Germany).
For the experiments with Hu BrnAggs, the cells were exposed to 1:50 dilution of a human brain homogenate containing either sCJD(VV2) made from thalamus of sCJD(VV2) case or normal brain (gift from Dr. William Seeley, UCSF Memory and Aging Center, San Francisco, CA) after 2 days in culture. Treatments were continued to 20 days in culture. Hu BrnAggs were harvested after 35 days. 20 BrnAggs were pooled for Western blot analysis and analyzed for PrPSc by Western blot using the anti-PrP HuM-P-HRP (1:10,000) antibody; for Aβ1-42 (Aβ42)-specific antibody diluted 1:500 (BioLegend Co., formerly Covance antibodies, SIG-39142, San Diego, CA); and the anti-tau oligomeric antibody diluted 1:1000 (Millipore, ABN454, Temecula, CA) overnight at room temperature.
To detect Aβ42, 5 Hu BrnAggs exposed to sCJD prions and 2 control BrnAggs not exposed to prions were fixed with 4% paraformaldehyde for 1 hour. The BrnAggs were then embedded in paraffin, cut at 8 microns, and mounted on glass slides. After deparaffinization, they were submerged in formic acid for 5 minutes for antigen retrieval and incubated with Aβ1-42 (Aβ42)-specific antibody diluted 1:500 overnight at room temperature. The secondary antibody, Alexafluor goat anti-mouse 488 (Life Technologies, A11029) conjugated to FITC, was applied for 2 hours. The slides were cover-slipped in mounting medium containing DAPI (Vector Laboratories, H-1200), analyzed, and photographed with a Leica DM IRB fluorescence microscope.
Quantification of Western Blots
Levela of Aβ were quantified with BioQuant Life Science software (Bioquant Image Analysis Corporation, Nashville, TN).
Statistical Analysis
Statistical analysis of human brain weights and mouse BrnAgg studies was assessed by the Student t-test. The χ2-squared test was used to compare the distribution of deaths by age group, and the prevalence of PrionD and AD. One-way Analysis of Variance (ANOVA) with multiple comparisons was used to compare the mean ages in the PrionD only, the AD only, and the PrionD+AD groups.
RESULTS
Overlap of PrionD and AZ
For CERAD staging of AD, we examined multiple brain regions including the hippocampus, entorhinal cortex, transentorhinal cortex, inferior temporal cortex, lateral frontal cortex areas 45–46, lateral parietal cortex areas 39–40, cingulate cortex area 24, and medial occipital cortex (14, 17). Of the 266 PrionD cases reviewed for the study, 46 (17%) contained both PrPSc and extracellular Aβ42 plaques and were designated as the “Prion-AD” group (Tables 1, 2). This group included 41 sCJD, 4 familial CJD, and 1 GSS cases. In 18 of the 46 Prion-AD cases, sufficient numbers of Aβ42 plaques were detected in all cerebral cortical and some subcortical regions to qualify for “definite” AD by CERAD criteria. In the remaining 28 Prion-AD cases, the numbers of Aβ42 plaques were insufficient to make a diagnosis of definite AD: therefore, we designated them as “incipient AD.” The 220 cases with PrPSc but no detectable Aβ42 plaques were designated as “Prion-only.”
Table 1.
Prion-Alzheimer Disease Patients
| Case No. | Age (y) | Sex | Diagnosis | Codon 129 | PrP mutation | PrP type | CERAD | Brain weight (g) |
|---|---|---|---|---|---|---|---|---|
| Study I: Diagnosis based on Bielschowsky preparation, and PrPSc and Aβ42 histoblots | ||||||||
| 1 | nd | nd | GSS | MV | Q217R | Type 1 | Incipient AD | nd |
| 2 | 71 | M | fCJD | MV | A117V | Type 1 | Def AD | 1120 |
| 3 | 68 | F | fCJD | MM | E200K | Type 1 | Incipient AD | nd |
| 4 | 62 | M | fCJD | MM | R208H | Type 1 | Incipient AD | nd |
| 5 | 69 | M | sCJD | MM | — | Type 1 | Incipient AD | 1190 |
| 6 | 60 | M | sCJD | VV | — | nd | Incipient AD | nd |
| 7 | 66 | F | sCJD | MV | — | Type 1–2 | Incipient AD | 1100 |
| 8 | 81 | F | sCJD | VV | — | Type 2 | Incipient AD | 1290 |
| 9 | 65 | F | sCJD | MM | — | Type 1 | Incipient AD | nd |
| 10 | 75 | F | sCJD | VV | — | Type 2 | Incipient AD | 1100 |
| 11 | 83 | F | sCJD | MM | — | Type 1 | Incipient AD | nd |
| 12 | 73 | F | sCJD | VV | — | Type 2 | Incipient AD | nd |
| 13 | 53 | F | sCJD | nd | nd | nd | Incipient AD | nd |
| 14 | 76 | F | sCJD | MV | silent 117 | Type 1 | Incipient AD | 1290 |
| 15 | 70 | NA | sCJD | MM | — | nd | Def AD | 1160 |
| 16 | 74 | F | sCJD | MM | — | Type 1 | Incipient AD | nd |
| 17 | 62 | F | sCJD | MM | — | Type 1 | Incipient AD | 1250 |
| 18 | 77 | F | sCJD | nd | nd | nd | Incipient AD | nd |
| 19 | 73 | F | sCJD | nd | nd | nd | Incipient AD | 1125 |
| 20 | 77 | M | sCJD | MM | — | Type 1 | Incipient AD | 1375 |
| 21 | 66 | M | sCJD | nd | nd | nd | Def AD | nd |
| 22 | 60 | M | sCJD | nd | nd | nd | Def AD | nd |
| 23 | 66 | F | sCJD | nd | nd | nd | Incipient AD | nd |
| 24 | 54 | M | sCJD | Type 2 | Def AD | 1330 | ||
| 25 | 57 | F | sCJD | nd | nd | nd | Incipient AD | nd |
| 26 | 65 | F | sCJD | MM | — | Type 1 | Def AD | nd |
| 27 | 66 | M | sCJD | VV | — | Type 2 | Incipient AD | 1400 |
| 28 | 69 | F | sCJD | nd | nd | nd | Incipient AD | 1175 |
| 29 | 73 | M | sCJD | MM | — | Type 1 | Def AD | nd |
| 30 | 79 | F | sCJD | MM | — | Type 1 | Incipient AD | 1350 |
| 31 | 60 | M | sCJD | nd | nd | nd | Def AD | nd |
| 32 | 73 | F | sCJD | MM | — | Type 1–2 | Incipient AD | 1300 |
| 33 | 52 | M | sCJD | VV | — | Type 2 | Incipient AD | nd |
| 34 | 52 | F | sCJD | MM | — | Type 1 | Incipient AD | 1280 |
| 35 | 70 | M | sCJD | nd | nd | nd | Def AD | nd |
| 36 | 51 | F | fCJD | MM | 6ORI | Type 1 | Def AD | nd |
| Study II: Diagnosis based on PrPSc, Aβ42, and abnormal tau immunohistochemistry | ||||||||
| 37 | 66 | F | sCJD | MM | — | Type 1 | Def AD | 1290 |
| 38 | 75 | M | sCJD | VV | — | Type 2 | Incipient AD | 1500 |
| 39 | 64 | M | sCJD | MM | — | Type 1 | Def AD | 1675 |
| 40 | 59 | M | sCJD | MM(MV) | — | Type 1–2 | Incipient AD | 1560 |
| 41 | 60 | F | sCJD | MM | — | Type 1–2 | Def AD | 1300 |
| 42 | 75 | M | sCJD | MM | — | Type 1 | Incipient AD | 1200 |
| 43 | 82 | F | sCJD | MV | — | Type 1 | Def AD | 1250 |
| 44 | 66 | F | sCJD | __ | Incipient AD | 1290 | ||
| 45 | 79 | M | sCJD | __ | Def AD | nd | ||
| 46 | 52 | M | sCJD | __ | Def AD | 1330 | ||
Alzheimer disease (AD) was classified according Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria using the density of Aβ plaques, as either definite AD (Def AD; varying) or incipient AD (sparse to moderate).
F, female; fCJD, familial Creutzfeldt-Jakob disease; GSS, Gerstmann-Sträussler-Scheinker disease; M, male; nd, not determined; PrPsc, sCJD, sporadic Creutzfeldt-Jakob disease.
Table 2.
Clinical Data
| A. Patient information | |||
|---|---|---|---|
| Prion-only | Prion-AD | AD-only | |
| sCJD patients (n) | 197 | 41 | 0 |
| fCJD patients1 (n) | 15 | 4 | 0 |
| GSS patients2 (n) | 6 | 1 | 0 |
| Total patients (n) | 218 | 46 | 127 |
| Mean age of death (years ± SD) | 62.8 ± 10.8 | 67.2 ± 8.8 | 76.7 ± 11.2 |
| Age of onset (years ± SD) | 59.7 ± 12.2 | 67.3 ± 7.8 | 69.7 ± 14.2 |
| Duration of disease (months ± SD) | 8.7 ± 12.2 | 9.3 ± 10.5 | 87.2 ± 67.3 |
| Brain weights (g ± SD) | 1316 ± 162 n = 49 |
1287 ± 146 n = 23 |
1121 ± 121 n = 15 |
| B. The number of female vs. male patients | ||||||||
|---|---|---|---|---|---|---|---|---|
| Prion-only | Prion-AD | Prion-only + Prion-AD | AD-only | |||||
| Female | Male | Female | Male | Female | Male | Female | Male | |
| Number of Patients | 95 | 106 | 25 | 19 | 120 | 125 | 73 | 54 |
| Female to Male Ratio | 0.90 | 1.32 | 0.96 | 1.35 | ||||
Mutations found: 7 E200K, 3 V210I, 1 R208H, 3 octapeptide insertions, 2 not available.
Mutations found: 1 P102L, 2 A117V, 2 Q217R.
The number of patients in Table B differs from that in Table A because of the absence of information regarding the sex of some patients.
AD, Alzheimer disease; fCJD, familial Creutzfeldt-Jakob disease; GSS, Gerstmann-Sträussler-Scheinker disease; sCJD, sporadic Creutzfeldt-Jakob disease.
Epidemiology
In the Prion-AD group, the mean age of death was 67.2 ± 8.8 years (n = 45 available ages) (Fig. 1B; Tables 1, 2). The age of death in the Prion-AD group was significantly older than in the Prion-only cases, 62.8 ±10.8 (n = 209 available ages) (Table 2), and significantly younger than the mean age in the 127 AD-only cases seen at UCSF, which was 76.8 ± 11.1 years (p = 0.005 and p < 0.001, respectively, ANOVA controlled for multiple comparisons) (Fig. 1B; Table 2).
Figure 1.
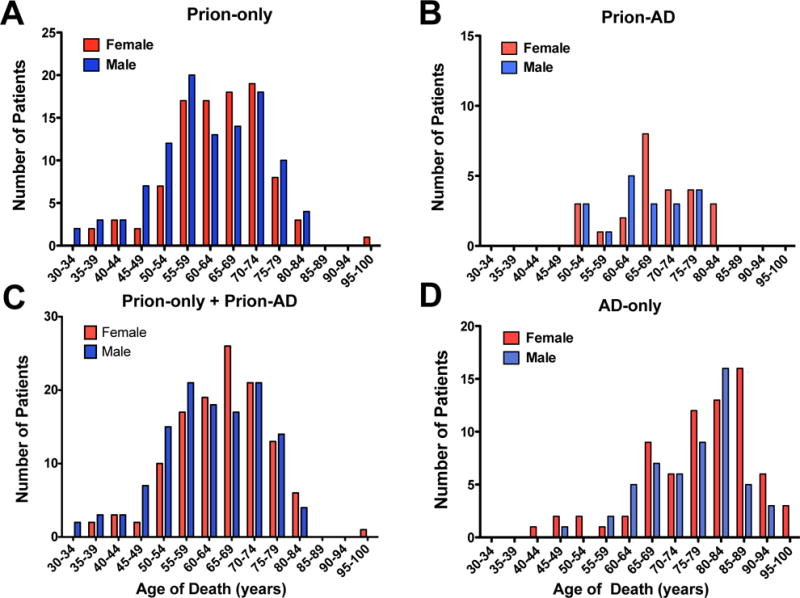
The numbers of female (red) and male (blue) patients with Prion-only, Prion-Alzheimer disease (Prion-AD), and AD-only are plotted as a function of the age of death. (A) The ages of death for the Prion-only group shows a predominance of males from 30 to 59 years of age, a predominance of females from ages of 60 to 74, and a predominance of males from 75 to 84 years. (B) The Prion-AD group has a noticeable predominance of females especially from the age of 65 to 70 and age 80. (C) Adding the number of females and males from the Prion-only group to the numbers in the Prion-AD group brings the ratio of females to males to near 1:1. Females predominate between the ages of 60 and 69 and between ages 80 to 84. Males tend to predominate at the younger ages, from 30 to 59. (D) In the AD-only group, females predominate at almost all ages.
The female to male ratio was calculated for each group because the AD literature indicates that women have an increased risk for AD (18, 19). We found that in our 127 cases of AD-only, the female to male ratio was 1.35, which is consistent with the literature (Fig. 1D; Table 2B). In 44 Prion-AD cases (the sex of 2 cases was not reported) the female to male ratio was 1.32, which was similar to AD-only cases. In contrast, the Prion-only cases had a male predominance, 106 males and 95 females, and the female to male ratio was 0.90 (Table 2B). We combined the number of females and males from the Prion-only group with the number from the Prion-AD group to test whether the predominance of men in the Prion-only cases was due to a shift of the females from the Prion-only group to the Prion-AD group. The totals, 120 females and 125 males, yielded a slight male predominance with a female to male ratio of 0.96 (Table 2B). Looking more closely at the data, in the Prion-AD group there were more females over 65 year of age and there were slightly more men between the ages of 50 to 64 than females (Fig. 1B). These data are similar to findings showing that AD is more common in older women than men.
PrPSc and Aβ42-Plaque Distribution
In the majority of patients, PrPSc was distributed either in all layers of the cortex or in the deep layers 4–6. Rarely the distribution of PrPSc was in the superficial layers 1–3. For cases in which PrPSc was deposited in the deeper cortical layers, such as an 81-year-old woman who was near the end of the age distribution for Prion-AD group (Fig. 2A), Aβ42 plaques tended to be distributed in the superficial layers 1–3 (Fig. 2B). In another case of a 53-year-old woman at the beginning of the Prion-AD age distribution, PrPSc staining was confined to layers 5–6 in the lateral frontal cortex (Fig. 2C) and Aβ42 plaques were located in layer 1 (Fig. 2D). In the parietal cortex of the same patient, PrPSc accumulated in all 6 cortical layers (Fig. 2E) and Aβ42-positive plaques were found in cortical layers 1–5 (Fig. 2F). This suggests that Aβ42-plaques are first formed in layer 1 of the cerebral cortex and subsequently progress in order from layer 2 through 6.
Figure 2.
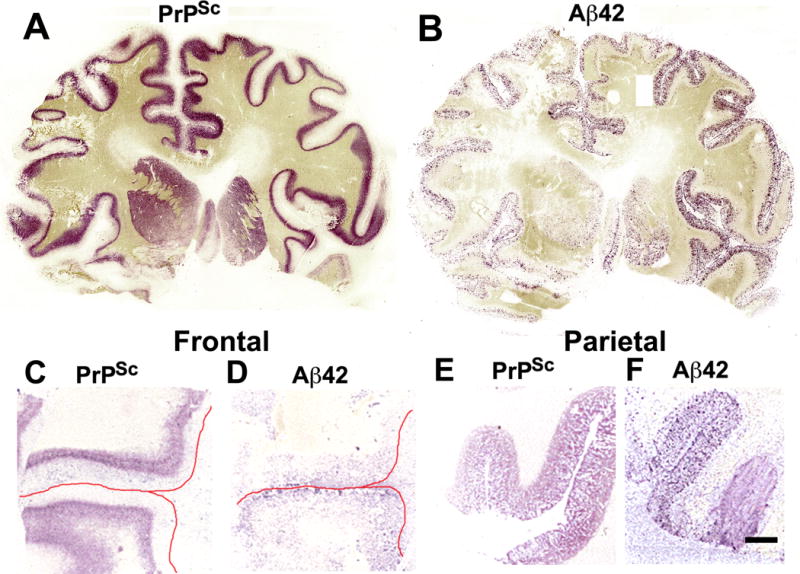
Scrapie-associated prion protein (PrPSc) and β-amyloid (Aβ42) histoblot analyses of the cerebrum in sporadic Creutzfeldt-Jakob disease (sCJD). (A, B) Full coronal sections from a sCJD case with incipient Alzheimer disease (AD) (case # 8, Table 1), not strictly fulfilling the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria for AD. The 3F4 immunostaining shows PrPSc located mostly in cortical layers 5–6 (A). The 4G8 immunostaining indicates that Aβ42 plaques are located mostly in layers 1–4 of the cerebral cortex. A few Aβ42 plaques are also located in the caudate nucleus and putamen, particularly on the right side (B). (C, D) Sections of the lateral frontal cortex in another sCJD case with incipient AD (case #13, Table 1). PrPSc also localizes to cortical layers 5–6 (C). A few Aβ42-positive plaques are located in layer 1. The red lines mark the location of the pial surface (D). (E, F) Sections of the parietal cortex of the same case shown in (C) and (D). PrPSc (E) and Aβ42 plaques (F) are located in layers 1–6. Scale bar in (F) represents 4 mm and also applies to (C–E).
Intracellular Aβ42-peptide Accumulation in Prion-only Patients Without Extracellular Aβ42 Plaques
To determine whether Aβ42 peptide was formed and accumulated in neurons in vivo in the absence of extracellular Aβ42-plaques, Aβ42 immunohistochemistry of the CA1 region of the hippocampus and the entorhinal cortex was performed on 14 recent Prion-only cases (13 sCJD and 1 GSS). Neuron counts in the CA1 were obtained by counting neurons in 5 consecutive 10× microscopic fields. We identified 3 groups of Prion-only cases (Table 3). In the first group, cases 1–5 had abundant intracellular Aβ42 in the CA1 (Fig. 3B). The neurons of the CA1 region appeared normal in size and shape, and the average number of neurons was 188 with only case number 1 showing a decrease of neurons to 131 (Fig. 3B; Table 3). Aβ42 also appeared to fill a few glial cells identified based on their small size compared to the size of neurons (Fig. 3B). In the second group, cases 6–12 had accumulation of Aβ42 in less than 5% of neurons in the CA1 region (Fig. 3D). In some of those cells (neuron or glial), Aβ42 appeared to fill the cytoplasm giving them a rounded appearance, although adjacent neurons appeared normal in size and contained only a small amount of Aβ42 (Fig. 3D). The average number of neurons was 127, which verified our visual impression of nerve cell loss (Table 3). The average of 188 neurons found in cases 1–5 and the average of 127 neurons in cases 6–12 were significantly different (T-test probability = 0.01). In the third group, cases 13–14 had low Aβ42 levels and had no nerve cell loss, showing an average of 212 neurons (Table 3). These data suggest the role of another factor in addition to Aβ42 and PrPSc determining neuron death.
Table 3.
Prion-only Cases
| Diagnosis | Sex | Age (y) | Codon 1291 | PrPSc Accumulation in CA1 Region and other neuropathologies | Aβ42 (%) | APOE-4 load | Neuron cell counts | |
|---|---|---|---|---|---|---|---|---|
| 1 | sCJD | F | 57 | MM2 | Prox CA1: Little finely granular PrPSc staining. Distal CA1: Moderate amount of finely granular PrPSc staining. | 90 | 28.0 | 131 |
| 2 | GSS | M | 49 | n.d. | CA4 to CA1: A background of dense finely granular PrPSc staining surrounds neurons and GSS plaques. | 80 | 21.0 | 217 |
| 3 | sCJD | M | 60 | MM2 | Prox CA1: Little or no PrPSc staining. Distal CA1: Moderate degree of PrPSc course deposits. | 80 | 68.0 | 200 |
| 4 | sCJD | M | 68 | MV2 | CA4 to CA1: dense finely granular PrPSc staining surrounding neurons. | 70 | 9.4 | 169 |
| 5 | sCJD | M | 57 | MV2 | Prox CA1: Very little PrPSc. Distal CA1: Moderate amount of finely granular PrPSc surrounding neurons. | 40 | 24.6 | 223 |
| 6 | sCJD | M | 74 | MV1-2 | CA1: Dense finely granular PrPSc; PrP plaques in the SR2 and SO2 and less in the SP2 surrounds nerve cells; a few nerve cell bodies and dendrites contain intracellular PrPSc. | <5 | 60.0 | 122 |
| 7 | sCJD | M | Anon. | MV1-2 | Same as case 6 with some exceptions: Dense, finely granular PrPSc and plaque-like deposits surround dendrites; PrPSc accumulation in nerve cell bodies. Distal: Severe vacuolation CA1 to the EC. | <5 | n.d. | 155 |
| 8 | sCJD | F | 66 | VV2 | All 3 cortical layers of the CA1 stain strongly for PrPSc with focally dense, finely granular PrPSc associated with neuronal cell membranes. | <5 | 35.4 | 105 |
| 9 | sCJD | M | 66 | MV1-2 | Prox CA1: No PrPSc. Distal: Moderate PrPSc in all layers if the CA1. | <5 | 55.2 | 113 |
| 10 | sCJD | F | 60 | MM2 | Prox CA1: Moderate amount of coarse PrPSc filling all 3 layers. | <5 | 56.4 | 179 |
| 11 | sCJD | F | 67 | MV1-2 | CA4 to CA1: Dense, finely granular PrPSc in all 3 layers. | <5 | 40.0 | 115 |
| 12 | sCJD | M | 72 | n.d. | CA1: Sparse amount of finely granular PrPSc in all 3 layers. | <5 | 60.0 | 100 |
| 13 | sCJD | M | 42 | n.d. | Prox CA1: No PrPSc. Distal CA1: ~15% of nerve cell bodies are filled with PrPSc. | <1 | 0.5 | 210 |
| 14 | sCJD | F | 56 | MM1 | CA4 to CA1: Little finely granular PrPSc in the SR; ~50% of CA1 neurons contain intracellular PrPSc; coarsely granular PrPSc is also scattered around dendrites; sparse in the SP. | <1 | 2.3 | 244 |
Comparison of Aβ42, APOE-4 and nerve cell loss in five 10x regions of the CA1 region of the hippocampus. The APOE-4 load is the number of APOE-4-positive neurons in the CA1 multiplied by the APOE-4 staining intensity estimated as 0-none, 1-mild, 2-moderate, and 3-severe. The t-test value for the APOE-4 loads in cases 1–5 and cases 6–12 is 0.07.
PRNP genotype and PrPSc phenotype
EC, entorhinal cortex; F, female; fCJD, familial Creutzfeldt-Jakob disease; GSS, Gerstmann-Sträussler-Scheinker disease; M, male; n.d., not determined; Prox, proximal; sCJD, sporadic Creutzfeldt-Jakob disease SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum.
Figure 3.
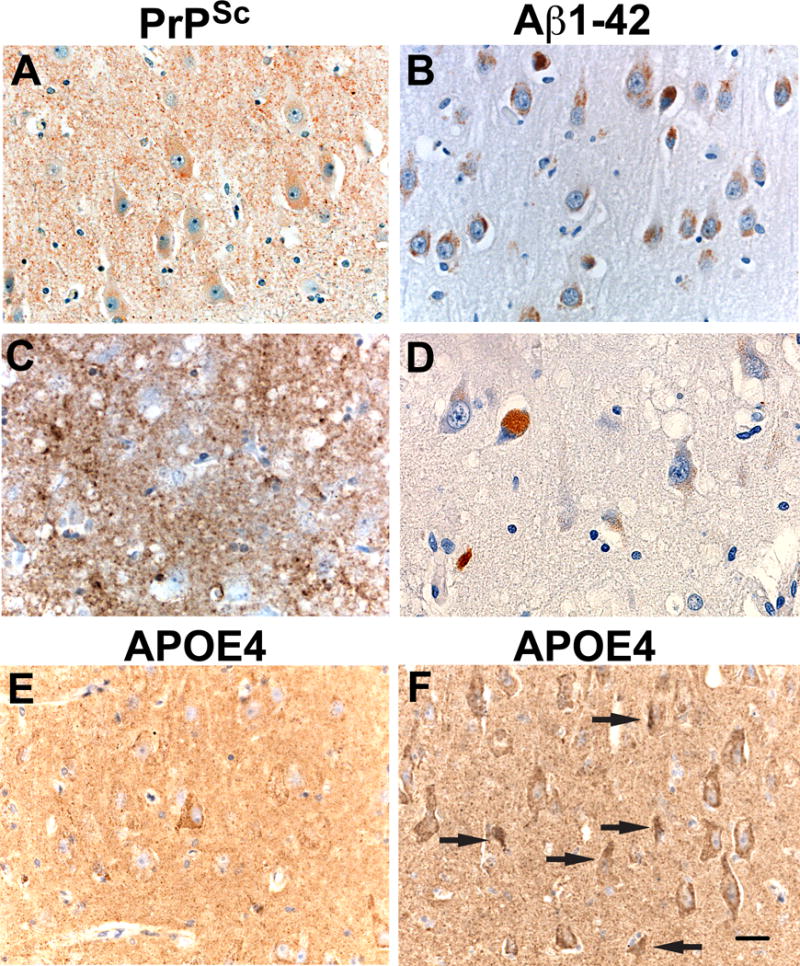
Intracellular β-amyloid (Aβ42) peptide and APOE-4 accumulation in neurons in cases of spontaneous Creutzfeldt-Jakob disease (sCJD): Two Prion-only autopsy cases: (A) Scrapie-associated prion protein (PrPSc) staining of case #1 (Table 3) shows finely granular deposits of PrPSc in the neuropil and nerve cell bodies of the CA1 region. (B) 90% of CA1 neurons containing Aβ42 cytoplasmic inclusions and 3 glial cells with intracellular Aβ42. (C) PrPSc immunohistochemistry in case #7 (Table 3) shows dense, coarse, and plaque-like PrPSc deposits filling the entire neuropil and surrounding and infiltrating neurons. (D) Aβ42 intracytoplasmic inclusions are strongly positive in a few cells that could be glial or neuronal. Three adjacent neurons contain very light brown peroxidase staining indicating Aβ42 positivity; it is admittedly difficult to differentiate the Aβ42 immunoreactivity inclusions from lipofuscin. Note there are also fewer neurons in the field compared to (B). (E) The same case (#1) as shown in (A) shows a single neuron at the center of the field that is APOE-4-immunopositive. There are a few scatted granular deposits of APOE-4 in the neuropil. (F) The same case (#7) as in (C) shows that almost all neurons in this field contain APOE-4. In addition, there are many punctate APOE-4 deposits in the neuropil, possibly resulting from degeneration of APOE-4-positive neurons. Arrows indicate apoptotic neurons. Scale bar in (F) represents 30 μm and applies to all panels.
Nerve Cell Loss Correlates with APOE-4
A factor more likely to cause nerve cell death is APOE-4. The APOE-4 allele is associated with an increased risk and earlier onset of AD; APOE-3/3 allele is associated with a significantly lower risk of AD; and APOE-2 allele is associated with a decreased risk of AD and delay of its onset (20–22). APOE is normally expressed in astrocytes but not in neurons (23). In conditions of stress, such as in mice treated with kainic acid, many hippocampal neurons express APOE-4. In another study, the expression of APOE-4 was associated with neuronal apoptosis (24).
We used APOE4-specific antibodies to stain the CA1 region in the 14 Prion-only cases (Table 3). The percentage of nerve cell bodies in the CA1 region containing Aβ42 and APOE-4 were quantified. Three markedly different results were obtained (Table 3). First, cases 1–5 had a low APOE-4 content in CA1 neurons and relatively few punctate APOE-4 deposits in the neuropil (Fig. 3E), which was associated with very little loss of CA1 neurons (Table 3). Second, cases 6–12 had a high content of intracellular neuronal APOE-4 (Fig. 3F), which was associated with a significant CA1 neuron loss and many APOE-4 deposits in the neuropil (Fig. 3F; Table 3). And third, cases 13 and 14 contained a very low amount of APOE-4 and no associated nerve cell loss (Table 3). APOE-4 accumulation in neurons resulting in apoptosis was reflected by the large numbers of shrunken neurons with degenerating nuclei (Fig. 3F arrows). We conclude that nerve cell loss was related to increased levels of APOE-4 and not to Aβ42.
sCJD(VV2) Induces Aβ in Hu BrnAggs
Hu BrnAggs were made from the brain of a 16-week human fetus, which did not contain extracellular Aβ42 plaques of AD. Starting from the second day in culture, BrnAggs were exposed either to a normal human brain homogenate or to human sCJD(VV2) brain homogenate, or not exposed to any brain substance (None). The BrnAggs were terminated at 35 days. We pooled groups of 20 Hu BrnAggs to test each condition by Western blot analysis. An increase in Aβ42 was visibly recognizable in the brain aggregates exposed to sCJD(VV2) prions but not in BrnAggs exposed to a normal brain homogenate or in the BrnAggs not exposed to any brain substance (None) (Fig. 4A). Densitometry of the Aβ-bands using BioQuant software produced density values of 542, 1506, and 450 for None, sCJD(VV2) and Normal Brain respectively (Fig. 4B), indicating that sCJD(VV2) induced a 3.35-fold increase in Aβ compared to the normal brain homogenate and a 2.78-fold increase compared to no treatment. To confirm the infection with sCJD(VV2), the samples were treated with proteinase K and subjected to Western blot analysis. PrP-specific antibodies yielded the signature 3 proteinase K resistant bands in Hu BrnAggs exposed to sCJD(VV2) but none in Hu BrnAggs exposed to the normal brain homogenate or in Hu BrnAggs not exposed to brain tissue (None) (Fig. 4A).
Figure 4.
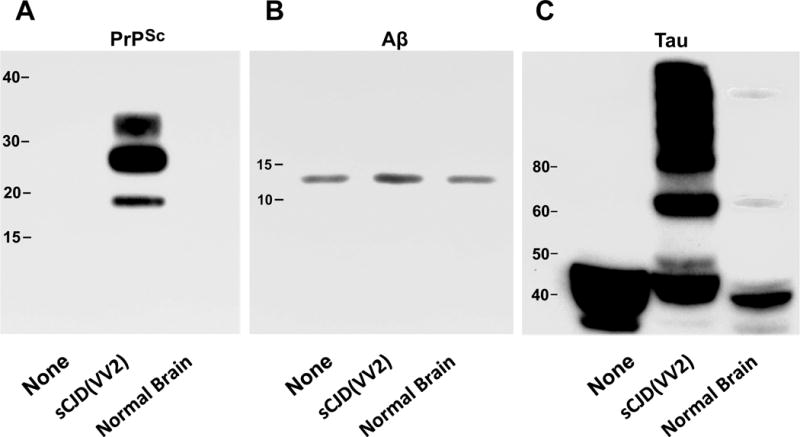
Western immunoblots stained for scrapie-associated prion protein (PrPSc), β-amyloid peptide (Aβ42), and tau protein in 20 pooled human brain aggregates (Hu BrnAggs). Each set of BrnAggs were either not treated (None), treated with a human brain homogenate from a patient with sporadic Creutzfeldt-Jakob disease (sCJD(VV2)), or treated with a normal human brain homogenate (Normal Brain). (A) All 3 of the treatments were exposed to Proteinase K to show protease resistant PrPSc. (B) Antibodies to Aβ peptide showed that sCJD(VV2) exposure resulted in an increased level of Aβ in the BrnAggs while None and Normal Brain exposure did not. (C) Antibodies to tau protein showed that sCJD(VV2) was the only treatment that caused a large amount of phosphorylated tau with molecular masses greater than 60 kDa to be generated. The Normal Brain homogenate generated very small bands at 60 kDa and greater than 100 kDa. Molecular weights are indicated in each immunoblot.
In a preliminary study, Hu BrnAggs were exposed to sCJD(VV2) prions for immunohistochemical localization of Aβ42. Prion exposure was begun on day 2 in culture and terminated at 35 days. The Hu BrnAggs were fixed with 4% paraformaldehyde. Immunostaining using anti-Aβ antibodies showed that all 5 of the Hu BrnAggs exposed to sCJD(VV2) were filled with large and small Aβ42 aggregates in nerve cell bodies and in the neuropil (data not shown because a normal age-matched Hu brain homogenate from a non-demented was not used as a control). When examined by double labeling, Aβ42 and the lysosomal marker cathepsin D, the Aβ42 deposits appeared to be mainly in the cytosol and only minimally in lysosomes. In contrast, BrnAggs not exposed to prions showed little or no Aβ42. These observations suggest that sCJD prions induced intraneuronal Aβ42 peptide accumulation relatively rapidly and without formation of extracellular Aβ42 plaques.
sCJD(VV2) Induces Hτ in Hu BrnAggs
Untreated Hu BrnAggs produced large bands of tau between 35 and 50 kDa (Fig. 4C). Exposure to a normal brain homogenate produced smaller bands between 35 and 45 kDa and 2 very weak bands at ~60 kDa and over 100 kDa (Fig. 4C). Exposure to sCJD(VV2) produced bands between 40 and 50 kDa and strong bands of phosphorylated tau from 60 to over 100 kDa (Fig. 4C).
In another preliminary study, we made BrnAggs from transgenic (Tg) mice (Mo) expressing human mutated tau(P301L). These Mo tau(P301L) BrnAggs spontaneously form small numbers of neurons containing Hτ in their nerve cell bodies and dendrites. When the Mo BrnAggs were exposed to a Aβ42-containing homogenate from a Tg(HuAPP695SWE)2576 mouse, the number of neurons containing abnormal tau was increased ~2-fold. In contrast, when the tau(P301L) BrnAggs were exposed to a brain homogenate from a wild-type mouse infected with RML scrapie prions containing PrPSc, the number of neurons and their processes containing abnormal tau was increased by 10-fold. These results were reproduced in triplicate. The findings argue that exposure to Aβ42 and PrPSc increases the levels of Hτ inclusions in the tau(P301L) BrnAggs. We did not use normal, age-matched human brain homogenate controls in these studies, which is why we classified them as preliminary (data not shown).
Hτ in 6 Age-matched Controls
Brains from 6 patients, aged 48 to 69 with no history of dementia were used as controls. One control had no AT8-positive Hτ (Fig. 5A). Three controls had varying amounts of AT8 positivity consisting of small numbers of Hτ bearing neuropil threads grouped into clusters (Fig. 5B–D arrows). Increasing amounts of Hτ positive neuropil threads mixed with intensely stained individual nerve cell bodies occurring in clusters were seen in 2 other controls (Fig. 5E, F). All of the Hτ existed as small single or multiple disconnected and patchy Hτ deposits in the presubiculum, entorhinal cortex, or transentorhinal cortex.
Figure 5.
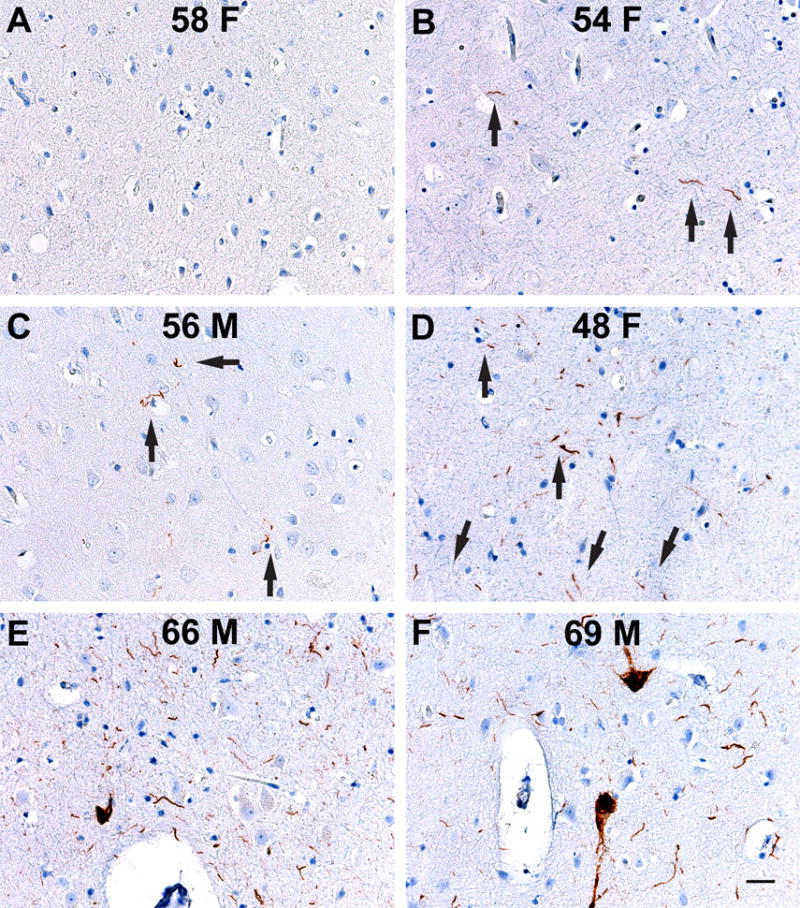
No β-amyloid (Aβ42) was found in the medial temporal lobe of six age-matched control cases without dementia or neurological disease stained for Aβ42 and for hyperphosphorylated tau. Abnormal tau was found in single or several clusters in the entorhinal cortex. The age (in years) and sex (F, female; M, male), of each control case are indicated. (A) No tau is found. (B–D) One to 3 microscopic foci of tau-positive neuropil threads are found. Arrows point to some of the neuropil threads. (E, F) Larger clusters of neuropil threads and occasional cell bodies contain abnormal tau. Clusters were isolated and not connected. Scale bar in (F) represents 50 μm and applies to all figures.
Hτ in the MTL of PrionD and AD cases
Unlike the controls in which abnormal tau staining was focal, Hτ staining of MTL in CJD-only (19 cases), a GSS-only (1 case), CJD-AD (7 cases) and AD-only (5 cases) was continuous or multifocal and extended from the entorhinal cortex into the transentorhinal cortex (Fig. 6C, F). CJD-only and CJD-AD cases showed strong positive and continuous staining in layers 1 and 2 of the entorhinal and transentorhinal cortices (Fig. 6B). A lower intensity of Hτ staining was also present in neuropil threads and neurons in deeper layers of the entorhinal cortex in CJD-only cases. CJD-AD cases showed intense immunostaining in the superficial layers and less intense in the deeper layers of the entorhinal cortex (Fig. 6E, F). In the AD-only cases, many Hτ threads and nerve cell bodies were stained (Fig. 6H, I). The most intense staining occurred in the GSS-only (Fig. 6K, L). In all of the groups, the presubiculum showed little Hτ staining. The hippocampal CA1 region and subiculum were intensely stained in CJD-AD and AD-only cases, while the GSS-only case had staining in portions of the CA1 region and the subiculum.
Figure 6.
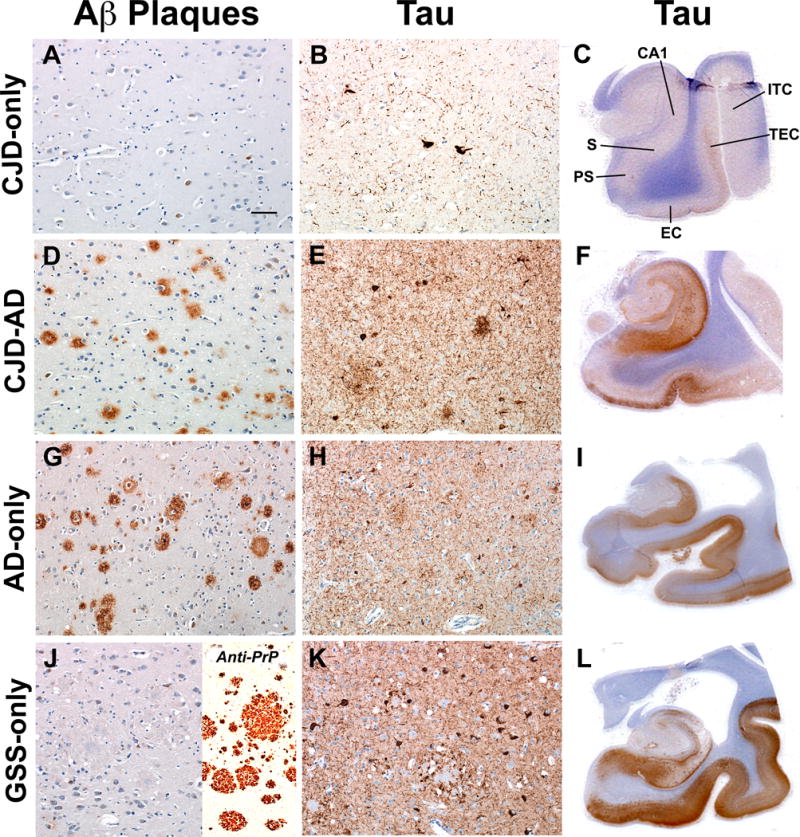
(A–L) The number and distribution of β-amyloid (Aβ) plaques and hyperphosphorylated tau protein (Hτ)-positive threads and nerve cell bodies are different from the controls in Prion-only cases (A–C), Prion-Alzheimer disease (CJD-AD) cases (D–F) and AD-only cases (G–I) in the medial temporal region. A single Gerstmann-Sträussler-Scheinker disease (GSS)-only case showed the most intense staining of neuropil threads and neuronal cell bodies and no Aβ plaques (J–L). GSS-plaques were identified with the 3F4 antibody (J, inset). Aβ-plaques were immunostained with 4G8 (left column), and Hτ immunostained with AT8 (middle and right columns). Microscopic sections in the left 2 columns were photographed with a 20× objective lens of a microscope. Tau-immunopositive cell bodies and neuropil threads were present in the entorhinal cortex in all groups of cases with the least in the Prion-only and the most in GSS-only cases (B, E, H, K). (C, F, I, L) Whole mounts of the medial temporal lobe are stained for Hτ (C, F, I, L). CA1, region of the hippocampus; S, subiculum; PS, presubiculum; EC, entorhinal cortex; TEC, transentorhinal cortex; and ITC, inferior temporal cortex. Scale bar in (A) represents 100 μm and applies also to other micrographs in the left and middle columns.
Hτ Quantification
The density and distribution of AT8-immunopositive Hτs were estimated in the regions of the LC/RN of the pons and in the MTL in 36 cases (Fig. 7). These cases included 20 Prion-only (19 sCJD and 1 GSS), 7 CJD-AD, 5 AD-only, and 4 “Other” (3 TDP-43 encephalopathy and 1 dementia lacking distinctive pathology). We compared the Hτ loads by estimating the intensity of Hτ staining multiplied by the area stained in the LC/RN and MTL. The highest densities of Hτ were in the MTL and the lowest in the LC/RN (Fig. 7A, B). The densities of Hτ in the MTL were significantly higher in the AD-only and CJD-AD groups compared to the Prion-only (p < 0.001, T-test; Fig. 7B). In the Prion-only cases, a score of 1 to 2 represents the presence of any focal Hτ staining in the entorhinal cortex; therefore, it may represent non-specific Hτ staining. A score of 3 or more represents continuous deposition of disease-related Hτ in the entorhinal and transentorhinal cortex (Fig. 6C). The one score of 48 occurred in the GSS case (Fig. 7A).
Figure 7.
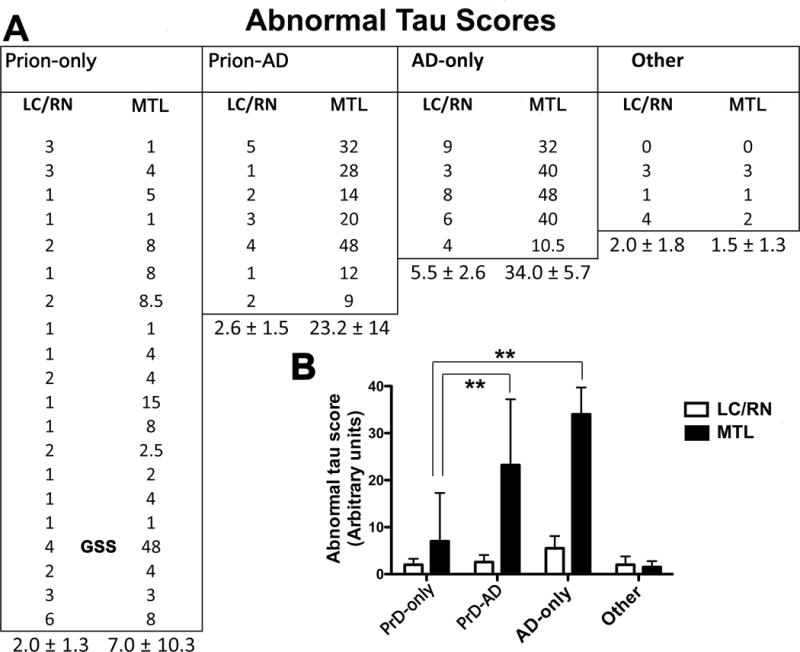
Quantification of hyperphosphorylated tau protein (Hτ) immunostaining in the locus coeruleus and raphe nuclei (LC/RN) and the medial temporal lobe (MTL) in autopsy subjects, 4 of which were shown in Figure 6. (A) The estimated amount of Hτ-containing neuropil threads, nerve cell bodies, and dystrophic neurites were measured by densitometry in 20 Prion disease-only (PrD-only), 7 Prion-Alzheimer disease (Prion-AD), 5 AD-only, and 4 “Other” (3 TDP-43 encephalopathies and 1 dementia lacking distinctive abnormalities) cases. Means and standard deviations are given for the values in each category. (B) Abnormal tau scores are shown as a function of each disease group (mean ± SD). When values in the MTL were compared, abnormal tau was significantly higher in the AD-only and Prion-AD groups vs. the Prion-only group (19 CJD and 1 GSS marked in the figure) (**p < 0.001, t-test). GSS, Gerstmann-Sträussler-Scheinker disease.
DISCUSSION
In examining 266 cases of PrionD we found that 46 patients (17%) also had AD and AD-like changes. In a previous study, Hainfellner et al looked at 110 cases of CJD using anti-PrP, anti-Aβ, and anti-tau immunostaining plus the Bielschowsky silver method to examine a single neocortical block of brain tissue. They found an overlap of PrionD and AD of 10.9% and proposed that AD and prion pathology are coincidental events (1). Like Hainfellner, we also used anti-PrP, anti-Aβ, and anti-tau immunostaining plus the Bielschowsky preparation but we examined multiple neocortical areas.
The yearly incidence of sCJD, which accounts for ~90% of PrionD, is 1 to 2 cases per million (25). The incidence of PrionD is similar to the prevalence because patients usually die within 1 year of diagnosis. The yearly incidence of AD may be as high as ~150,000 cases per million (26), but patients may live 10 years or longer so the prevalence is much greater than the incidence. We could not find adequate data on the prevalence of AD by age to allow us to determine whether or not our findings were coincidental or mechanistic, but the proportion of Prion-AD cases found in both studies strongly suggest that large size (25%–30%) of the preclinical and definite AD populations overlap with many populations including the population of PrionD. Because we found large quantities of Aβ inside neurons in Prion-AD cases, our results are consistent with the concepts that the mechanism of AD in PrionD starts with PrPSc stimulation of the synthesis of Aβ and that its accumulation inside neurons is not included in determining the proportion of preclinical and definite AD. This hypothesis is supported by several factors linking PrionD and AD.
One factor that led us to think that PrionD might induce AD or AD-like changes was the age of patients in the Prion-AD group. In our series, the findings of concomitant Prion-AD changes began abruptly at about the age of 50 and appeared to end just as abruptly at the age of 84, except for 1 outlier who died at 93 years of age. The upper age of 84 years for the Prion-AD patients coincides with the end of the Prion-only group. By contrast, Aβ-associated pathology in AD-only cases continues to increase in the brain until the last AD patients die at age of ~100 years (7, 27). The implication of these data is that AD does not induce PrionD whereas it appears that PrionD induces AD and AD-like pathology. These observations led us to look further to see if we could identify mechanisms by which PrionD could cause or accelerate the onset of AD in susceptible patients.
Another factor that links PrionD with AD is the formation and neuronal accumulation of PrPSc that increases levels of Aβ (28), and the subsequent formation and accumulation of APOE-4 in neurons (Fig. 3; Table 3). Both events also occur in AD-only cases without PrPSc stimulation. We saw low levels of PrPSc, high intraneuronal levels of Aβ, and low levels of intraneuronal APOE-4 in PrionD patients who died without apparent nerve cell loss in the CA1 area of the hippocampus. We saw high levels of PrPSc, low levels of intracellular Aβ, and the transition to high levels of APOE-4 in patients who demonstrated nerve cell loss (Table 3). The study by Umeda et al supports the role of Aβ oligomers in causing nerve cell death by an endoplasmic reticulum stress mechanism (29); however, the authors of that study did not mention APOE-4 activation, a factor that most likely links Aβ pathology with apoptosis of nerve cells (24). APOE-4 binds tightly to Aβ42 to clear it from neurons (30). As a result, soluble oligomeric Aβ levels are increased by APOE-4 (31). We found that concomitant changes of PrionD and AD occurred in patients who were between the ages of 50 and 85, the time when the APOE-4, a known risk factor for AD (32, 33), and neprilysin-α (34), the predominant degrading enzyme of Aβ40 and Aβ42 (35), exert their maximal effects on the development of AD (36).
APOE alleles are not a risk factor for PrionD (37), but APOE-4 contributes to nerve cell death in patients with PrionD. In our Prion-only cases, cases 1–5 had low PrPSc levels, high intraneuronal levels of Aβ42, low numbers of neurons expressing APOE-4, and little neuronal loss in the CA1 (Table 3). The Prion-only cases (cases 6–12) that had high PrPSc levels, low Aβ42 levels, and high APOE-4 levels had an average loss of 40% of CAI neurons. In cases 6–12 we also saw punctate APOE-4 immunostaining in the neuropil between nerve cell bodies, findings that were not observed in the cases 1–5. We believe that the neuropil APOE-4 resulted from nerve cell death releasing APOE-4 into the extracellular space. Neuronal death was an apoptotic event based on the number of shrunken neurons with fragmented nuclei containing APOE-4 seen in the section (Fig. 3F). Therefore, it appears that increasing PrPSc levels triggered increasing APOE-4.
The one exception to this pattern was case 2 (Table 3), a GSS case associated with a 9-octapeptide repeat insertion in the PRNP gene. This patient had a 10-year history of tremor that progressed to a severe mental disorder during the last 2 years of life; 80% of his CA1 neurons contained Aβ42 peptide and the PrPSc deposits were densely granular and associated with GSS-plaques. Nerve cell loss was not identified in the CA1 although APOE-4 levels were not particularly low (Table 3).
The literature indicates that homozygosity at codon 129 of the PRNP gene, either M/M or V/V, is associated with an increased risk of developing sporadic CJD, iatrogenic CJD, or variant CJD (38–40). In regard to AD, in the German population, M/M homozygosity at codon 129 is a risk factor for an early onset of AD, but not for late-onset AD (41). In the Dutch population, the risk of developing early onset of AD is higher for V/V homozygotes than for M/M homozygotes (42). In our study, an overrepresentation of codon-129 homozygosity (84%) was found in the PrionD-only and Prion-AD groups compared to the general US population (49%) (Table 3). However, the PrionD and AD association is not exclusively related to homozygosity because a small percentage of Prion-AD cases in our cohort were heterozygous (M/V).
Our results suggest that being female is a risk factor for the simultaneous occurrence of PrionD and AD. It has been reported that women have a 2-fold higher risk of developing AD than men. This increased risk has been attributed to the loss of estrogen after menopause (18, 19, 43), and to the fact that women tend to live longer than men (44). In our Prion-AD patients, the female-to-male ratio was 1.32, which is similar to the ratio in the AD-only group of 1.35, but different than the Prion-only group, in which it was only 0.90. Because nearly all of our Prion-only patients died in less than 1 year after onset, the longevity of females was not an issue in Prion-AD cases. In contrast, the sex distribution associated with intracellular Aβ42 accumulation in the absence of Aβ42 plaques in our Prion-only cases was predominantly male (9 of 14 cases) (Table 3). This observation suggests that the emergence of amyloid plaques of AD has a greater association with females. It is interesting that 3 out of 4 MV Prion-AD patients were female (Table 1).
The second classical neuropathological feature of AD is the accumulation of Hτ. Several clinical studies have identified increased levels of total tau and phosphorylated tau in the cerebrospinal fluid of patients with CJD (45–47). Our autopsy study appears to be the first to demonstrate increased amounts of abnormal tau (Hτ) and Aβ42 peptides by immunohistochemistry and biochemically in Prion-only, Prion-AD, and AD-only cases vs. “Other” neurodegenerative disease cases, including TDP-43 encephalopathy and dementia lacking distinct histology (Fig. 7). The highest levels of Hτ were present in the AD-only group with progressively less Hτ in the Prion-AD and Prion-only groups. In the Prion-only group (Fig. 7), 4 tau loads in the MTL were given a tau score of 1, which did not represent continuous tau immunostaining in the entorhinal and transentorhinal cortex; rather, it represents Hτ staining that was similar to our six controls. The “Other” group of neurodegenerative disease had the least Hτ score from 0 to 3.
In summary, we found that in some patients, Prion D induces pathology consistent with AD with increased numbers of extracellular Aβ42 plaques, increased intracellular Aβ42 peptide accumulation, and increased deposition of Hτ. PrionD are able to induce AD changes but AD does not appear to induce PrionD. Thus, we propose that PrionD are disorders of four aberrant protein conformers, i.e. PrPSc, Aβ42, APOE-4 and Hτ and that each of these proteins contributes to the pathogenesis of the PrionD.
Does it matter whether or not PrionD induces or accelerates AD? It certainly does not to the patient who dies of PrionD within a year. A better understanding of the combined effects of PrPSc, Ab42, APOE-4, and Hτ in PrionD, however, may provide potential targets for prion therapy. And, more importantly, understanding how PrionD can incite AD-like changes may lead to discovery of additional triggers and clues to control the growing epidemic of AD.
Acknowledgments
The authors thank Dr. Pierluigi Gambetti at the Institute of Pathology (Neuropathology), Case Western Reserve University (Cleveland, OH) for PRNP genotyping and the Western analyses of PrPSc in the human cases; Dr. Stanley B. Prusiner at UCSF for attracting all of the prion disease cases, developing PrP specific antibodies, and bringing funding to UCSF; Dr. George Carlson of the McLaughlin Research Institute, Great Falls, MT, for pregnant mice expressing human tau P301L; Dr. Todd E. Golde, Center for Translational Research in Neurodegenerative Disease, University of Florida, for clarification of Aβ levels in normal subjects and Alzheimer disease and reagents; Hang Nguyen for editing the final draft of the paper. We must also acknowledge Shu-Lian Yang who performed the histoblot analysis on numerous human prion disease cases.
This research was supported by AG02132, AG010770, AG021601 and AG023501 (ADRC).
References
- 1.Hainfellner JA, Wanschitz J, Jellinger K, et al. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 1998;96:116–22. doi: 10.1007/s004010050870. [DOI] [PubMed] [Google Scholar]
- 2.Lauren J, Gimbel DA, Nygaard HB, et al. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–32. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parkin ET, Watt NT, Hussain I, et al. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc Natl Acad Sci USA. 2007;104:11062–7. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kovacs GG, Seguin J, Quadrio I, et al. Genetic Creutzfeldt-Jakob disease associtiated with the E220K mutation: characterization of a complex proteinopathy. Acta Neuropathol. 2011;121:39–57. doi: 10.1007/s00401-010-0713-y. [DOI] [PubMed] [Google Scholar]
- 5.Thal DR, Rüb U, Orantes M, et al. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 6.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braak H, Thal DR, Ghebremedhin E, et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–9. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 8.Boyle PA, Yu L, Wilson RS, et al. Relation of neuropathology with congnitive decline among older persons without dementia. Front Aging Neurosci. 2013;5:1–8. doi: 10.3389/fnagi.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–81. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bajsarowicz K, Ahn M, Ackerman L, et al. A brain aggregate model gives new insights into the pathobiology and treatment of prion diseases. J Neuropathol Exp Neurol. 2012;71:449–66. doi: 10.1097/NEN.0b013e3182544680. [DOI] [PubMed] [Google Scholar]
- 11.Taraboulos A, Jendroska K, Serban D, et al. Regional mapping of prion proteins in brain. Proc Natl Acad Sci USA. 1992;89:7620–4. doi: 10.1073/pnas.89.16.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeArmond SJ, Yang S-L, Lee A, et al. Three scrapie prion isolates exhibit different accumulation patterns of the prion protein scrapie isoform. Proc Natl Acad Sci USA. 1993;90:6449–53. doi: 10.1073/pnas.90.14.6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jendroska K, Poewe W, Daniel SE, et al. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 1995;90:461–6. doi: 10.1007/BF00294806. [DOI] [PubMed] [Google Scholar]
- 14.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 15.Ramsden M, Kotilinek L, Forster C, et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–47. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westerman MA, Cooper-Blacketer D, Mariash A, et al. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2002;22:1858–67. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mirra SS, Hart MN, Terry RD. Making the diagnosis of Alzheimer’s disease. A primer for practicing pathologists [see comments] Arch Pathol Lab Med. 1993;117:132–44. [PubMed] [Google Scholar]
- 18.Rocca WA, Amaducci LA, Schoenberg BS. Epidemiology of clinically diagnosed Alzheimer’s disease. Ann Neurol. 1986;19:415–24. doi: 10.1002/ana.410190502. [DOI] [PubMed] [Google Scholar]
- 19.Gao S, Hendrie HC, Hall KS, et al. The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Arch Gen Psych. 1998;55:809–15. doi: 10.1001/archpsyc.55.9.809. [DOI] [PubMed] [Google Scholar]
- 20.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 21.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–4. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 22.Martinez M, Campion D, Brice A, et al. Apolipoprotein E epsilon4 allele and familial aggregation of Alzheimer disease. Arch Neurol. 1998;55:810–6. doi: 10.1001/archneur.55.6.810. [DOI] [PubMed] [Google Scholar]
- 23.Xu Q, Bernardo A, Walker D, et al. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene of the ApoE locus. J Neurosci. 2006;26:4985–94. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimoto Y, Jiang H, Niikura T, et al. Neuronal apoptosis by apolipoprotein E4 through low-density lipoprotein receptor-related protein and heterotrimeric GTPases. J Neurosci. 2000;20:8401–9. doi: 10.1523/JNEUROSCI.20-22-08401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holman RC, Khan AS, Belay ED, et al. Creutzfeldt-Jakob disease in the United States, 1979–1994: using national mortality data to assess the possible occurrence of variant cases. Emerging Inf Dis. 1996;2:333–7. doi: 10.3201/eid0204.960409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rocca WA, Petersen RC, Knopman DS, et al. Trends in the incidence and prevalence of Alzheimer’s disease, dementia, and cognitive impairment in the United States. Alzheimers Dementia. 2011;7:80–93. doi: 10.1016/j.jalz.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mar J, Soto-Gordoa M, Arrospide A, et al. Fitting the epidemiology and neuropathology of the early stages of Alzheimer’s disease to prevent dementia. Alzheiners Res Ther. 2015:1–8. doi: 10.1186/s13195-014-0079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reiniger L, Lukic A, Linehan J, et al. Tau, prions and Aβ: the triad of neurodegeneration. Acta Neuropathol. 2011;121:5–20. doi: 10.1007/s00401-010-0691-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Umeda T, Tomiyama T, Sakama N, et al. Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci. 2011;89:1031–42. doi: 10.1002/jnr.22640. [DOI] [PubMed] [Google Scholar]
- 30.Roses AD. Apolipoprotein E affects the rate of Alzheimer disease expression: beta-amyloid burden is a secondary consequence dependent on APOE genotype and duration of disease. J Neuropathol Exp Neurol. 1994;53:429–37. doi: 10.1097/00005072-199409000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Tai LM, Mehra S, Shete V, et al. Soluble apoE/Aβ complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol Neurodegener. 2014;9:1–14. doi: 10.1186/1750-1326-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele e4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 33.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–56. [PubMed] [Google Scholar]
- 34.Shirotani K, Tsubuki S, Iwata N, et al. Neprilysin degrades both amyloid β peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J Biol Chem. 2001;276:21895–901. doi: 10.1074/jbc.M008511200. [DOI] [PubMed] [Google Scholar]
- 35.Grimm MOW, Mett J, Stahlmann CP, et al. Neprilysin and Aβ clearance: impact of the APP intracellular domain in NEP regulation and implications in Alzheimer’s disease. Front Aging Neurosci. 2013;5:1–27. doi: 10.3389/fnagi.2013.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blacker D, Haines JL, Rodes L, et al. ApoE-4 and age at onset of Alzheimer’s disease: the NIMH genetics initiative. Neurology. 1997;48:139–47. doi: 10.1212/wnl.48.1.139. [DOI] [PubMed] [Google Scholar]
- 37.Chapman J, Cervenakova L, Petersen RB, et al. APOE in non-Alzheimer amyloidoses: transmissible spongiform encephalopathies. Neurology. 1998;51:548–53. doi: 10.1212/wnl.51.2.548. [DOI] [PubMed] [Google Scholar]
- 38.Palmer MS, Dryden AJ, Hughes JT, et al. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature. 1991;352:340–2. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- 39.Collinge J, Sidle K, Meads J, et al. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–90. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 40.Brown P, Will RG, Bradley R, et al. Bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease: background, evolution, and current concerns. CDC. 2001;7:1–13. doi: 10.3201/eid0701.010102. http://www/cdcgov/ncidod/EID/vol7no1/brownhtm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riemenschneider M, Klopp N, Xiang W, et al. Prion protein codon 129 polymorphism and risk of Alzheimer disease. Neurology. 2004;63:364–6. doi: 10.1212/01.wnl.0000130198.72589.69. [DOI] [PubMed] [Google Scholar]
- 42.Dermaut B, Croes EA, Rademakers R, et al. PRNP Val129 homozygosity increases risk for early-onset Alzheimer’s disease. Ann Neurol. 2003;53:409–12. doi: 10.1002/ana.10507. [DOI] [PubMed] [Google Scholar]
- 43.Paganini-Hill A, Henderson VW. Estrogen deficiency and risk of Alzheimer’s disease in women. Am J Epidemiol. 1994;140:256–61. doi: 10.1093/oxfordjournals.aje.a117244. [DOI] [PubMed] [Google Scholar]
- 44.Hebert LE, Scherr PA, McCann JJ, et al. Is the risk of developing Alzheimer’s disease greater for women than for men? Am J Epidemiol. 2001;153:132–6. doi: 10.1093/aje/153.2.132. [DOI] [PubMed] [Google Scholar]
- 45.Otto M, Wiltfang J, Cepek L, et al. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology. 2002;58:192–7. doi: 10.1212/wnl.58.2.192. [DOI] [PubMed] [Google Scholar]
- 46.Skillbäck T, Rosén C, Asztely F, et al. Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in Creutzfeldt-Jakob disease: results from the Swedish Mortality Registry. JAMA Neurol. 2014;71:476–83. doi: 10.1001/jamaneurol.2013.6455. [DOI] [PubMed] [Google Scholar]
- 47.Wang G-R, Gao C, Shi Q, et al. Elevated levels of tau protein in cerebrospinal fluid of patients with probable Creutzfeldt-Jakob disease. Am J Med Sci. 2010;340:291–5. doi: 10.1097/MAJ.0b013e3181e92a1f. [DOI] [PubMed] [Google Scholar]