

Abstract
Combined administration of certain doses of opioid compounds with a non-steroidal anti-inflammatory drug can produce additive or supra-additive effects while reducing unwanted effects. We have recently reported that co-administration of metamizol with tramadol produces antinociceptive effect potentiation, after acute treatment. However, none information about the effect produced by the combination after chronic or repeated dose administration exists. The aims of this study were to investigate whether the antinociceptive synergism produced by the combination of metamizol and tramadol (177.8 + 17.8 mg/kg, s.c. respectively) is maintained after repeated treatment and whether the effects observed are primarily due to pharmacodynamic interactions or may be related to pharmacokinetics changes. Administration of metamizol plus tramadol acute treatment significantly enhanced the antinociceptive effect of the drugs given alone (P < 0.05). Nevertheless, this effect decreased about 53% after the chronic treatment (3 doses per day, for 4 days). No pharmacokinetic interaction between metamizol and tramadol was found under acute treatment (P > 0.05). The mechanism involved in the synergism of the antinociceptive effect observed with the combination of metamizol and tramadol in single dose cannot be attributed to a pharmacokinetic interaction, and other pharmacodynamic interactions have to be considered. On the other hand, when metamizol and tramadol were co-administered under repeated administrations, a pharmacokinetic interaction and tolerance development occurred. Differences found in metamizol active metabolites’ pharmacokinetics (P < 0.05) were related to the development of tolerance produced by the combination after repeated doses. This work shows an additional preclinical support for the combination therapy. The clinical utility of this combination in a suitable dose range should be evaluated in future studies.
Keywords: Metamizol, Tramadol, Tolerance, Pharmacokinetics, Pharmacodynamics
1. Introduction
Opioid drugs remain the common choice for the treatment of pain of moderate to severe intensity. However, the usefulness of these drugs in treating chronic pain is limited due to the development of tolerance to the analgesic effect that occurs after repeated administrations, resulting in escalation of the dose administered and therefore to an increased incidence of adverse effects (Gammaitoni et al., 2003, Domínguez-Ramírez et al., 2010). A common strategy to maintain adequate analgesic effects and to reduce the adverse effects is to combine doses of opioid compounds with nonsteroidal anti-inflammatory drugs (NSAIDs). In certain combinations of these drugs, it has been shown that it is not only possible to reduce the risk of incidence of adverse effects associated with the administration of high doses of the individual drugs, but also the antinociceptive effects are increased (Hernández-Delgadillo et al., 2003, López-Muñoz et al., 2004). Although some clinical and preclinical studies showing that combinations between opioids and NSAIDs can produce analgesic potentiation, little is known about the antinociceptive effects of repeated administrations. Some studies have shown good efficacy of the combination of tramadol and metamizol under preclinical conditions (Planas et al., 2003, Poveda et al., 2003). Metamizol and tramadol are analgesic drugs with complex mechanisms of action, extensively used in combination in the management of acute postoperative pain in humans (Poveda et al., 2003). The pharmacodynamic mechanism for the interaction between metamizol and tramadol could be attributed partially to their participation in the opioidergic system (Vasquez and Vanegas, 2000, López-Muñoz et al., 2013a). Other mechanisms such as the l-arginine-NO-cyclic GMP pathway and interaction with N-methyl d-aspartic acid receptors could be proposed to explain the antinociceptive synergism observed with the combination of such drugs. In a previous study, 25 different combinations of metamizol and tramadol were analyzed using the model of “pain-induced functional impairment in rat” (PIFIR) after a single dose administration, and the results as antinociceptive effects were additive or potentiative, for all the combinations studied (López-Muñoz et al., 2013a).
Tramadol is a central analgesic drug with a low affinity for opioid receptors. Tramadol is metabolized in the liver by two principal pathways: O-demethylation to O-desmethyltramadol (M1) by CYP2D6 and N-desmethylation to N-desmethyltramadol (M2) by CYP2B6 and CYP3A4. Only one of tramadol metabolites, M1, is pharmacologically active. Its selectivity for μ receptors has recently been demonstrated, showing a higher affinity for opioid receptors than the parent drug (Scott and Perry, 2000). Metamizol is a nonsteroidal anti-inflammatory drug that acts as an effective analgesic and antipyretic agent. Metamizol is a pyrazolone derivative that inhibits the synthesis of prostaglandins at central and peripheral levels (Alves and Duarte, 2002, Ortiz et al., 2003), and it is also known that its antinociceptive effects are mediated at least in part by central mechanisms (Hernández and Vanegas, 2001). Metamizol is a pro-drug that undergoes hydrolysis to yield 4-methylaminoantipyrine (MAA), which is transformed in the liver by cytochrome CYP3A4 to 4-aminoantipyrine (AA) and by oxidation to 4-formylaminoantipyrine (FAA). AA is acetylated to 4-acetylaminoantipyrine (AAA). MAA and AA produce a dose-dependent antinociceptive effect in arthritic rats (PIFIR model), whereas the other metabolites are inactive (López-Muñoz et al., 2013b).
It may be possible that metamizol and tramadol could compete for the same enzymes, causing changes in the concentrations of metabolites of metamizol and consequently in the pharmacological effects produced. The aims of this study were to investigate the antinociceptive synergism produced by the combination of metamizol and tramadol (17.8 + 177.8 mg/kg, s.c. respectively) in an acute and chronic administration schedules and if the effects produced are mainly due to pharmacodynamic interactions or may be related to pharmacokinetic changes in the two main active metabolites of metamizol.
2. Materials and methods
2.1. Animals
Male Wistar rats [Crl (WI)fBR] from the Production Unit of Laboratory Animal Species of the Metropolitan Autonomous University, weighing 180–220 g, were used. Animals were housed in an animal room with controlled temperature (22 ± 2 °C) under a light–dark cycle of 12 h. Rats were provided with standard chow (Purina Laboratory Rodent Diet 5001, Pet Food, México City, México) and water ad libitum. In the 12 h before the experiments, food was withheld, leaving only free access to water. Experiments were performed during the light phase and animals were used only once.
All experimental procedures were approved by the local Institutional Animal Care and Use Committee in accordance with the Mexican federal regulations for the care and use of laboratory animals NOM-062-ZOO-1999 (Mexican Ministry of Health), the Guidelines on Ethical Standards for Investigations of Experimental Pain in Animals (Zimmermann, 1983), the Committee for Research and Ethical Issues of the International Association for the Study of Pain (Covino et al., 1980) and adhere to the Guide for Care and Use of Laboratory Animals, Washington, D.C. (2011). The number of experimental animals was kept to a minimum. At the conclusion of the study, rats were euthanized with CO2 to avoid unnecessary suffering.
2.2. Drugs
Tramadol hydrochloride was obtained from Rimsa Laboratories (Guadalajara City, Mexico); Metamizol was purchased from Sanofi-Aventis (Mexico City, Mexico). Uric acid was obtained from Sigma Chemical Co (St. Louis, MO, USA) and suspended in mineral oil. Either metamizol or tramadol alone, or in combination was administered subcutaneously (s.c.), using isotonic saline solution as vehicle (0.9% w/v). The drug solutions were freshly prepared and administered at volume of 2-mL/kg body weight for metamizol or tramadol. The doses mentioned in the text refer to salts of these substances.
2.3. Study design
Animals were randomly distributed into three groups of 12 animals each. Group I was used for studying the pharmacodynamics and pharmacokinetics of metamizol alone at different administration schedules; animals were randomly distributed into two subgroups of six each in which the antinociceptive effects and the pharmacokinetics of MAA and AA metabolites were studied after single-dose (MET) and after multiple-dosing treatments (3 doses per day) during a period of 4 days (MET-4D). On the day of the experiment, rats under this treatment received a single dose of 177.8 mg/kg of metamizol. The MET-4D group received three daily doses of metamizol (177.8 mg/kg, per dose) during a period of 4 days. Pharmacokinetics of MAA and AA and the antinociceptive effects using the PIFIR model (López-Muñoz et al., 1993) were determined in the same animal. Blood samples were drawn in the group after administration of the drug, immediately after the antinociceptive effect was measured.
Group II was treated with the combination of metamizol and tramadol following the same administration schedules used for Group I. Animals in this group were randomly distributed into two subgroups of six rats each and the pharmacodynamics and pharmacokinetics of metamizol were studied after the simultaneous administration of metamizol (177.8 mg/kg) and tramadol (17.8 mg/kg) in a single dose (MET+TRA) and after repeated dosing of metamizol and tramadol (177.8 + 17.8 mg/kg, 3 times/day) during a total period of 4 days (MET+TRA-4D). Pharmacokinetics of MAA and AA metabolites and the antinociceptive effects of metamizol plus tramadol groups using the PIFIR model were determined in the same animal and blood samples were drawn following the same sampling scheme as in Group I. In the MET+TRA-4D group, pharmacodynamic and pharmacokinetic studies were performed after the last dosing was concluded.
Group III consisted of 12 animals divided into two subgroups of six rats each, in which the antinociceptive effects of tramadol administered alone, were studied after single-dose (TRA) and after multiple-dosing treatments during a period of 4 days (TRA-4D). On the day of the experiment, rats in TRA group received a single dose of 17.8 mg/kg of tramadol. TRA-4D group received three daily doses of tramadol (17.8 mg/kg, per dose) during a period of 4 days. No pharmacokinetic studies were conducted for this group.
2.4. Pharmacodynamic analysis
2.4.1. Arthritis induction
Antinociception was assessed using the PIFIR model in rat (López-Muñoz et al., 1993). Detailed methodology has been previously described. Briefly, under isoflurane anesthesia, rats were injected with 50 μL of uric acid (30%) into the right knee joint to induce nociception. Immediately afterward, an electrode was attached to each hind-paw of the animals. Rats were allowed to recover from anesthesia and then placed on a stainless steel cylinder of 30 cm diameter. This cylinder was rotated at 4 rpm for periods of 2 min every 30 min in order to force the animals to walk. The time of electrode contact on the cylinder was recorded with a computer controlled data acquisition system.
2.4.2. Behavioral assessment
After uric acid injection, rats developed progressive dysfunction of the injured limb. The time of contact of the injured hind limb reached a zero value 2.5 h after injection with uric acid. At this time, metamizol, tramadol or metamizol plus tramadol, previously dissolved in 0.9% saline solution, was subcutaneously (s.c.) administered to the animals. The time of electrode contact was recorded immediately before blood sampling at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, and 4 h after drug(s) administration. Antinociception was evaluated as the recovery of the contact time of the injured limb. Data were expressed as the functionality index percent (FI%), i.e., the time of contact of injected limb divided by the time of contact of the control left paw × 100. For the purpose of this study, inducing nociception in the experimental animals was unavoidable. However, care was taken to avoid unnecessary suffering. Rats were euthanized 8 h after administration of the drug(s) when the last blood sample was taken.
2.5. Pharmacokinetic analysis
2.5.1. Blood sampling
On the day of the study, rats were cannulated approximately 30 min before uric acid injection and 3 h before the drugs were administered. Rats were anesthetized as previously described and the caudal artery was cannulated with a PE-10 cannula (Clay Adams, Parsippany, NJ, USA) connected to a PE-50 cannula. The cannula was kept patent with heparinized saline solution and stopped with a needle.
Rats were allowed to recover from anesthesia and the drugs were administered subcutaneously. Blood samples were withdrawn from the caudal artery at 0 h (before the administration of the drug) and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6 and 8 h after administration of the drug. The cannula was withdrawn and the animal was sacrificed after the 8 h sample.
Blood samples were transferred to heparinized polypropylene tubes. Plasma was separated by centrifugation at 3500 rpm for 10 min at 4 °C and stored at −20 °C until further analysis. The total volume of blood taken from each animal did not exceed 1.8 mL.
2.5.2. Sample preparation
Extraction of main active metabolites of metamizol from plasma samples was conducted using a Solid Phase Extraction technique (SPE). SPE Cartridges were preconditioned by flushing with 6 mL of methanol and 6 mL of distilled water. 50–100 μL of plasma sample was loaded into the cartridge and allowed to stand for 5 min, washed with 0.4 ml of water and then dried under vacuum. The analytes were eluted with 3 mL of methanol, at a flow rate of 1 mL/min. The eluate was evaporated to dryness in a water bath at 45 ± 5 °C under a gentle stream of nitrogen. The residue was reconstituted in 50–100 μL of mobile phase and 20 μL was injected onto the HPLC system for analysis.
2.5.3. Chromatographic conditions
Metabolite MAA and AA plasma concentrations were determined by high performance liquid chromatography (HPLC) with UV detection (Domínguez-Ramírez et al., 2012). The chromatographic system consisted of a Knauer high performance liquid chromatograph (Berlin, Germany) equipped with a Smartline pump 100, a Smartline PDA detector 2800 and a Smartline auto sampler 3950. The chromatographic station ClarityChrom V2.6.xx software was used for acquisition and processing of data. The separation was performed on an Alltech Platinum C18 column (5 μm) 250 × 4.6 mm (Alltech Associates, Deer-field, IL, USA); a Phenomenex security guard column (4 × 0.3 mm C18 cartridge, Torrance, CA, USA) was used before the analytical column. The mobile phase consisted of a mixture of water–methanol–triethylamine–acetic acid (70.9:27.7:0.9:0.5, v/v/v/v) at pH 5, degassed before use, and the flow rate was 1 mL/min. Detection wavelength was set at 254 nm. All the analyses were carried at room temperature (25 °C). This method was proved to be linear (R2 > 0.99), precise (CV < 9%), reproducible (CV < 9%) and accurate (RE < 6%) within a range of 1–100 μg/ml, for MAA and AA metabolites.
2.6. Data analysis and statistics
2.6.1. Pharmacodynamic data analysis
Temporal effect courses for tramadol, metamizol, or the combination of metamizol and tramadol was constructed by plotting the antinociceptive effect (FI%) vs. time (h) for each treatment and the area under the effect–time curve (AUCe) was calculated by the trapezoidal rule (Gibaldi, 1991). Comparisons were established using analysis of variance (ANOVA) followed by Tukey’s test. A significant difference between the means was indicated by a value of P < 0.05.
2.6.2. Pharmacokinetic data analysis
MAA and AA metabolite concentration-time curves were constructed and evaluated by non-compartmental analysis. From the time courses obtained, the following pharmacokinetic parameters were determined: area under the curve from time zero until 8 h after drug administration (AUC0–8h), area under the curve from time zero to infinity (AUC0−∞), rate constant of the terminal phase of elimination (β), half-life of elimination (t½ β), maximum plasma concentration achieved (Cmax) and total clearance (Cl/F). All parameters were obtained from the individual curves for each metabolite and for each treatment and the geometric mean was calculated. Non-compartmental analysis was performed using WinNonlin v. 4.1 (Pharsight, Mountain View, CA, USA). Treatment effects on pharmacokinetic parameters were assessed by one-way ANOVA after logarithmic transformation followed by Tukey’s test. Statistical analysis was performed using SPSS software v. 17.0 (SPSS Inc., Chicago, IL, USA). A P < 0.05 was considered statistically significant.
2.6.3. Relationship between pharmacokinetics of the MAA and AA metabolites and the antinociceptive effect
In order to investigate the existent relationship between MAA and AA plasma concentrations and the antinociceptive effect observed under different experimental treatments, mean FI% was plotted against mean plasma concentrations of MAA or AA metabolite at each time during a 4 h period after the administration of the drug(s). Finally, cumulative area under the effect–time curve (AUCe) vs. cumulative AUC plasma concentrations (AUCp) of each metabolite after the different administration treatments were fitted to an Emax sigmoid model according to the Hill’s equation:
where E is the observed overall effect (AUCe), Emax is the theoretical maximal effect that can be attained (387.5 FI% h), C is the area under the metabolite (MAA or AA) plasma concentration-time curve (AUCp), EC50 is the area under MAA or AA plasma concentration-time curve that induces an effect equivalent to 50% of Emax and γ is the response factor (Hill coefficient). The fit model for the relation between plasma concentration and antinociceptive effect was carried out using GraphPad Prism program v. 4.0 (GraphPad Software, Inc., San Diego, CA, USA).
3. Results
3.1. Pharmacodynamic analysis
The intra-articular injection of uric acid resulted in the complete dysfunction of the right paw in a period of approximately 2.5 h, which corresponded to FI% = 0. At this time (zero), drugs were administered. At the doses of metamizol and tramadol used, no adverse effects that could interfere with the course of the study or the recording of the data were observed. The cumulative antinociceptive effect during the observation period (4 h) was determined as AUCe in order to analyze the whole antinociception effect elicited by TRA or MET or by the MET+TRA and MET+TRA-4D schedules. Fig. 1 shows the antinociceptive effects elicited by tramadol, metamizol and the combination of metamizol and tramadol under different schedules.
Figure 1.
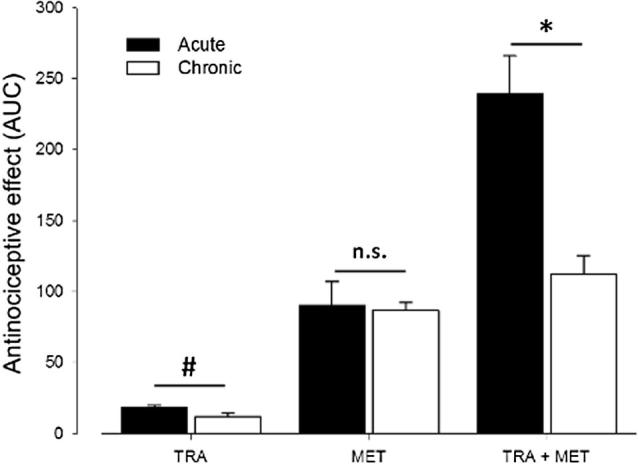
Overall antinociceptive effects after a single dose of 177.8 mg/kg of metamizol (MET), 17.8 mg/kg of tramadol (TRA), administered alone or the combination of the drugs (MET+TRA) administered in single dose and in repeated once-daily doses for 4 days in arthritic rats. Data from temporal courses of the effect were transformed in AUCe (FI%h). Values are mean AUCe ± S.E.M. (n = 6). #P < 0.05, TRA acute vs. chronic treatment; ∗P < 0.05, MET+TRA acute vs. MET+TRA-4D; n.s. P > 0.05, chronic vs. acute treatment of MET.
Table 1 shows the pharmacodynamic parameters obtained after subcutaneous administration of tramadol (17.8 mg/kg), metamizol (177.8 mg/kg) and the combination of metamizol and tramadol under acute and chronic administrations (3 times/day/4 days). The MET group showed an estimated maximal effect of 89.95 ± 17.2 FI% h, whereas the value of 86.70 ± 5.6 FI% h for MET-4D demonstrates no apparent tolerance development (P > 0.05). However, when tramadol was administered alone (acute administration), the global effect was 18.33 ± 1.6%IF h and the antinociceptive effect at 4 h after administration was 0.83 ± 0.7 > %IF. The global effect diminished significantly to 11.9 ± 2%IF and the antinociceptive effects diminished to zero respectively (P < 0.05), showing a tolerance development to the antinociceptive effect of tramadol.
Table 1.
Pharmacodynamic parameters obtained after subcutaneous administration of tramadol (17.8 mg/kg), metamizol (177.8 mg/kg) and the combination of metamizol and tramadol under acute and chronic administrations (3 times/day/4 days).
| Treatment | Global efficacy ABCe (%h) | Tmax (h) | Emax (%IF) | E4h (%IF) |
|---|---|---|---|---|
| Acute administration | ||||
| TRA 17.8 | 18.33 ± 1.6 | 1.00 | 14.00 ± 1.3 | 0.83 ± 0.7 |
| MET 177.8 | 89.95 ± 17.2 | 0.75 | 59.56 ± 11.6 | 7.00 ± 6.2 |
| MET+TRA | 239.17 ± 26.7⁎ | 1.00 | 85.56 ± 7.9 | 34.69 ± 5.9 |
| Chronic administration (3 times/day/4 days) | ||||
| TRA 17.8 | 11.90 ± 2.0# | 0.50 | 14.00 ± 2.7 | 0.00 ± 0.0 |
| MET 177.8 | 86.70 ± 5.6 | 0.50 | 52.33 ± 6.6 | 0.83 ± 0.6# |
| MET+TRA | 112.00 ± 13.2# | 0.75 | 66.33 ± 5.8# | 5.88 ± 3.8# |
P ⩽ 0.05 with respect to the theoretical sum of the individual effects.
P ⩽ 0.05 with respect to the acute treatment.
In the MET+TRA schedule, a higher AUCe compared to that obtained with the MET or TRA schedules was observed (239.17 ± 26.7 FI% h) as a result of a supra-additive effect (P < 0.05). However, after the administration of MET+TRA-4D treatment the effect was approximately 53% lower (112.00 ± 13.2 FI% h) than the overall effect obtained with the MET+TRA treatment (P < 0.05), evidenced that tolerance development may occur with the drug combination under this administration schedule. However, the effect of the MET+TRA-4D treatment continues to be higher than the effect produced by MET-4D (P < 0.05).
3.2. Pharmacokinetics of the metabolites of metamizol
Mean plasma concentrations of MAA and AA, found after administration of metamizol alone (177.8 mg/kg) or in combination with tramadol (17.8 mg/kg) in single or repeated administration, were plotted as a function of time. No parent drug (metamizol) concentrations were detected.
3.2.1. MAA pharmacokinetics
Time courses for MAA plasma concentrations are shown in Fig. 2A and the corresponding pharmacokinetic parameters are included in Table 2. No significant differences between the pharmacokinetic parameters for MAA obtained after the administration of metamizol alone and in combination with tramadol under acute treatments were found (P > 0.05). However, significant differences were observed in AUC0−∞ after the MET-4D schedule compared with the MET+TRA-4D treatment. Values decreased from 150.1 ± 25.4 μg h/mL for MET-4D to 88.1 ± 4.8 μg h/mL for the MET+TRA-4D treatment (P < 0.01), about a 60% of the initial value. β value increased from 0.66 ± 0.13 h−1 for the MET-4D treatment to 1.2 ± 0.09 h−1 for MET+TRA-4D (P < 0.05). No significant pharmacokinetic differences (P > 0.05) were found when MET-4D was compared with MET treatment. However, significant differences in both the AUC0−∞ and AUC0–8h were observed when the combination MET+TRA was compared with the MET+TRA-4D treatment: the first, with a decrease from 166.6 ± 23.5 μg h/mL to 88.1 ± 4.8 μg h/mL (P < 0.01) and the second, from 133.8 ± 16.8 μg h/mL to 83.2 ± 4.7 μg h/mL. Similar differences in β and t½ β were observed: the value of β increased from 0.28 ± 0.06 h−1 to 1.2 ± 0.09 h−1. With respect to the t½ β value, a significant decrease from 3.3 ± 0.7 h for the acute treatment to a value of 0.6 ± 0.1 h for the repeated dosing treatment was observed. It should be noted that differences were also found in the corresponding clearance (Cl/F) values. The values ranged from 1.2 ± 0.2 L/h/kg in the case of MET+TRA to 2.0 ± 0.1 L/h/kg for the MET+TRA-4D treatment.
Figure 2.

Panel A: MAA plasma concentration–time curves. Panel B: AA plasma concentration–time curves. Values represent mean ± S.E.M of six individual readings (n = 6).
Table 2.
Pharmacokinetic parameters for the two main active metabolites of metamizol after subcutaneous administration of 177.8 mg/kg of metamizol alone (MET) or in combination with 17.8 mg/kg of tramadol (MET+TRA), under acute or chronic treatment for 4 days.
| Parameter | MET | MET+TRA | MET-4D | MET+TRA-4D |
|---|---|---|---|---|
| 4-methyl-amino-antipyrine (MAA) | ||||
| ABC0−∞ (μg h/mL) | 145.3 ± 14.5 | 166.6 ± 23.5ns | 150.1 ± 25.4ns | 88.1 ± 4.8⁎,⁎⁎ |
| ABC0–8h (μg h/mL) | 125.2 ± 11.0 | 133.8 ± 16.8ns | 140.6 ± 25.4ns | 83.2 ± 4.7⁎,⁎ |
| β (h−1) | 0.42 ± 0.06 | 0.28 ± 0.06ns | 0.66 ± 0.13ns | 1.20 ± 0.09⁎⁎,⁎⁎⁎ |
| t½β (h) | 1.9 ± 0.3 | 3.3 ± 0.7ns | 1.4 ± 0.4ns | 0.6 ± 0.1ns,⁎ |
| Cmax (μg/mL) | 85.5 ± 12.8 | 64.7 ± 11.1ns | 78.2 ± 5.3ns | 88.5 ± 7.2ns,ns |
| Cl/F (L/h/kg) | 1.3 ± 0.1 | 1.2 ± 0.2ns | 1.4 ± 0.3ns | 2.0 ± 0.1ns,⁎⁎ |
| 4-amino-antipyrine (AA) | ||||
| ABC0−∞ (μg h/mL) | 153.5 ± 15.0 | 120.9 ± 14.3ns | 86.0 ± 13.4⁎⁎ | 87.5 ± 9.0ns,ns |
| ABC0–8h (μg h/mL) | 98.2 ± 6.1 | 80.8 ± 10.4ns | 63.0 ± 11.2⁎ | 65.5 ± 10.1ns,ns |
| β (h−1) | 0.15 ± 0.02 | 0.18 ± 0.03ns | 0.31 ± 0.07ns | 0.30 ± 0.02ns,⁎ |
| t½β (h) | 4.9 ± 0.7 | 4.8 ± 1.0ns | 2.9 ± 0.6ns | 2.4 ± 0.2ns,⁎ |
| Cmax (μg/mL) | 19.2 ± 1.0 | 16.6 ± 2.4ns | 18.3 ± 1.9ns | 18.4 ± 2.1ns,ns |
| Cl/F (L/h/kg) | 1.2 ± 0.1 | 1.6 ± 0.2ns | 2.4 ± 0.5ns | 2.1 ± 0.2ns,ns |
P ⩾ 0.05.
P ⩽ 0.01.
P ⩽ 0.001.
P > 0.05 between the groups of comparison (MET vs MET+TRA; MET-4D vs MET+TRA-4D; MET vs MET-4D and MET+TRA vs MET+TRA-4D) from each metabolite. Data are expressed as the mean ± S.E.M. (n = 6).
3.2.2. AA pharmacokinetics
Time courses for AA plasma concentrations are shown in Fig. 2B and the corresponding pharmacokinetic parameters are shown in Table 2. No significant differences (P > 0.05) were observed in the pharmacokinetic parameters of AA after MET vs. MET+TRA. Likewise, no significant differences (P > 0.05) in any AA pharmacokinetic parameter were found in the comparison of MET-4D vs. MET+TRA-4D treatments.
After the MET schedule, the AUC0−∞ for AA was recorded as 153.5 ± 15.0 μg h/mL, which decreased significantly to 86.0 ± 13.4 μg h/mL for MET-4D. AUC0–8h also showed a significant reduction from 98.2 ± 6.1 μg h/mL to 63.0 ± 11.2 μg h/mL (P < 0.05).
With the comparison of MET+TRA with the MET+TRA-4D treatment, significant differences were also found in β and t½ β values for AA metabolite when comparing MET+TRA with the MET+TRA-4D treatment; β values increased from 0.18 ± 0.03 h−1 for MET+TRA to 0.30 ± 0.02 h−1 for the MET+TRA-4D treatment (P < 0.05). Consequently, t½ β values, a decrease from 4.8 ± 1.0 h for the MET+TRA to a value of 2.4 ± 0.2 h for the MET+TRA-4D treatment were observed (P < 0.05).
3.3. Relationship between pharmacokinetics of the MAA and AA metabolites and the antinociceptive effects
In order to establish the relationship between the pharmacokinetics of the metabolites MAA and AA and the antinociceptive effect, FI% vs. each metabolite plasma concentrations graphs were constructed. Results from different treatment schedules after administration of metamizol alone or in combination with tramadol are shown in Figure 3, Figure 4 respectively.
Figure 3.
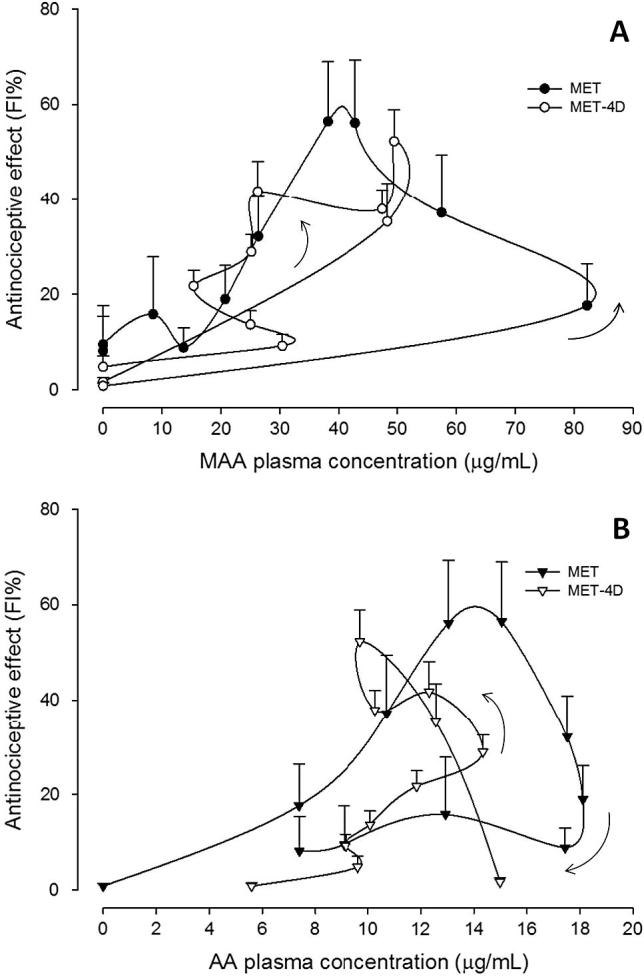
Mean plasma concentration–antinociceptive effect relationship for MAA (panel A) and AA (panel B) metabolites after s.c. administration of a single dose of metamizol (MET) and chronic treatment for 4 days (MET-4D). Values are mean ± S.E.M. (n = 6).
Figure 4.

Mean plasma concentration–antinociceptive effect relationship of MAA (panel A) and AA (panel B) metabolites after s.c. administration of metamizol plus tramadol in a single dose (MET+TRA) and chronic treatment for 4 days (MET+TRA-4D). Values are mean ± S.E.M. (n = 6).
For the MET and MET-4D schedules, the relationship between the antinociceptive effect (FI%) and MAA metabolite plasma concentrations was characterized by a counter clockwise hysteresis loop when data were connected in sequence (Fig. 3A). On the other hand, the relationship between the effect and AA metabolite plasma concentrations showed a mixed behavior, whereas the MET schedule exhibits a clockwise hysteresis and the MET-4D treatment showed a counter clockwise hysteresis (Fig. 3B).
Meanwhile, for the MET+TRA and MET+TRA-4D schedules, the relationship was characterized by a clockwise hysteresis loop for both MAA and AA metabolites (Fig. 4A and B), except for the MET+TRA schedule where a counter clockwise hysteresis loop was observed between the effect and the MAA plasma concentrations (Fig. 4A).
Finally, the cumulative areas under the effect–time curve were plotted against the cumulative areas under the curve of metabolite plasma concentrations (Figure 5, Figure 6). Data for each treatment were fitted to an Emax sigmoid model since in all cases a good fit (R2 > 0.99) was observed.
Figure 5.

Data obtained after administration of metamizol alone in a single and chronic treatment fitted to the Emax model. Symbols represent mean MAA (panel A) and AA (panel B) plasma concentrations of six rats ± S.E.M.
Figure 6.

Data obtained after administration of metamizol plus tramadol in a single and chronic treatment fitted to the Emax model. Symbols represent mean MAA (panel A) and AA (panel B) plasma concentrations of six rats ± S.E.M.
Fig. 5 shows the relationship between mean overall effect (AUCe) and mean metabolite (MAA or AA) plasma concentrations vs. time area under the curve (AUCp) up to 4 h after the administration of metamizol alone under different administration schedules (Group I). AUCe for the MAA metabolite in a single dose attained a maximum value of 93.75 FI% h for an AUCp of ∼90 μg h/mL (Fig. 5A). The overall effect was sustained during chronic treatments. Fig. 5B shows the relationship between the effects observed (AUCe) and the AA metabolite concentrations (AUCp) after the administration of the drug under the two different administration schedules (MET and MET-4D).
Fig. 6 shows the relationship between AUCe and AUCp up to 4 h after the administration of metamizol in combination with tramadol also under different administration schedules (Group II). The overall effects in this comparison decreased significantly in 51% of the effect during the MET+TRA-4D treatments.
4. Discussion
4.1. Antinociceptive effect of the combination of metamizol and tramadol
The combination of metamizol and tramadol (177.8 + 17.8 mg/kg) was selected from a total of 25 combinations studied (López-Muñoz et al., 2013a) because this proportion gives an adequate antinociceptive effect and presents fewer side effects than other combinations. The results obtained after administration of the combination of metamizol and tramadol after a single dose, confirmed the potentiation of the antinociceptive effect elicited by the drugs given alone (P < 0.05). Similar results were found with this combination using the “plantar test model” (Moreno-Rocha et al., 2012).
Development of tolerance to antinociceptive effect of tramadol was observed when the drug was administered alone under repeated doses (4-day treatment); the same was observed with metamizol alone under the same conditions; however in this case, although the duration of the effect was diminished, descending to a value near zero at 2 h after the administration of the drug on the last treatment day, the maximal effect was the same as for metamizol given alone in single dose. The development of tolerance to the antinociceptive effect was significantly increased with the drug co-administration of both drugs. An increase in the rate of development of tolerance to the antinociceptive effect of metamizol co-administered with tramadol was previously reported using the plantar-test model (Moreno-Rocha et al., 2012). Nevertheless, when the effect attained by the combination after chronic treatment was compared with the effect of the individual drugs after the 4-day treatment, an increase in the global effect was observed (P < 0.05), which is equivalent to the sum of the individual effects (additive effect).
The pharmacodynamic mechanism for the interaction between metamizol and tramadol could be partially attributed to the participation of mechanisms such as the l-arginine-NO-cyclic GMP pathway and interaction with N-methyl d-aspartic acid receptors (López-Muñoz et al., 2013a). Additionally, several reports indicate that the antinociceptive effects produced by metamizol are due, at least in part, to the release of endogenous opioids in the descending pain pathways (Hernández and Vanegas, 2001), and this could also explain the development of tolerance observed. However, other pharmacokinetic interactions could be present, which are discussed afterward.
4.2. Pharmacokinetics of the main active metabolites of metamizol
In this study, possible changes in the pharmacokinetics of MAA or AA metabolites in rats, which could explain the pharmacological effects observed (antinociceptive and tolerance development) after administration of metamizol alone and in combination with tramadol, under acute and chronic treatments, were investigated. To achieve this, the pharmacokinetics of MAA and AA metabolites, under the four treatments described, were followed. After the administration of the MET schedule, pharmacokinetic parameters for MAA and AA were calculated from data obtained by a non-compartmental analysis (Table 1). This is consistent with earlier studies in which the metabolite pharmacokinetics have been described (Vlahov et al., 1990). Under these conditions, the pharmacokinetic of metamizol in rats, is similar in some aspects with some results obtained in studies conducted in humans; i.e., the maximum plasma concentration of MAA after a single administration of MET was 5–10 times higher compared to other metabolites (Nogami et al., 1970). It can be said that the pharmacokinetics of metamizol in acute treatment vary among species in parameter values obtained, but not in the relative proportion of the metabolites formed. Pharmacokinetic parameters obtained in this study in rats could not be compared with other studies, as scarce information was found.
When comparing the parameters obtained with the administration of the MET with the data of the MET+TRA schedule, no differences in the pharmacokinetic parameters for MAA or AA, between both treatments were found (P > 0.05), showing that tramadol does not modify the pharmacokinetics of metamizol when administered together in a single dose (Table 1). It can be observed that although there is an increase in MAA metabolite’s elimination half-life, together with a decrease in β value, there is not enough evidence to show that a significant change in the pharmacokinetics of this metabolite exists (P > 0.05). Similar results were obtained after administration of the combination of morphine and metamizol, in acute treatment (Domínguez-Ramírez et al., 2012). So, it can be said that the synergism of the antinociceptive effect observed with the combination of metamizol and tramadol (acute treatment) cannot be attributed to a pharmacokinetic interaction. Other pharmacodynamic interactions as the involvement of the l-arginine-nitric oxide-GMPc pathway and the activation of opioidergic system should be considered (Moreno-Rocha et al., 2012).
Although no statistical differences were found for MAA pharmacokinetics, between MET and MET-4D schedules, in the case of AA metabolite, which is formed by oxidation of MAA, via CYP2D6 system, the areas under the curve decreased significantly (P < 0.05), even when the decrease in β or half-life time, was not statistically significant (P > 0.05). This may partly explain the decrease in the effect of metamizol in chronic treatment since it is known that AA shows a similar antinociceptive activity to that of MAA (López-Muñoz et al., 2013b).
Currently there are no pharmacokinetic studies including the pharmacokinetics of metamizol in rats after a chronic treatment schedule, so this study appears to be the first to make this comparison. Nevertheless, a study carried out in human volunteers, with single (0.75, 1.5 and 3 g) and multiple doses (1 g/3 times/day/7 days) administrations, showed a change in the metabolism of MAA, to preferably form AA, instead of FAA (Levy et al., 1995). It could be assumed that the increase in the elimination process of AA is possibly due to the induction of this particular metabolic pathway by a mechanism that has not been clarified yet.
The decreases in the areas under the curve of the active metabolite AA after repeated treatment of metamizol may also explain the decreased antinociceptive effect of metamizol under the dosing schedule employed, since the decrease may be accompanied by the formation of inactive metabolites (mainly AAA), which is finally excreted by the kidneys.
When the combination of metamizol and tramadol was administered in single dose, no significant differences were found when comparing the pharmacokinetic parameters of metamizol alone. Unlike the above, the comparison of the combined treatment using the multiple dose schedule showed significant differences in the pharmacokinetics of the active metabolites, compared to the single dose treatment. This was observed in most of the pharmacokinetic parameters for the two main active metabolites (MAA and AA) of metamizol. β and Cl/F values of MAA metabolite increased significantly, resulting in a significant decrease in the AUC0–8, AUC0−∞ and t½ β values. This should be explained by an induction of the biotransformation mechanisms of metamizol, as well as the development of tolerance observed with the combination in a chronic treatment schedule. In this regard, Saussele et al., in 2007 demonstrated an increase in the expression of some cytochromes (CYP2B6 and CYP3A4) in patients treated with metamizol for a long time, suggesting that there is a potential mechanism of induction of metamizol and its active metabolite AA similar to that found in phenobarbital over some drugs or its own metabolism (Saussele et al., 2007). The development of tolerance observed with the combination could therefore be explained by the induction of these elimination pathways not only for metamizol itself but also on the metabolism of tramadol.
Finally, it should be mentioned that the frequency of the administration impacts directly on the appearance of the development of tolerance of metamizol either alone or combined with this particular opioid drug. Apparently there is no rule that indicates whether the combination of metamizol with an opioid invariably will result in the development of tolerance. All depend on the type of opioid, the proportions employed and the administration schedule. Added to this, various pharmacodynamic mechanisms of the compounds contribute to the variability of responses. Up to this moment, however, the mechanisms underlying antinociceptive potentiation and tolerance development for the metamizol plus tramadol combination are not fully clear and deserve further investigation.
4.3. Relationship between pharmacokinetics of the main active metabolites of metamizol and the antinociceptive effects
When the antinociceptive effect (FI%) was plotted against metabolite plasma concentrations after acute treatment, a hysteresis was displayed (Figure 3, Figure 4). As seen, a counter-clockwise loop was observed, which indicates the lack of a direct relationship. It can be assumed that the metabolite appearance is delayed into the pharmacodynamic effect site at a slower rate than that in which it appears in plasma. Several explanations have been proposed for this performance, including the formation of active metabolites, or an effect compartment different from those detected by conventional pharmacokinetic analysis (Remington and Gennaro, 2006, Louizos et al., 2014). The possibility of active metabolites formation cannot be discarded since the AA metabolite that is formed from MAA, also shows a high pharmacological activity (López-Muñoz et al.,, 2013b). On the other hand, the relationship between functionality index (FI%) and metabolite plasma concentration shown by metamizol plus tramadol combination groups, showed a clockwise hysteresis loop that may be the result of tolerance development by the presence of antagonistic metabolites that are formed as the drug is metabolized, or down-regulation of receptors and feedback regulation (Kwon, 2001).
Finally, when the relationship between the AUCe and AUCp of MAA and AA metabolites was fitted to the Emax sigmoid model, for all metamizol and combination groups, a satisfactory correlation (R2 > 0.99) was found. AUCe is considered to reflect the overall antinociceptive effect for the whole experimental observation, whereas AUCp is referred to the total amount of drug (or metabolite) in systemic circulation along the same time interval. Both parameters can be adequately related in the case of drugs with indirect effects (Domínguez-Ramírez et al., 2010). The slope for MET+TRA-4D treatment curves decreased slightly when compared to MET+TRA (Fig. 4A) for AA but the difference was not significant (P > 0.05). The maximum effect estimated from de Emax model was near 250 FI% h for MET+TRA for both metabolites and diminished about to 100 FI% h after the MET+TRA-4D schedule, which shows the relationship between the diminution of metabolites concentration and the effect observed, it is to say, the development of tolerance.
5. Conclusions
The results indicate that when the combination metamizol and tramadol (177.8 + 17.8 mg/kg) is administered in single dose, a potentiation of the antinociceptive effect is observed, while the repeated administration of metamizol and tramadol leads to the development of tolerance to the antinociceptive effect of the drugs. Although less pronounced than that observed with the co-administration of both drugs, a development of tolerance was found for metamizol and tramadol given individually, under repeated administration. No pharmacokinetic interaction was observed between metamizol and tramadol when co-administered in single dose. On the contrary, when metamizol and tramadol were given in combination under repeated doses administration, a pharmacokinetic interaction was observed.
It is possible that changes in the antinociceptive effect of the combination after chronic treatment are due in part to an induction in the elimination process (biotransformation and renal excretion) of metamizol. It is likely that the decrease in plasma concentrations of the active metabolites MAA and AA may be related to the decrease in the antinociceptive effect of the combination of tramadol and metamizol after repeated doses.
An increased tolerance development for the antinociceptive effect of tramadol was confirmed after the administration of metamizol (177.8 mg/kg) under chronic treatment for 4 days. Pharmacokinetic changes were found after chronic treatments and also contributed to the rapid onset of tolerance development of the antinociceptive effect of tramadol.
This work shows additional preclinical support for the combination therapy, especially of combinations involving the use of NSAIDs whose use is widespread. It requires caution when selecting a particular combination, especially for chronic pain treatment. Overall, the usefulness of the combination depends on the treatment scheme used and the balance between effectiveness and the occurrence of adverse effects observed. The clinical utility of this combination in a suitable dose range should be evaluated in future studies.
Acknowledgment
The authors wish to thank L. Oliva and F. Sánchez for technical assistance.
Footnotes
Peer review under responsibility of King Saud University.
References
- Alves D.P., Duarte I.D.G. Involvement of ATP-sensitive K+ channels in the peripheral antinociceptive effect induced by dipyrone. Eur. J. Pharmacol. 2002;444:47–52. doi: 10.1016/s0014-2999(02)01412-7. [DOI] [PubMed] [Google Scholar]
- Covino B.G., Dubner R., Gybels J., Kosterlitz H.W., Liebeskind J.C., Sternbach R.A., Vyclicky L., Yamamura H., Zimmermann M. Ethical standards for investigations of experimental pain in animals. The Committee for Research and Ethical issues of the International Association for the Study of Pain. Pain. 1980;9:141–143. doi: 10.1016/0304-3959(80)90002-0. [DOI] [PubMed] [Google Scholar]
- Domínguez-Ramírez A.M., Cortés-Arroyo A.R., Hurtado y de la Peña M., Medina-López J.R., López-Muñoz F.J. Effect of metamizol on morphine pharmacokinetics and pharmacodynamics after acute and subchronic administration in arthritic rats. Eur. J. Pharmacol. 2010;645:94–101. doi: 10.1016/j.ejphar.2010.07.019. [DOI] [PubMed] [Google Scholar]
- Domínguez-Ramírez A.M., Calzadilla P.C., Cortés-Arroyo A.R., Hurtado M., Medina-López J.R., Gómez-Hernández M., López-Muñoz F.J. High-performance liquid chromatographic assay for metamizol metabolites in rat plasma: application to pharmacokinetic studies. J. Pharm. Biomed. Anal. 2012;71:173–178. doi: 10.1016/j.jpba.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Gammaitoni A.R., Fine P., Álvarez N., McPherson M.L., Bergmark S. Clinical application of opioid equianalgesic data. Clin. J. Pain. 2003;19:286–297. doi: 10.1097/00002508-200309000-00002. [DOI] [PubMed] [Google Scholar]
- Gibaldi M. Estimation of area under the curve. In: Gibaldi M., editor. Biopharmaceutics and Clinical Pharmacokinetics. forth ed. Lea & Febiger; Pennsylvania, USA: 1991. pp. 377–378. [Google Scholar]
- Guide for Care and Use of Laboratory Animals, 2011. eighth ed. Washington, DC: National Academic Press.
- Hernández N., Vanegas H. Antinociception induced by PAG-microinjected dipyrone (metamizol) in rats: involvement of spinal endogenous opioids. Brain Res. 2001;896:175–178. doi: 10.1016/s0006-8993(01)02085-6. [DOI] [PubMed] [Google Scholar]
- Hernández-Delgadillo G.P., López-Muñoz F.J., Salazar L.A., Cruz S.L. Morphine and dipyrone co-administration delays tolerance development and potentiates antinociception. Eur. J. Pharmacol. 2003;469:71–79. doi: 10.1016/s0014-2999(03)01727-8. [DOI] [PubMed] [Google Scholar]
- Kwon Y. Kluwer Academic/Plenum Publishers; New York: 2001. Handbook of Essential Pharmacokinetics, Pharmacodynamic, and Drug metabolism for Industrial Scientists. [Google Scholar]
- Levy M., Zylber-Katz E., Rosenkantz B. Clinical pharmacokinetics of dipyrone and its metabolites. Clin. Pharmacokinet. 1995;28:216–234. doi: 10.2165/00003088-199528030-00004. [DOI] [PubMed] [Google Scholar]
- López-Muñoz F.J., Salazar L.A., Castañeda-Hernández G., Villarreal J.E. A new model to assess analgesic activity: pain-induced functional impairment in the rat (PIFIR) Drug Dev. Res. 1993;28:169–175. [Google Scholar]
- López-Muñoz F.J., Díaz-Reval M.I., Terrón J.A., Déciga-Campos M. Analysis of the analgesic interactions between ketorolac and tramadol during arthritic nociception in rat. Eur. J. Pharmacol. 2004;484:157–165. doi: 10.1016/j.ejphar.2003.11.005. [DOI] [PubMed] [Google Scholar]
- López-Muñoz F.J., Moreno-Rocha L.A., Bravo G., Guevara-López U., Domínguez-Ramírez A.M., Déciga-Campos M. Enhancement of antinociception but not constipation by combinations containing tramadol and metamizole in arthritic rats. Arch. Med. Res. 2013;44:495–503. doi: 10.1016/j.arcmed.2013.09.004. [DOI] [PubMed] [Google Scholar]
- López-Muñoz F.J., Soria-Arteche O., Medina-López J.R., Hurtado M., Lozada-García C., Moreno-Rocha L.A., Domínguez-Ramírez A.M. Antinociceptive activity of metamizol metabolites in a rat model of arthritic pain. Drug Dev. Res. 2013;74:332–338. [Google Scholar]
- Louizos C., Yáñez J.A., Forrest M.L., Davies N.M. Understanding the hysteresis loop conundrum in pharmacokinetic/pharmacodynamic relationships. J. Pharm. Pharm. Sci. 2014;17:34–91. [PMC free article] [PubMed] [Google Scholar]
- Moreno-Rocha L.A., Domínguez-Ramírez A.M., Cortés-Arroyo A.R., Bravo G., López-Muñoz F.J. Antinociceptive effects of tramadol in co-administration with metamizol after single and repeated administrations in rats. Pharmacol. Biochem. Behav. 2012;103:1–5. doi: 10.1016/j.pbb.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Nogami H., Hamano M., Awazu S., Imaoka K. Studies on absorption and excretion of drugs (XVI): determination of sulpyrin and its metabolites in the rat urine. Yakagaku Zasshi. 1970;90:378–383. doi: 10.1248/yakushi1947.90.3_378. [DOI] [PubMed] [Google Scholar]
- Ortiz M.I., Castañeda-Hernández G., Granados-Soto V. Possible involvement of potassium channels in peripheral antinociception induced by metamizol: lack of participation of ATP-sensitive K+ channels. Pharmacol. Biochem. Behav. 2003;74:465–470. doi: 10.1016/s0091-3057(02)01023-7. [DOI] [PubMed] [Google Scholar]
- Planas E., Poveda R., Sánchez S., Romero A., Puig M.M. Non-steroidal anti-inflammatory drugs antagonize the constipating effects of tramadol. Eur. J. Pharmacol. 2003;482:223–226. doi: 10.1016/j.ejphar.2003.09.048. [DOI] [PubMed] [Google Scholar]
- Poveda R., Planas F., Pol O., Romero A., Sánchez S., Puig M.M. Interaction between metamizol and tramadol in a model of acute visceral pain in rats. Eur. J. Pain. 2003;7:439–448. doi: 10.1016/S1090-3801(03)00003-X. [DOI] [PubMed] [Google Scholar]
- Remington J.P., Gennaro A.R. 21 ed. Mack Publishing; Easton, PA: 2006. Remington: The Science and Practice of Pharmacy. [Google Scholar]
- Saussele T., Burk O., Blievernicht J.K., Klein K., Nussler A., Nussler N., Hengstler J.G., Eichelbaum M., Schwab M., Zanger U.M. Selective induction of human hepatic cytochromes P-450 2B6 and 3A4 by metamizol. Clin. Pharm. Ther. 2007;82:265–274. doi: 10.1038/sj.clpt.6100138. [DOI] [PubMed] [Google Scholar]
- Scott L.J., Perry C.M. Tramadol: a review of its use in perioperative pain. Drugs. 2000;60:139–176. doi: 10.2165/00003495-200060010-00008. [DOI] [PubMed] [Google Scholar]
- Vasquez E., Vanegas H. The antinociceptive effect of PAG microinjected dipyrone in rats is mediated by endogenous opioids of the rostral ventromedical medulla. Brain Res. 2000;854:249e252. doi: 10.1016/s0006-8993(99)02303-3. [DOI] [PubMed] [Google Scholar]
- Vlahov V., Badian M., Verho M., Bacracheva N. Pharmacokinetics of metamizol metabolites in healthy subjects after a single oral dose of metamizol sodium. Eur. J. Clin. Pharmacol. 1990;38:61–65. doi: 10.1007/BF00314805. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]