

Abstract
Hepcidin, a peptide produced in the liver, decreases intestinal iron absorption and macrophage iron release by causing degradation of the iron exporter, ferroportin. Because its levels are inappropriately low in patients with iron overload syndromes, hepcidin is a potential drug target. We previously conducted a chemical screen that revealed ipriflavone, an orally available small molecule, as a potent inducer of hepcidin expression. To evaluate ipriflavone's effect on iron homeostasis, we placed groups of 5-week old wild type or thalassemia intermedia (HbbTh3+/−) mice on a soy-free, iron-sufficient diet, AIN-93G containing 220 mg iron and 0-750 mg ipriflavone per kg of food for 50 days. Ipriflavone 500 mg/kg significantly reduced liver iron stores and intestinal ferroportin expression in WT mice, while increasing the ratio of hepcidin transcript levels to liver iron stores. Ipriflavone supplementation in HbbTh3+/− mice failed to alleviate iron overload and was associated with a milder reduction in intestinal ferroportin and a failure to alter the ratio of hepcidin transcript levels to liver iron stores or splenic expression of the hepcidin-regulatory hormone, erythroferrone. These data suggest that dietary supplementation with ipriflavone alone would not be sufficient to treat iron overload in thalassemia intermedia.
Introduction
As humans do not actively excrete iron, maintaining iron homeostasis requires modulation of intestinal iron uptake to reflect the body's iron stores. Hepcidin is a transcriptionally regulated peptide hormone [1] that is produced primarily in the liver and excreted in the urine. Hepcidin transcription increases in response to inflammation [2, 3] or iron overload [4] and decreases in in the setting of increased erythropoiesis, iron deficiency, or hypoxia [2]. Hepcidin regulates iron uptake by enterocytes and iron delivery to erythrocytes by binding the iron exporter ferroportin1 (fpn), which results in internalization and degradation of fpn [5].
In patients with iron overload disorders such as β-thalassemia and hereditary hemochromatosis, regulation of iron absorption and metabolism is disrupted, resulting in accumulation of iron in body tissues such as the liver and spleen. Thalassemias are inherited hemoglobinopathies resulting in impaired synthesis of hemoglobin and are among the most common genetic diseases in the world. [1, 2]. Patients with thalassemia major (TM), most commonly those homozygous for β-globin gene deletions (β0-thalassemia), exhibit severe anemia resulting in dependence on blood transfusions to maintain normal development and limit progression of liver and spleen enlargement and skeletal deformities caused by extramedullary hematopoiesis [1]. Patients with thalassemia intermedia (TI) have less severe abnormalities in globin production and do not usually require blood transfusions, although they are prone to increased intestinal iron absorption [1]. While life expectancy for thalassemia major (TM) patients has improved in recent decades, heart failure remains the leading cause of mortality [3, 4]. Improved control of iron overload, as assessed by serum ferritin levels, correlates with a lower probability of heart disease and death [4].
HbbTh3, subsequently referred to as Th3, mice have a targeted deletion in the mouse βminor and βmajor gene. Th3 homozygote mice, a model of thalassemia major, lack all β globin genes and are nonviable unless rescued by blood transfusions. Th3 heterozygotes (Th3+/−), a model for thalassemia intermedia, are viable without blood transfusions, but exhibit moderate anemia, ineffective erythropoiesis, and iron overload, particularly in the liver and spleen [5]. Th3+/− exhibit increased intestinal fpn expression and inappropriately low hepatic transcript levels of hepcidin [5].
Because its levels are inappropriately low in patients with iron overload syndromes[6] and experimental over-expression of hepcidin or injection of synthetic hepcidin improves iron overload in mouse models[7, 8], Hepcidin is a potential drug target for the treatment of tissue iron accumulation. While patients with hereditary hemochromatosis may be treated with therapeutic phlebotomy, individuals with iron-loading anemias, such as thalassemia rely on chelation therapy rather than phlebotomy. Despite the widespread use of chelators, the majority of thalassemia major patients die of iron-related organ failure, which is incompletely resolved by chelation therapy [9, 10], thus we would like to identify small molecules that modify Hepcidin expression, reduce tissue iron overload, and may synergize with iron chelators.
We recently conducted a chemical screen in human hepatocytes (HepG2 cells) for small molecules that regulate Hepcidin expression [11, 12]. We first identified genistein as a flavone molecule that increased hepcidin expression in human hepatocytes in vitro and in zebrafish embryos in vivo[12]. On further screening[11], we identified ipriflavone, an isoflavone with estrogenic properties, as an upregulator of Hepcidin expression that increases Hepcidin luciferase activity and Hepcidin mRNA transcript abundance at 1 μM concentration, which is ten-fold more potent than genistein. Ipriflavone increased expression of the BMP-dependent gene ID3 [11], suggesting it activates the BMP-6 pathway to modulate Hepcidin transcription.
Because ipriflavone is a relatively nontoxic, orally available small molecule that has been approved as a drug for osteoporosis in Europe and Japan, we hypothesized that it could be useful in the treatment of iron overload syndromes in vivo. Ipriflavone is a synthetic isoflavone that is derived from daizdein, a molecule that is abundant in soybeans. While ipriflavone has estrogenic effects, it is less stimulatory to the endometrium than estradiol [13]. Ipriflavone, taken as an oral supplement, has been shown to increase bone mineral density in postmenopausal women [14] and to have antioxidant properties [15].
In this study we investigated the effect of dietary supplementation with ipriflavone on iron homeostasis in wild type (WT) mice and in a mouse model of thalassemia intermedia, HbbTh3+/− (Th3+/−). We found that short-term gavage or dietary supplementation with ipriflavone increased serum hepcidin levels in WT mice. Long-term dietary supplementation with ipriflavone significantly reduced liver iron stores in WT, but not in Th3+/− mice. Furthermore, dietary supplementation with ipriflavone at 500 mg/kg decreased intestinal fpn expression in WT and Th3+/− mice and significantly increased the ratio of hepcidin transcript levels to liver iron stores in WT mice.
Materials and Methods
Mouse Strains and Diet
All mouse strains were maintained on the C57BL/J6 background and included WT C57BL/J6 (Jackson Laboratories, Bar Harbor, ME) and a thalassemia intermedia model (HbbTh3+/− (Th3+/−) a gift of Dr. Seth Alper). In the experiments, WT mice were males, while Th3+/− mice included both males and females, housed separately. All experiments were approved by the Animal Care and Use Committee of Beth Israel Deaconess Medical Center. At 4 weeks of age, the mice were equilibrated for 1 week on a soy-free rodent diet (AIN-93G with 2.5 mg of iron per kg of food, BioServ, Flemington, NJ) that was supplemented with ferrous gluconate hydrate (Sigma Aldrich, St. Louis, MO) to achieve a total iron content of 35 mg of elemental iron per kilogram of food for short-term experiments or 220 mg of elemental iron per kilogram of food, which is typical for a laboratory mouse diet, for long-term experiments. For 4-hour experiments, ipriflavone (#I6068, LKT Laboratories, St. Paul, MN) dissolved in 90% phosphate buffered saline/10% DMSO was administered by oral gavage at 120 mg/kg body weight for one dose followed by nonterminal blood collection four hours later. The diet was then supplemented with 0, 250, 500, or 750 mg ipriflavone per kg of food and maintained for 7 or 50 days. All groups were fed and watered ad libitum and food intake and body weight were monitored throughout. Animals were maintained on a 12 h light-dark schedule. After 7 or 50 days, the mice were anesthetized by intraperitoneal injection of tribromoethanol and underwent terminal cardiac exsanguination and tissue harvest. Genotypes of the Th3+/− animals were confirmed by PCR amplification using the primers in Supplementary Table 1.
Hematologic Analysis
Blood was collected from the heart using a heparinized sterile 1 ml tuberculin syringe and 25-gauge needle and then transferred to a test tube with 3.6 mg EDTA. Complete blood counts, white blood cell differential, and reticulocyte counts were performed in the Boston Children's Hospital Department of Laboratory Medicine on an Advia 120 (Siemens Healthcare, Malvern, PA).
Quantitative Real Time RT-PCR and Quantitation of Serum Hepcidin Levels
Fresh tissues were immediately stabilized in RNAlater solution (Thermo Fisher, Cambridge, MA) and stored at 4°C. Thirty mg sections of tissue were removed from the stabilization reagent, flash frozen in liquid nitrogen, and disrupted by grinding with a mortar and pestle. RNA was extracted using the RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. cDNA was generated and quantitative realtime RT-PCR was performed according to the method as described [12]. Serum hepcidin protein level was measured using the Hepcidin Murine-Compete ELISA Kit (Intrinsic Lifesciences, La Jolla CA).
Non-Heme Tissue Iron Assay
Fresh aliquots, 200 mg each, of liver and spleen were weighed and frozen at −20° C. Nonheme iron content was determined using the bathophenanthroline method, as described[16]. Data shown are μg iron per gram of tissue. All standards and samples were evaluated in triplicate.
Histology and Immunohistochemistry
The ileum and a lobe of the liver from each mouse were fixed in phosphate buffered paraformaldehyde (4%, pH 7.4) and embedded in paraffin, sectioned, and mounted on glass slides.
Diaminobenzidine (DAB)-enhanced Perls’ Staining for nonheme iron
Deparaffinized and rehydrated liver and ileum slides were incubated in 1% potassium ferrocyanide/0.12M HCl for 30 minutes, washed 3 times in PBS, quenched in 0.3% H2O2/Methanol for 20 minutes, washed 3 times in PBS, and incubated in 3,3′-Diaminobenzidine tetrahydrochloride solution (SIGMA FAST™ DAB with Metal Enhancer Tablet, #D0426, 1 tablet set per 5 mL solution, Sigma Aldrich) for 30 minutes. Slides were then washed 3 times in PBS, prior to dehydration and placement of a coverslip.
Immunohistochemistry
Immunohistochemical staining of ileum slides with an anti-ferroportin (anti-fpn) antibody (#MTP11-A, Alpha Diagnostic International, San Antonio, TX) was performed using the Dako EnVision+ System-HRP (#K4010, Dako, Carpinteria, CA). Control slides were stained using a mouse fpn control peptide (MTP11-P, Alpha Diagnostic International) with 10 μg/ml antibody in 0.05 M Tris-HCl-1% BSA, or 0.05 M Tris-HCl-1% BSA alone.
Flow Cytometry
To assess the effects of ipriflavone treatment on erythropoiesis, flow cytometry was performed to determine the percentage of mature and immature erythrocytes in the spleen and bone marrow using the method as described [5] with the following antibodies, APC-conjugated anti-Ter119 (#557909, BD Biosciences, San Jose, CA) and PE- conjugated anti-CD71 (#AB22393, Abcam, Cambridge, MA). Cells were counted and analyzed using a Gallios flow cytometer and the Kaluza acquisition and analysis programs (Beckman Coulter, Inc., Indianapolis, IN).
Transferrin-bound Iron Uptake Assay
Human hepatoma cells, HepG2 (American Type Tissue Collection, Manassas, VA), were plated at a density of 106 cells/well in 2 mL of α-minimal essential medium/10% endotoxin-free fetal bovine serum/1% penicillin/streptomycin (Life Technologies, Grand Island, NY) at 37° C, 5% CO2. Twenty-four hours after plating, the media was changed to α-MEM/1% FBS/1% penicillin/streptomycin. The following day, the media was supplemented with 6 nM 55Fe-transferrin and vehicle only (1% DMSO) or 1-100 μM ipriflavone. 55Fe Transferrin-bound iron uptake assay was performed as described [12].
Statistical Analysis
Data shown are means ± standard errors unless otherwise indicated. The Kruskal-Wallis method was to generate a global p-value using Prism 6.0c (Graphpad, San Diego, CA). Where the global p-value was <0.05, pairwise Student's t-tests were performed. P<0.05 was considered a significant result on the Student's t-test.
Results
Ipriflavone increases serum hepcidin levels in wild-type mice
We had previously identified ipriflavone as a potent inducer of Hepcidin transcript levels in vitro, but had not evaluated its effect on iron metabolism in vivo. To avoid the confounding effects of soy-derived flavones in standard mouse diets, we used the soy-free diet, AIN 93G (Bioserv). To evaluate ipriflavone's short-term effects, we equilibrated 4-week old wild-type, C57/BL6 mice, to an iron-sufficient AIN 93G diet containing 35 mg of iron per kg of food for 1 week and then delivered ipriflavone (120 mg/kg of body weight) or vehicle alone by oral gavage. Four hours later (Figure 1A), we measured serum hepcidin levels and found an increase in hepcidin levels in the ipriflavone-gavaged mice (550.3+51.1 vs. 348.8+83.79, p=0.05). We then treated the mice for 7 days on the iron-sufficient diet supplemented with 500 mg ipriflavone/kg of food. At the end of 7 days, the mice exhibited a significant increase in serum hepcidin levels (351.7+44.2 vs 151.4+16.1, p=0.001), however there was no significant change in liver hepcidin transcript levels, liver, or spleen iron stores (Figure 1B-D).
Figure 1. Treatment with ipriflavone increases serum hepcidin levels in wild type mice.

Four-week old C57/BL6 mice received an iron-sufficient diet (35 mg of iron/kg of food) for 1 week and then underwent a single gavage of ipriflavone (120 mg/kg in 90%PBS/10% DMSO) or vehicle alone. Mice then continued the iron-sufficient diet with or without ipriflavone (500 mg/kg of food) for 7 days. (A) Serum hepcidin levels of WT mice after a single gavage treatment or after 7 day dietary treatment with ipriflavone. (B-D) Evaluation of RNA transcript levels for liver hepcidin (B) and nonheme iron quantification of the liver (C) and spleen (D) after 7 days of dietary supplementation with ipriflavone (500 mg/kg of food). N=7-8 mice per group. * Signifies p=0.05, while ** signifies p=0.001 by Student's t-test compared to 0 mg/kg control group.
Ipriflavone effectively reduced nonheme iron level in the livers of wild-type mice
We were particularly interested in whether longer-term treatment of the mice would prevent hepatic iron loading. To that end, we fed wild-type or Th3+/− mice diets including higher amounts of elemental iron (220 mg iron/kg food), supplemented with 0, 250, 500, and 750 mg ipriflavone/kg food. The WT mice achieved mean daily doses of 0, 56.30±2.71, 104.0±6.28, and 186±14.4 mg of ipriflavone per kg of body weight, while the Th3+/− mice, which ate more than the WT mice but gained less weight, ingested mean doses of 0, 108.9±3.25, 151.4±7.07, or 322.3±10.3 mg of ipriflavone per kilogram of body weight (Supplementary Table 2).
We found that dietary supplementation with ipriflavone decreased liver nonheme iron stores (Figure 2A,B) in WT mice. We observed a significant decrease in hepatic iron stores of WT mice treated with ipriflavone at 500 mg/kg or at 750 mg/kg, compared to those that did not receive ipriflavone supplementation (39.2±2.06 and 44.1±2.58 μg iron/g tissue, respectively, compared to 57.8±3.39 μg iron/g tissue, p=0.003 and p=0.012, respectively). In contrast, spleen iron concentration did not change significantly with ipriflavone treatment. Blinded evaluation of diaminobenzidine (DAB)-enhanced Perls’ staining (Figure 2C,D), also revealed a decrease in hepatic iron stores in WT mice treated with ipriflavone 500 mg/kg or 750 mg/kg.
Figure 2. Dietary supplementation with ipriflavone decreased hepatic iron stores in wild type mice.

C57/BL6 mice received diets supplemented with ipriflavone in mg/kg of food at the following doses: 0 mg/kg (n=5), 250 mg/kg (n=5), 500 mg/kg (n=4), and 750 mg/kg (n=5) starting at 5 weeks of age for a total of 50 days followed by tissue harvest for quantification of nonheme iron levels in the liver (A) and spleen (B), reported as μg iron per g of tissue. Data shown are means ± standard errors. By Kruskal-Wallis test, p=0.027 for liver comparisons and 0.099 for spleen comparisons. * Signifies p<0.05, ** signifies p<0.01 by Student's t-test compared to 0 mg/kg controls. (C) Diaminobenzidine (DAB)-enhanced Perls’ staining for nonheme iron in liver tissue. (D) Blind analysis was used to score the staining levels of each slide from 1+ (weak) to 4+ (strong).
To evaluate whether ipriflavone is effective at relieving iron overload in a mouse model of thalassemia, we treated Th3+/− mice with the same regimens that we had used in the WT mice. The untreated Th3+/− mice exhibited levels of liver and spleen iron of 286.8±41.24 and 1565±86.87 μg iron/mg of tissue, respectively (Figure 3A,B), which were nearly 5-fold greater than untreated WT liver iron and nearly 6-fold greater than WT spleen iron levels. Treatment with ipriflavone at 250-750 mg/kg failed to reduce significantly the mean level of iron loading in the liver (p=0.379) or spleen (p=0.824) or the extent of liver iron staining (Figure 3C,D).
Figure 3. Dietary supplementation with ipriflavone did not decrease iron stores in the livers or spleens of Th3+/− mice.

Th3+/− mice on a C57/BL6 background received diets supplemented with ipriflavone in mg/kg of food at the following doses: 0 mg/kg (n=3), 250 mg/kg (n=3), 500 mg/kg (n=4), and 750mg/kg (n=2) starting at 5 weeks of age for a total of 50 days followed by tissue harvest for quantification of nonheme iron levels in the liver (A) and spleen (B), reported as μg iron per g of tissue. Data shown are means ± standard errors. P=0.379 by Kruskal-Wallis for liver comparisons and 0.824 for spleen comparisons. (C) Diaminobenzidine (DAB)-enhanced Perls’ staining for nonheme iron in liver tissue. (D) Blind analysis was used to score the staining levels 1+ (weak) to 4+ (strong).
Th3+/− mice have previously been shown to exhibit high levels of intestinal fpn[5], which contributes to the development of iron overload. In fact, we found that Th3+/− mice exhibit high levels of fpn expression even in the distal small intestine, where fpn expression is usually low (Supplementary Figure 1, Figure 4A,B). Enterocyte fpn expression in WT mice declined with increasing ipriflavone dosage (60% of mouse intestines were strongly stained at 0 mg/kg, compared to 33% at 500 mg/kg and 20% at 750 mg/kg), while the intensity of fpn staining in Th3+/− mice decreased slightly at 500 mg/kg and 750 mg/kg, but remained much higher than in WT mice treated with equivalent doses of ipriflavone (Figure 4A,B).
Figure 4. Dietary supplementation with ipriflavone decreased intestinal fpn protein expression in WT mice.

A. Representative immunohistochemical staining for fpn in cross-sections of intestine from WT and Th3+/− mice treated with ipriflavone: 0, 500, or 750 mg/kg of food. B. Blinded evaluation of staining intensity from 1+ (weak) to 4+ (strong) staining.
Ipriflavone's effect on erythropoiesis
To evaluate the effect of ipriflavone on erythropoiesis in WT and Th3+/− mice, we measured the hemoglobin level, hematocrit, mean corpuscular volume (MCV), and reticulocyte percentage (Figure 5A-D). Interestingly, in WT mice, the mean MCV decreased significantly with ipriflavone treatment at all doses, from 59.86±0.29 untreated, to 58.7±0.41 at 250 mg/kg, 56.83±0.67 at 500 mg/kg, and 58.26 at 750 mg/kg, (p=0.049, 0.003, and 0.019, respectively, in comparison to untreated). The reticulocyte percentage decreased significantly at the highest dose of ipriflavone (750 mg/kg) from 3.1±0.16 to 1.9±0.15 (p=0.001), suggesting a toxic effect at this high dose. We found that Th3+/− mice exhibited a phenotype consistent with ineffective erythropoiesis that was not altered by ipriflavone treatment. The Th3+/− mice exhibited lower hemoglobin and hematocrit levels and erythrocyte mean corpuscular volumes (MCV's) than WT mice, while their reticulocyte percentages were consistently higher than WT mice (Figure 5A-D).
Figure 5. Ipriflavone treatment decreased mean corpuscular volume (MCV) in wild type mice.

Erythrocyte indices for WT and Th3+/− (mice after 50 days of treatment with ipriflavone 0-750 mg/kg of food. A. Hemoglobin measurements in g/dL, B. Hematocrit, C. MCV, D. Reticulocyte percentage. * Denotes p<0.05 and ** p<0.01 for WT treated vs WT untreated. ++ Denotes p< 0.01 and +++ denotes p<0.001 for Th3+/− vs WT untreated.
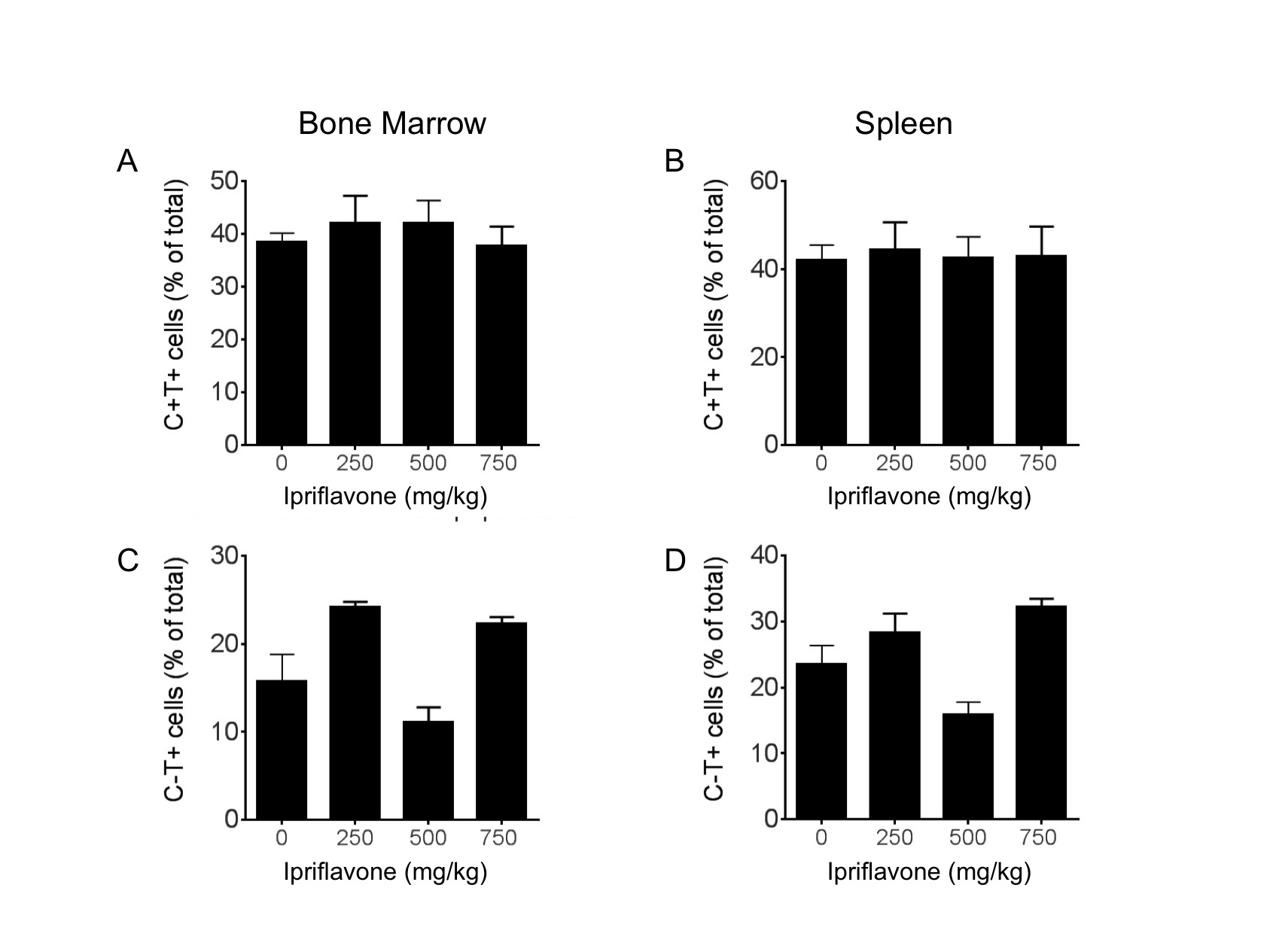
To determine whether ipriflavone has any effect on the distribution of erythroid progenitors in Th3+/− mice, we performed fluorescence-activated cell sorting (FACS) for CD71 and Ter119 to identify immature and mature erythroid populations in the bone marrow and spleens. We found no significant effect of ipriflavone treatment on the percentage of erythroid progenitor cells or mature erythrocytes in the bone marrow or spleen in Th3+/− mice (Supplementary Figure 2).
Ipriflavone's effect on hepcidin, DMT1, fpn, and erythroferrone transcript levels and transferrin-dependent iron uptake
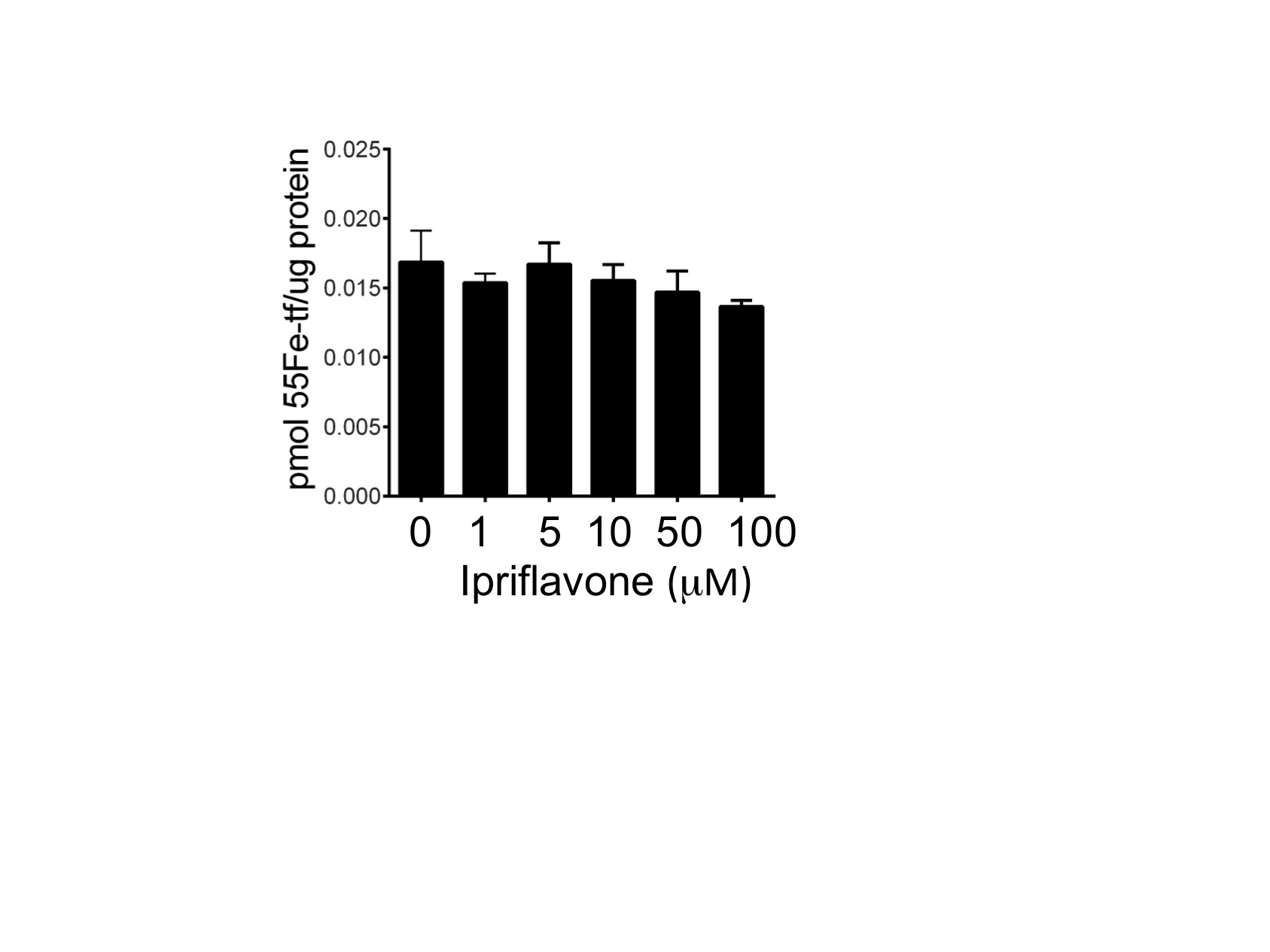
To assess the effect of ipriflavone on gene transcription, we evaluated liver hepcidin transcript levels and intestinal transcript levels of the iron transporters, DMT1 and fpn. The liver hepcidin:G3PD transcript levels did not change significantly between treated and untreated mice. The Th3+/− mice exhibited higher hepcidin transcript levels than WT mice whether treated with ipriflavone or not (Figure 6A). Treatment with ipriflavone 250 or 500 mg/kg, however, significantly increased the ratio of hepatic hepcidin transcript levels to liver iron stores in WT mice (Figure 6B) (0.024±0.003 and 0.02±0.002, respectively, compared to 0.017±0.001, p=0.049 and p=0.021, respectively). Transcript levels for intestinal DMT1 decreased significantly at 750 mg/kg in WT mice (0.632±0.04 vs 1.04±0.11, p=0.017), but were otherwise unaffected in both WT and Th3+/− mice at all doses of ipriflavone. To determine if ipriflavone affected the transcript levels of the recently identified hormone, erythroferrone, we measured transcript levels in the spleens of the treated and untreated Th3+/− mice, but found no significant difference in expression (p=0.96) (Supplementary Figure 3). We also considered that ipriflavone may be lowering hepatic iron stores by interfering with transferrin-dependent iron uptake by hepatocytes. To evaluate this hypothesis, we co-treated human hepatocytes, hepG2 cells, with ipriflavone and 55Fe-transferrin for four hours. We found no significant difference in 55Fe-transferrin uptake in ipriflavone-treated cells compared to cells treated with vehicle only (p=0.46) (Supplementary Figure 4).
Figure 6. Ipriflavone treatment increased the ratio of hepcidin transcript level to liver iron level in WT mice.

Evaluation of RNA transcript levels for liver hepcidin (A), ratio of liver hepcidin to liver non-heme iron stores (B), intestinal DMT1 expression (C), and intestinal fpn expression for mice treated for 50 days with ipriflavone 0-750 mg/kg of food. * Denotes p<0.05 for WT treated vs WT untreated. + Denotes p<0.05, ++ p< 0.01, and +++ denotes p<0.001 for Th3+/− vs WT untreated.
Discussion
In this study we evaluated the in vivo effects of ipriflavone, an orally available synthetic flavone, that we recently identified as a potent inducer of Hepcidin expression in a high throughput screen in human hepatocytes (HepG2 cells) for small molecules that regulate Hepcidin expression [11, 12]. In the current study, we found that oral administration of ipriflavone by gavage or dietary supplementation increased serum hepcidin levels (Figure 1). We also found that dietary supplementation with ipriflavone for 50 days lowered hepatic iron stores and increased hepcidin transcript levels relative to liver iron stores in WT, but not Th3+/− mice, which are a mouse model of thalassemia intermedia. The Th3+/− mice actually ingested more ipriflavone per kg of body mass than WT mice (Supplementary Table 2), but also ingested more iron per kg from their diet. Furthermore, baseline hepatic hepcidin expression was higher in the Th3+/− mice than the WT mice, thus the Th3+/− mice may require a stronger hepcidin inducer than ipriflavone.
The decrease in intestinal fpn protein expression in WT mice treated with ipriflavone 500 mg/kg may be secondary to the increase in the ratio of hepcidin transcript level to iron stores, however recent studies indicate that fpn down-regulation can occur in a hepcidin-independent manner. In mice, stimulation of toll-like receptors (TLR2 or 6) reduces fpn transcript and protein expression in macrophages, liver, and spleen without increasing macrophage hepcidin expression.[17] However, unlike the model of TLR-stimulation, we did not observe a decrease in intestinal fpn transcript levels, with ipriflavone treatment (Figure 6), thus we do not think that ipriflavone's effect is caused by TLR-stimulation. We did observe a decrease in intestinal DMT1 expression in WT mice treated with ipriflavone at the 750 mg/kg dose. This may have helped to limit intestinal iron absorption in these animals.
While ipriflavone structurally resembles estradiol, ipriflavone was effective in WT male mice (Figure 1,2), thus it does not appear that ipriflavone requires feminine gender for its effect. Ipriflavone is known to have effects that are independent of estrogen-receptor, as it has been shown to inhibit osteoclast activity by increasing intracellular calcium levels [18] in an effect that is not antagonized by estradiol treatment [19]. We have previously shown that estradiol does not induce hepcidin expression in vitro [12], while others have shown that estradiol inhibits hepcidin transcription[20], thus we think it is unlikely that ipriflavone is increasing hepcidin expression in an estrogen-receptor dependent manner.
Other investigators have been seeking to treat iron overload in mouse models by daily injection of minihepcidins, chemically stabilized derivatives of hepcidin, in hepcidin-knockout mice [8]. Injection of a minihepcidin, PR65, in hepcidin knockout mice caused a more profound reduction in liver iron stores than we observed with ipriflavone treatment in WT mice. Treatment with PR65 was accompanied by a decrease in hemoglobin level and an increase in splenic iron, which is consistent with iron-restricted erythropoiesis [8]. While ipriflavone treatment in WT mice did not produce anemia or a significant increase in splenic iron, we found that it was associated with a modest decrease in erythrocyte mean corpuscular volume (MCV) (Figure 5C) that could be associated with mildly iron-restricted erythropoiesis. Interestingly, pre-loading hepcidin-knockout mice with iron and then treating with minihepcidins was less effective at reducing liver iron stores than treating the mice while they simultaneously received the high iron diet [8]. By 5 weeks of age, when we started ipriflavone as a hepcidin inducing treatment, Th3+/− mice may already be “pre-loaded” with iron, as well.
We are intrigued by the fact that ipriflavone did not significantly increase the absolute hepcidin transcript levels or reduce the hepatic iron stores in the Th3+/− mice. The hormone, erythroferrone, has recently been identified as an erythropoietic iron sensor that suppresses hepcidin expression in the Th3+/− mouse model of thalassemia[21]. We found that erythroferrone (ERFE) transcript levels did not change in Th3+/− mice in response to ipriflavone treatment (Supplementary Figure 3). It may be necessary to find a small molecule or combination of molecules that can suppress ERFE activity in order to increase hepcidin expression further in Th3+/− mice. In future experiments, we hope to identify more potent, yet nontoxic, small molecule hepcidin inducers that could be used in combination with iron chelators to treat thalassemia-related iron overload more effectively.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This research was supported by R01 DK085250, R01 GM083198, and the Basil O'Connor Scholar Award of the March of Dimes Birth Defects Foundation (PGF). The funding agencies did not influence the design or interpretation of the studies. We would like to thank Prof. Seth Alper, Beth Israel Deaconess Medical Center, for providing the HbbTh3+/− mouse strain.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005;353(11):1135–46. doi: 10.1056/NEJMra050436. [DOI] [PubMed] [Google Scholar]
- 2.Gu X, Zeng Y. A review of the molecular diagnosis of thalassemia. Hematology. 2002;7(4):203–9. doi: 10.1080/1024533021000024102. [DOI] [PubMed] [Google Scholar]
- 3.Borgna-Pignatti C, et al. Survival and disease complications in thalassemia major. Ann N Y Acad Sci. 1998;850:227–31. doi: 10.1111/j.1749-6632.1998.tb10479.x. [DOI] [PubMed] [Google Scholar]
- 4.Borgna-Pignatti C, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187–93. [PubMed] [Google Scholar]
- 5.Gardenghi S, et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109(11):5027–35. doi: 10.1182/blood-2006-09-048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papanikolaou G, et al. Hepcidin in iron overload disorders. Blood. 2005;105(10):4103–5. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicolas G, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34(1):97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 8.Ramos E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120(18):3829–36. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunningham MJ, et al. Complications of beta-thalassemia major in North America. Blood. 2004;104(1):34–9. doi: 10.1182/blood-2003-09-3167. [DOI] [PubMed] [Google Scholar]
- 10.Wood JC, et al. The effect of deferasirox on cardiac iron in thalassemia major: impact of total body iron stores. Blood. 2010;116(4):537–43. doi: 10.1182/blood-2009-11-250308. [DOI] [PubMed] [Google Scholar]
- 11.Gaun V, et al. A chemical screen identifies small molecules that regulate hepcidin expression. Blood Cells Mol Dis. 2014;53(4):231–40. doi: 10.1016/j.bcmd.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhen AW, et al. The small molecule, genistein, increases hepcidin expression in human hepatocytes. Hepatology. 2013;58(4):1315–25. doi: 10.1002/hep.26490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, Zhu Y, Wu J. Effects of estrogen and estrogenic compounds on cognition in ovariectomized rats. Climacteric. 2008;11(3):212–20. doi: 10.1080/13697130802162855. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, et al. Effects of ipri fl avone on postmenopausal syndrome and osteoporosis. Gynecol Endocrinol. 2010;26(2):76–80. doi: 10.3109/09513590903184159. [DOI] [PubMed] [Google Scholar]
- 15.Delarmelina JM, Dutra JC, Batitucci Mdo C. Antimutagenic activity of ipriflavone against the DNA-damage induced by cyclophosphamide in mice. Food Chem Toxicol. 2014;65:140–6. doi: 10.1016/j.fct.2013.12.028. [DOI] [PubMed] [Google Scholar]
- 16.Fraenkel PG, et al. Ferroportin1 is required for normal iron cycling in zebrafish. J Clin Invest. 2005;115(6):1532–41. doi: 10.1172/JCI23780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guida C, et al. A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood. 2015;125(14):2265–75. doi: 10.1182/blood-2014-08-595256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albanese CV, et al. Ipriflavone directly inhibits osteoclastic activity. Biochem Biophys Res Commun. 1994;199(2):930–6. doi: 10.1006/bbrc.1994.1318. [DOI] [PubMed] [Google Scholar]
- 19.Miyauchi A, et al. Novel ipriflavone receptors coupled to calcium influx regulate osteoclast differentiation and function. Endocrinology. 1996;137(8):3544–50. doi: 10.1210/endo.137.8.8754785. [DOI] [PubMed] [Google Scholar]
- 20.Yang Q, et al. 17beta-estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site. Endocrinology. 2012;153(7):3170–8. doi: 10.1210/en.2011-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kautz L, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678–84. doi: 10.1038/ng.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuchiya H, et al. Suppressive effects of retinoids on iron-induced oxidative stress in the liver. Gastroenterology. 2009;136(1):341–350. e8. doi: 10.1053/j.gastro.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 23.Anderson ER, Xue X, Shah YM. Intestinal hypoxia-inducible factor-2alpha (HIF-2alpha) is critical for efficient erythropoiesis. J Biol Chem. 2011;286(22):19533–40. doi: 10.1074/jbc.M111.238667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang F, et al. Normalizing genes for real-time polymerase chain reaction in epithelial and nonepithelial cells of mouse small intestine. Anal Biochem. 2010;399(2):211–7. doi: 10.1016/j.ab.2009.12.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.