

Preface
The recent genomic characterization of cancers has revealed recurrent somatic point mutations and copy number changes affecting genes encoding RNA splicing factors. Initial studies of these ‘spliceosomal mutations’ suggest that the proteins bearing these mutations exhibit altered splice site and/or exon recognition preferences relative to their wild-type counterparts, resulting in cancer-specific mis-splicing. Such changes in the splicing machinery may create novel vulnerabilities in cancer cells that can be therapeutically exploited using compounds that can influence the splicing process. Further studies to dissect the biochemical, genomic, and biological effects of spliceosomal mutations are critical for the development of cancer therapies targeted to these mutations.
Splicing of mRNA precursors is required for the maturation of almost all human mRNAs, and furthermore is a key step in the regulation of expression of many genes. Alternative splicing, wherein a pre-mRNA can be processed into different mature mRNAs via splice site selection, allows multiple potential protein products to be generated from a single gene and thereby expands the cellular proteome. Although the functional roles of most isoforms generated by alternative splicing are unknown, specific isoforms have been identified that are selected in cancer due to their ability to promote neoplastic transformation, cancer progression, and/or therapeutic resistance (reviewed in1,2). In some cases, specific splicing changes are promoted or repressed by recurrent somatic point mutations near splice sites, which may promote cancer by inducing mis-splicing of genes encoding tumor suppressors3,4.
In addition to differential splicing of specific genes, cancers may also exhibit global splicing abnormalities. Genome-wide analyses of cancer transcriptomes5–7 have revealed widespread splicing alterations such as inefficient intron removal in neoplastic tissues relative to their normal counterparts, but the functional consequences of these differences are unknown.
Finally, recurrent somatic mutations within genes encoding core spliceosomal proteins and associated RNA splicing factors are present at high frequencies in several cancers8–12. These mutations provide a direct genetic link between dysfunction of the splicing machinery and cancer. Both the genetic spectrum of mutations and functional studies of their consequences indicate that RNA splicing factors can act as proto-oncoproteins and tumor suppressors.
In this Review, we outline the current genetic and functional links between dysregulated and/or mutated RNA splicing factors and cancer. We discuss how recurrent mutations affecting splicing factors might promote the development and/or maintenance of cancer. We describe the challenges inherent in connecting mutations in spliceosomal proteins to specific downstream splicing changes, as well as the importance of testing whether mutated splicing factors dysregulate biological processes other than splicing itself. Finally, we discuss how determining the mechanistic consequences of mutated splicing factors may facilitate the identification of novel therapeutic opportunities to selectively target cancers with spliceosomal mutations.
RNA splicing catalysis and regulation
RNA splicing is essential for processing pre-mRNA transcribed from the >90% of human protein-coding genes that contain more than one exon into mature mRNAs prior to translation into proteins13,14. The primary function of splicing is the removal of non-coding introns, a process carried out by the large macromolecular machineries known as the major spliceosome and minor spliceosome (reviewed in15). The major spliceosome consists of five small nuclear ribonucleoprotein complexes (snRNPs, pronounced “snurps”), U1, U2, U4, U5 and U6, and it is responsible for the excision of >99% of human introns. The minor spliceosome contains the U5 snRNP, along with the U11, U12, U4atac and U6atac snRNPs, which are the functional analogues of the corresponding snRNPs in the major spliceosome (reviewed in16). Each constituent snRNP contains a different short non-coding RNA, an Sm or Sm-like protein complex that is required for the formation of the mature snRNP complex and proteins specific to each snRNP (reviewed in15,17).
Intron excision is facilitated by short sequence motifs in the pre-mRNA, in particular at boundaries between the upstream exon and intron (the 5′ splice site) and the intron and downstream exon (the 3′ splice site) (Fig. 1a). Although splicing itself is catalyzed by RNA18, the proper recognition of splice sites relies upon RNA:RNA, RNA:protein and protein:protein interactions. U1 snRNP recognizes and binds to the 5′ splice site, whereas U2 snRNP interacts with the branch site region adjacent to the 3′ splice site, facilitated by interactions with U2 auxiliary factors (U2AFs, which form the U2AF complex) that bind the 3′ splice site. Following recruitment of the U4/U6.U5 tri-snRNP, the assembled spliceosome enters its active conformation and splicing proceeds via two sequential transesterification reactions (reviewed in15,17) (Fig. 1b).
Figure 1. Simplified model of constitutive and alternative splicing.
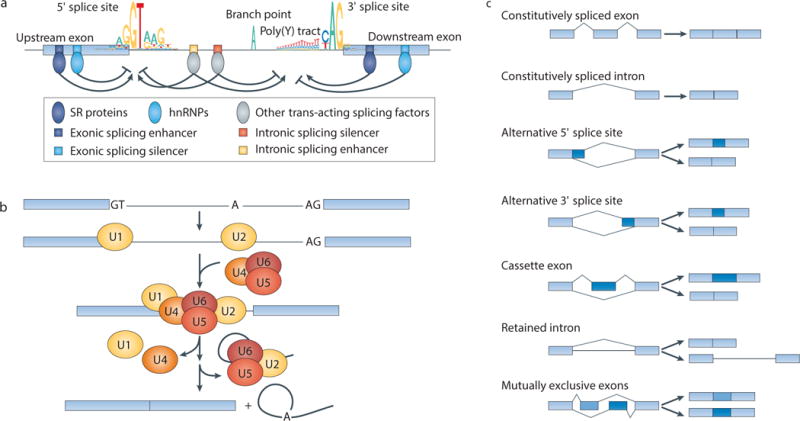
(a) The key sequence features that govern splicing are shown, including consensus sequences of the 5′ and 3′ splice sites and sequence motifs bound by trans-acting splicing factors (serine/arginine-rich (SR) proteins, heterogeneous nuclear ribonucleoproteins (hnRNPs) and others). Sequence elements required for assembly of the spliceosome onto the pre-mRNA, including the splice sites themselves, polypyrimidine (poly(Y)) tract and branch point, frequently follow the illustrated consensus motifs, whereas the sequences of enhancer or silencer elements depend upon the specific RNA-binding protein that recognizes them. Consensus motifs are illustrated as sequence logos, where the height of each nucleotide corresponds to its approximate genome-wide frequency in bits. Sequence motifs are illustrated as genomic DNA sequence rather than pre-mRNA sequence (“T” instead of “U”). Note that splicing factors such as SR proteins and hnRNPs frequently play context-dependent regulatory roles. (b) Simplified schematic of intron excision and ligation of two adjacent exons. The steps shown are: recognition of the 5′ and 3′ splice sites by the U1 and U2 small nuclear ribonucleoprotein complexes (snRNPs), assembly of the snRNPs into the active spliceosome, the excision of the intron lariat and the ligation of the two exons. (c) Schematic of constitutive and alternative splicing events. Light blue: constitutive sequence that always forms part of the mature mRNA; dark blue: alternative sequence that can be either included or excluded in the mature mRNA.
Splice sites are typically categorized as constitutive splice sites or alternative splice sites, depending on whether they are always (constitutive) or only sometimes (alternative) recognized as splice sites and spliced into the mature mRNA. Splicing of both categories of splice sites is catalyzed by the same molecular machinery, although the efficient recruitment of spliceosomal proteins to alternative splice sites frequently depends on the binding of additional trans-acting factors. Although most splicing reactions in some eukaryotes such as Saccharomyces cerevisiae are constitutive — meaning that the same 5′ and 3′ splice sites are always ligated together — splicing is more complex in mammals.
Transcripts from almost all human multi-exon genes are alternatively spliced, where a particular 5′ splice site can be joined to different 3′ splice sites (or vice versa), frequently in a regulated fashion13,14. Alternative splicing events can be further classified based on how they affect the exonic structure of the mature mRNA (Fig. 1c). The distinction between constitutive and alternative splicing is empirical, and so can vary over time as additional transcript sequencing data becomes available. For example, high-throughput sequencing has transformed the study of RNA splicing by enabling the rapid and accurate quantification of genome-wide splicing patterns (Box 1). This has led to the realization that many splice sites and/or exons that were previously classified as constitutive are actually alternatively spliced (for example, in distinct cell types or in response to particular stimuli).
Box 1. Methods to determine differential RNA splicing using RNA-seq data.
Accurate detection and quantification of differential splicing remain bioinformatic challenges178,179. Important aspects of the methods used to measure splicing are described below and include: the quality and origins of the genomic annotation of splicing events, whether to consider full-length isoforms or single splice sites (or single exons) in isolation and the choice of statistical method to identify differences between samples.
Genome-wide splicing annotations are available from both general-purpose genomic databases180–182 and specialized software packages183. These annotations are frequently based on exon-exon junctions inferred from expressed sequence tags (ESTs) and full-length cDNAs, limiting the annotation to the extent that such sequences are available for a given cell type or organism. For example, there are >2.5-fold more alternative splicing events annotated in humans than in mice183, which is a product of the more complete human genome annotation as well as increased rates of alternative splicing in human versus mouse tissues184. As comparatively few splicing events have been annotated in many model organisms, detecting unannotated splicing (or novel splicing) with RNA-seq is frequently necessary. Novel splicing detection can involve searching for reads overlapping new combinations of known 5′ and 3′ splice sites of a gene, detecting alternate 5′ or 3′ ends of exons that constitute novel splice junctions185,186 or conducting de novo isoform reconstruction187,188 (methods ordered by increasing complexity). These approaches are more computationally intensive than using existing annotations, and experimental validation of novel splicing events is crucial.
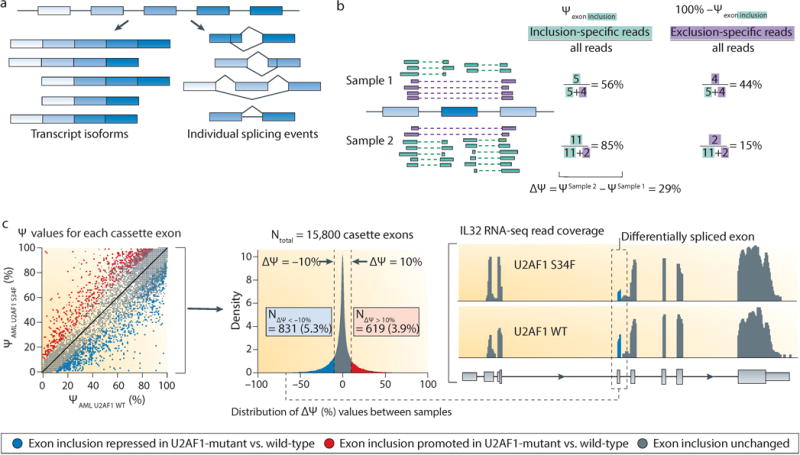
Differential splicing can be analyzed at the level of differentially expressed full-length isoforms or at the level of single splicing events (for example, the inclusion or exclusion of a particular cassette exon; see panel (a) of the figure). Full-length isoforms are the biologically relevant entities that encode the final protein products189, but the exon composition of each isoform must be statistically inferred due to the short read lengths of most high-throughput sequencing platforms. Conversely, focusing on single splicing events may be beneficial when analyzing splicing mechanisms and regulation—for example, the altered motif preferences induced by U2AF1 and SRSF2 mutations72,76,88,98—or when a particular alternatively spliced region has an important impact on gene function. Many such studies focus on single events and then infer full-length isoforms for downstream functional studies. In either case, splicing is quantified using a ‘percent spliced in’ value (PSI, or ψ value) ranging from 0 to 100%14,183, defined as the percentage of all mRNAs that correspond to the isoform or alternatively spliced sequence of interest relative to all transcripts of the parent gene (see panel (b) of the figure), independent of gene expression. (For simplicity, only junction-spanning reads are illustrated in the computation of ψ.)
Quantifying differences in splicing, whether of full-length isoforms or of single events, also involves statistical choices with biological implications. Fold-change in ψ values can be used to quantify differential splicing, but may not correlate well with biological importance. For example, consider an isoform with ψ values of 0.1% and 1% in two samples. This corresponds to a fold-change of 10, yet the isoform is low-abundance relative to other isoforms of that gene in both samples. Therefore, many studies instead measure absolute changes in splicing as Δψ, the difference in ψ value between two samples, and apply thresholds on Δψ to identify potentially important changes in splicing14,183 (for example, using U2AF1-mutant (U2AF1 S34F, shown on the y axis) versus wild-type (U2AF WT, shown on the x axis) acute myeloid leukemia (AML) samples, with exons satisfying Δψ ≥ 10% (red) or Δψ ≤ 10% (blue) highlighted; see panel (c) of the figure). This measure is more statistically robust, but may ignore differential splicing of low-abundance yet important isoforms, such as a novel isoform that confers gain-of-function or dominant-negative activity.
Ultimately, the optimal splicing analysis strategy depends upon the specific aims of a given study and the downstream data interpretation plan. Three common goals of splicing studies are described below. First, many studies seek to identify mechanistic changes in the splicing process itself, such as alterations caused by spliceosomal mutations or global differences between tumor and normal samples5,7. In such cases, meta-analyses created by averaging over many individual splicing events typically yield the highest statistical power. Second, a study may focus on specific isoforms of biological importance, such as ligand-independent isoforms of the androgen receptor190,191, in which case quantifying entire isoforms is essential. Third, a study may seek to identify differentially spliced isoforms that contribute to a known biological phenotype, such as impaired hematopoiesis or enhanced cell migration. Although the biologically relevant entities are whole isoforms, combining single-event and whole-isoform analysis is frequently useful given the exploratory nature of such studies.
Alternative splicing is frequently regulated by trans-acting splicing factors, which bind to sequence motifs that are associated with the promotion (enhancers) or repression (silencers) of splicing. These motifs lie in both exons and introns, and frequently have the strongest effect when in close proximity to a splice site19 (Fig. 1a; reviewed in20,21). Splicing enhancers and silencers are bound by diverse RNA-binding proteins, exemplified by the serine/arginine-rich proteins (SR proteins) and heterogeneous nuclear ribonucleoproteins (hnRNPs; reviewed in22–25). SR proteins contain one or two copies of an RNA recognition motif (RRM) domain at the N-terminus that provides RNA-binding specificity and a C-terminal arginine/serine-rich (RS) domain that contributes to protein-protein interactions26,27. hnRNPs similarly contain both RNA-binding domains and relatively unstructured domains that likely contribute to protein-protein interactions28.
SR proteins and hnRNPs canonically promote and repress splicing, respectively, in a sequence-specific manner22. However, recent studies have revealed further nuances to these roles. The regulatory consequences of SR protein or hnRNP binding within pre-mRNA is frequently context-specific, giving rise to complex regulatory relationships (reviewed in21). For example, SR proteins can promote or repress splicing, depending on where they bind within a pre-mRNA29,30. Similarly, canonical splicing repressors such as the hnRNP polypyrimidine tract binding protein 1 (PTBP1; also known as PTB), can activate splicing in a context-dependent manner31,32. Some hnRNPs primarily act as splicing repressors, whereas others activate exon inclusion33. SR proteins and hnRNPs regulate splicing in diverse ways, including facilitating recruitment of the U1 or U2 snRNP, occluding a splice site, ‘looping out’ an exon and other mechanisms (reviewed in20,21).
Many other RNA-binding proteins regulate splicing in addition to the SR proteins and hnRNPs. These include the CUG-BP- and ETR-3-like (CELF) proteins (reviewed in34), Muscleblind-like (MBNL) proteins (reviewed in35), RBFOX proteins, Signal transduction and activation of RNA (STAR) proteins, NOVA proteins36, epithelial splicing regulatory protein (ESRP) families, T-cell-restricted intracellular antigen-1 (TIA1) and TIA1-like (TIAL1), and many others (reviewed in21). Furthermore, alterations in the levels of core spliceosomal components such as the snRNP proteins SmB/B′ (also known as snRNP-B)37 can regulate splicing. Mass spectrometry studies indicate that the spliceosome is associated with >170 proteins38, and computational studies of exon recognition suggest that hundreds of sequence motifs contribute to the regulation of splicing39. Although the total number of splicing factors is unknown, the above studies suggest that hundreds of proteins may have a role in splicing regulation.
Dysregulation of splicing in cancer
Pro- and anti-tumorigenic splicing factors
Just as regulation of alternative splicing has essential roles in cellular growth, differentiation and tissue development, dysregulated splicing can give rise to protein isoforms that contribute to tumor establishment, progression, and resistance to therapy. Many studies have found links between the altered expression and/or activity of splicing factors, cancer-associated splicing and transformation (reviewed in1,2) (Table 1). SR proteins, hnRNPs and other splicing factors can act as both oncoproteins and tumor suppressors.
Table 1.
Unmutated splicing factors that function as proto-oncogenes or tumour suppressors
| Splicing factor | Downstream dysregulated isoforms that are functionally linked to cancer |
|---|---|
| ESRP1 and ESRP2 | Promotes an epithelial splicing program to regulate EMT62,63. |
| hnRNP A1 | Contributes to aerobic glycolysis in cancer by promoting the expression of specific isoforms of pyruvate kinase (PKM2 isoform)47,48 and MAX (the Delta MAX isoform)49. |
| hnRNP A2 | Contributes to aerobic glycolysis in cancer by promoting the expression of a specific isoform of pyruvate kinase (PKM2 isoform)47,48. Increases breast cancer cell invasion by promoting the expression of a specific isoform of TP53INP2197. |
| hnRNP A2/B1 | Acts as an oncogenic driver in glioblastoma by regulating splicing of several tumor suppressors and oncogenes, including RON198. |
| hnRNP H | Contributes to survival of gliomas as well as invasion by promoting the expression of specific isoforms of IG20 and RON, respectively199. |
| hnRNP K | Serves as a tumor suppressor in leukemia. Deletion is associated with aberrant p21 and C/EBP expression (although mechanistic links to splicing and gene expression are unclear)52. |
| hnRNP M | Contributes to EMT in breast cancer and increases metastasis in mice by promoting the expression of a specific isoform of CD44 (CD44s)200. |
| PRPF6 | Promotes cell proliferation in colon cancer by altering splicing of genes associated with growth regulation, including the kinase gene ZAK201. |
| PTB (PTBP1) | Contributes to aerobic glycolysis in cancer by promoting the expression of a specific isoform of pyruvate kinase (PKM2 isoform)47,48. Also promotes the expression of an isoform of the deubiquitinating enzyme-encoding gene USP5202 that has been shown to promote glioma cell growth and mobility (although the mechanism underlying this phenotypic association is not resolved). |
| QKI | Acts as a tumor suppressor by regulating alternative splicing of NUMB in lung cancer cells54. |
| RBFOX2 | Promotes a mesenchymal splicing program to regulate EMT63. |
| RBM4 | Acts as a tumor suppressor by promoting the pro-apoptotic isoform BCL-XS of BCL2L1 and opposing the pro-tumorigenic effects of SRSF1 on mTOR activation61. |
| RBM5, RBM6 and RBM10 | RBM5 modulates apoptosis by regulating alternative splicing of CASP2203 and FAS204. RBM5 or RMB6 depletion has an opposite effect to RBM10 depletion, as these factors antagonistically regulate the alternative splicing of NUMB59,60. |
| SRSF1 | Promotes an isoform of the kinase MNK2 that promotes eIF4E phosphorylation independently of MAP kinase signaling40. In the context of breast cancer, SRSF1 overexpression promotes alternative splicing of BIM and BIN141 to promote the expression of isoforms that lack pro-apoptotic functions. |
| SRSF3 | Regulates alternative splicing of TP5344 such that SRSF3 loss promotes expression of p53β, an isoform of p53 that promotes p53-mediated senescence. |
| SRSF6 | Promotes expression of isoforms of the extracellular matrix protein tenascin C that are characteristic of invasive and metastatic skin cancer45, contributing to epithelial cell hyperplasia. |
| SRSF10 | Promotes cell proliferation and colony formation in vitro and increases tumorigenic capacity of colon cancer cells in mice by inducing expression of a specific isoform of BCLAF1 (BCLAF1-L)205. |
BCLAF1, Bcl-2-associated transcription factor 1; EMT, epithelial to mesenchymal transition; ESRP, epithelial splicing regulatory protein; hnRNP, heterogeneous nuclear ribonucleoprotein; NMD, nonsense-mediated decay; PRPF, pre-mRNA-processing factor; PTB, polypyrimidine tract-binding protein 1; QKI, quaking; RBM, RNA binding motif protein; SRSF, serine/arginine-rich splicing factor.
Some SR proteins can act as oncoproteins when overexpressed in the correct cellular context. For example, SR splicing factor 1 (SRSF1; also known as ASF/SF2) is upregulated in cancers including lung, colon and breast cancer40,41. Modest overexpression of SRSF1 drove the immortalization of murine fibroblasts and human and mouse mammary epithelial cells40,41. SRSF1 promotes transformation in part by inducing mis-splicing of MNK2 and S6K1 and activating the mTOR pathway, which is required for SRSF1-mediated transformation40,42, and promoting the expression of BIM (also known as BCL2L11) and bridging integrator 1 (BIN1) protein isoforms without pro-apoptotic functions41. A recent study proposed that SRSF3 is also an oncoprotein when overexpressed43. Consistent with this, SRSF3 downregulation promoted p53-mediated cellular senescence in part by promoting the expression of the p53β isoform44. SRSF6 has also been characterized as a proto-oncogene that is frequently overexpressed in human skin cancer45. Transgenic overexpression of SRSF6 from the collagen type Iα1 (Col1a1) locus in mice induced hyperplasia of skin sensitized by shaving or wounding, partially through aberrant alternative splicing of tenascin C (Tnc)45. SRSF6 may act as an oncoprotein in lung and colon cancer as well46.
hnRNPs have been implicated in cancer in both pro- and anti-tumorigenic capacities. For instance, two studies reported that MYC-mediated upregulation of specific hnRNPs (hnRNP A1, A2 and PTB) resulted in exclusion of exon 9 of pyruvate kinase muscle (PKM), thus promoting expression of the cancer-associated embryonic PKM2 isoform and aerobic glycolysis in glioma47,48. Expression of the constitutively active variant of epidermal growth factor receptor (EGFR), EGFRvIII, in gliomas was shown to upregulate hnRNP A1 which, in turn, contributed to alternative splicing of MYC associated factor X (MAX) to produce delta MAX and further promote glycolytic gene expression and proliferation49. The splicing factor hnRNP A2/B1 also acts in a pro-tumorigenic capacity. hnRNP A2/B1 is overexpressed in gliomas, where it correlates with poor prognosis, and its overexpression transforms cells in vitro50.
Conversely, other hnRNPs may act as tumor suppressors. Motivated by the observation that HNRNPK (among other genes) lies within a chromosomal locus that is recurrently deleted in acute myeloid leukemia (AML)51, one recent mouse study52 found that deletion of one allele of Hnrnpk resulted in myeloid hematologic transformation. However, it remains unknown whether the observed myeloid transformation phenotype is due to changes in splicing or other biological pathways. hnRNP K has been implicated in many biological processes in addition to RNA splicing, including cellular proliferation and cellular senescence through its effects on p5353 and p2152, as well as myeloid differentiation through its effects on CCAAT/enhancer-binding protein-α (C/EBPα) and C/EBPβ52.
Many splicing factors other than SR proteins and hnRNPs have also been implicated in cancer in pro- as well as anti-tumorigenic capacities. For example, the STAR family protein quaking (QKI) may act as a tumor suppressor in lung cancer, where it is commonly downregulated54. QKI overexpression inhibits the proliferation of lung cancer cells in vitro and in vivo, in part by regulating the alternative splicing of NUMB54. Other splicing factors are also linked to cancer by their regulation of NUMB. RNA binding motif protein 5 (RBM5), RBM6 and RBM10, which encode homologous RNA-binding proteins, are commonly deleted, mutated, and/or under- or overexpressed in lung and other cancers55–59. RBM5 or RBM6 depletion has an opposite effect to RBM10 depletion on in vitro colony formation. This is partially due to the antagonistic functions of these factors in regulating the alternative splicing of NUMB60. RBM4 is also downregulated in multiple cancers and has been characterized as a tumor suppressor that regulates BCLX (also known as BCL2L1) splicing and antagonizes the pro-tumor activity of SRSF161. The epithelial to mesenchymal transition (EMT) is regulated by splicing factors including epithelial splicing regulatory protein 1 (ESRP1; also known as RBM35A) and ESRP2 (also known as RBM35B), which promote epithelial splicing programs in breast cancer, and RBFOX2, which promotes mesenchymal splicing programs in breast cancer62,63. MBNL and CELF proteins and hnRNPs have also been implicated in EMT63, highlighting the complexity of cancer-associated splicing dysregulation.
As described above, both functional and prognostic data indicate that many splicing factors play pro- or anti-tumorigenic roles. In a few cases, such as for hnRNP K, RBM5, RBM6 and RBM10, genetic changes such as chromosomal deletions may alter the expression of splicing factors. However, in general, the splicing factors described above are not known to be subject to recurrent, high-frequency mutations in cancer. Although these data suggest that dysregulated expression of splicing factors plays important roles in tumor development or progression, thus far there is limited functional evidence that altered levels of splicing factors alone can drive cancer initiation or that altered levels of splicing factors are required for cancer maintenance.
Recurrent mutations in spliceosomal genes in cancer
The discovery of recurrent somatic mutations in genes encoding core spliceosomal proteins throughout diverse cancer types provided the first genetic evidence directly linking RNA splicing regulation to cancer. These spliceosomal mutations were initially discovered in hematological malignancies including myelodysplastic syndromes (MDS)8–10 and other myeloid neoplasms as well as chronic lymphocytic leukemia (CLL)11,12 (Fig. 2a and Table 2). More recently, recurrent spliceosomal mutations have also been found in several solid tumor types, including uveal melanoma (18.6%)64,65, pancreatic ductal adenocarcinoma (4%)66, lung adenocarcinoma (3%)56 and breast cancers (1.8%)67–70. Most reported spliceosomal mutations are concentrated in four genes: splicing factor 3B, subunit 1 (SF3B1), serine/arginine-rich splicing factor 2 (SRSF2), U2 small nuclear RNA auxiliary factor 1 (U2AF1) and zinc finger, RNA-binding motif and serine/arginine-rich 2 (ZRSR2)8–10.
Figure 2. Commonly mutated spliceosomal proteins and their associations with specific cancer types.
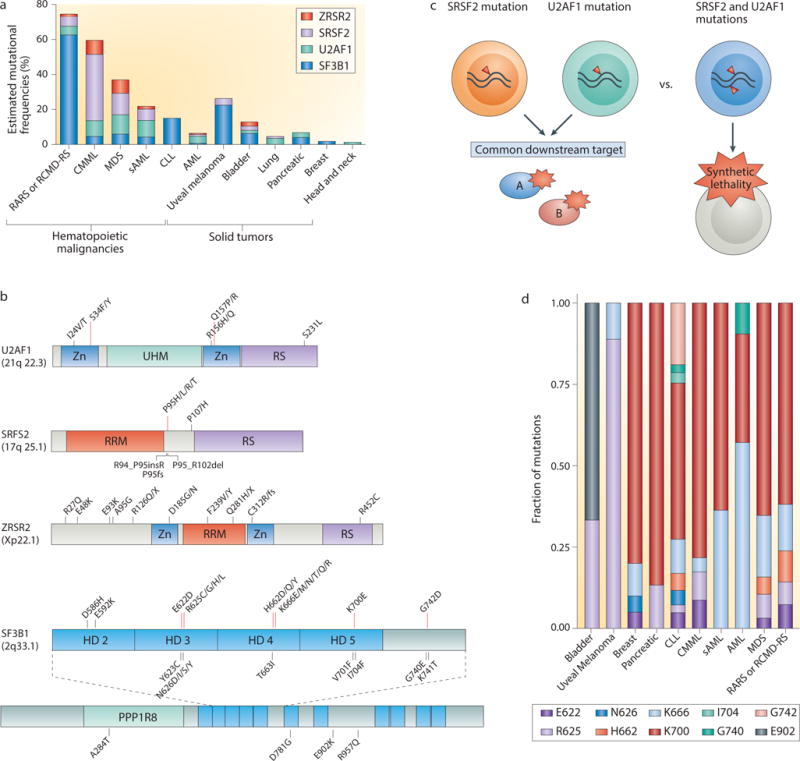
(a) The incidence and cancer distribution of the most frequently mutated spliceosomal genes are shown. (b) The spectrum of mutations that have been identified in U2AF1 (U2 small nuclear RNA auxiliary factor 1), SRSF2 (serine/arginine-rich splicing factor 2), ZRSR2 (zinc finger, RNA-binding motif and serine/arginine-rich 2) and SF3B1 (splicing factor 3B, subunit 1). Mutations shown in bold text occur at hotspots; other illustrated mutations are recurrent but rare. As ZRSR2 mutations do not occur at hotspots, very rare or private mutations are shown as examples. (c) Schematic illustrating the potential reasons that spliceosomal gene mutations are mutually exclusive with one another: they either converge on a common downstream target or result in synthetic lethality. (d) The frequency of specific mutations in SF3B1 across various histological subtypes of cancer. AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CMML, chronic myelomonocytic leukemia; HD, HEAT domain; PPP1R8, binding site for protein phosphatase 1 regulatory subunit 8; RARS, refractory anemia with ringed sideroblasts; RCMD-RS, refractory cytopenia with multilineage dysplasia and ringed sideroblasts; RRM, RNA recognition motif; RS, arginine/serine-rich domain; sAML, secondary AML; UHM, U2AF homology motif; ZN, zinc finger domain.
Table 2.
Recurrent mutations affecting splicing factors in cancer and their disease associations.
| Splicing factor | Diseases | Mutated residue* | Frequencies | Clinical associations |
|---|---|---|---|---|
| SF3B1 | RARS | K700, K666, H662, R625, E622 | 48 – 75.3% | Increased event-free or overall survival, lower risk of transformation to AML10 |
| CMML | K700, K666, R625, E622 | 4.3 – 4.7% | ||
| MDS | K700, K666, H662, R625, E622 | 4.4 – 7% | ||
| sAML | K700, K666 | 3.7 – 4.8% | ||
| AML | K700, K666, G740 | 2.6 – 7.3% | ||
| CLL | K700, K666, H662, G742 | 15% | Reduced treatment-free or overall survival, faster disease progression11,12,79 | |
| Pancreatic | K700, R625 | 4% | – | |
| Breast | K700, K666, E622, N626 | 1.80% | – | |
| Uveal melanoma | R625, K666 | 15 – 20% | Relatively favorable prognosis64,65 | |
| Bladder | R625, E902 | 4% | – | |
| U2AF1 | RARS | S34, Q157 | 5% | Less favorable overall survival, higher risk of transformation to AML9 |
| CMML | 8 – 11% | |||
| MDS | 10.3 – 11.6% | |||
| sAML | 9.7% | |||
| AML | 1.3% | |||
| Lung | S34 | 3% | Reduced progression-free survival56 | |
| SRSF2 | RARS | P95 | 5.5% | Unfavorable prognostic factor for AML transformation, reduced overall survival206 |
| CMML | 28 – 47% | |||
| MDS | 11.6% | |||
| sAML | 6.5% | |||
| AML | 0.7% | |||
| ZRSR2 | RARS | No evident mutational hotspot (presumed loss-of-function) | 1.4–10% | Higher risk of transformation to AML207 |
| CMML | 8.0% | |||
| MDS | 7.7% | |||
| sAML | 1.6% |
AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CMML, chronic myelomonocytic leukemia; MDS, myelodysplastic syndromes; RARS, refractory anemia with ring sideroblasts; sAML, secondary AML; SF3B1, splicing factor 3B, subunit 1; SRSF2, serine/arginine-rich splicing factor 2; U2AF1, U2 small nuclear RNA auxiliary factor 1.
Only residues recurrently affected by somatic mutations are listed.
Several features of spliceosomal gene mutations immediately suggested their potential functional consequences. First, with the exception of ZRSR2 mutations, these mutations affect highly restricted amino acid residues in an exclusively heterozygous state with the wild-type allele (Fig. 2b). These data suggest that mutations in most spliceosomal genes likely confer gain or alteration of function, except ZRSR2 mutations, which follow a pattern consistent with loss of function. Second, splicing factor mutations are mutually exclusive of one another, which may be due to either functional redundancy or synthetic lethality, possibilities that have not yet been explored in published studies (Fig. 2c).
These genetic observations suggest that SF3B1, SRSF2 and U2AF1 may be proto-oncogenes, as they are subject to highly specific missense mutations are suggestive of gain or alteration of function. In contrast, ZRSR2 may play a tumor suppressor role, as ZRSR2 mutations frequently introduce in-frame stop codons or disrupt the reading frame, likely inactivating the gene and/or protein. Functional evidence supporting pro-oncogenic versus tumor suppressor roles for these four proteins is described below.
Although each of these four proteins have some impact on 3′ splice site recognition, it is not clear why they are the targets of recurrent mutations in cancer. The >30 other proteins which comprise the U2 snRNP, the U2AF complex or the SR protein family are not recurrently mutated despite having similarly important roles in 3′ splice site and exon recognition. The highly specific nature of spliceosomal mutations suggests that these mutations, and presumably not others, alter RNA:protein and/or protein:protein interactions in a manner that promotes a specific set of downstream changes that are critical for oncogenesis.
Specific splicing factors are most frequently mutated in distinct cancer subtypes (Fig. 2a). For example, SF3B1 is the only splicing factor that has been identified as a common mutational target in breast cancer67–70 (based on cohorts sequenced to date), whereas U2AF1 is the most commonly mutated factor in non-small cell lung cancer56. Moreover, in diseases such as MDS in which multiple splicing factors are commonly mutated, specific splicing factor mutations associate with distinct MDS subtypes. Mutations in SF3B1 are highly enriched in refractory anemia with ringed sideroblasts (RARS), a form of MDS characterized by a typically indolent course, anemia, and the accumulation of erythroid precursor cells with abnormally iron-loaded mitochondria8,10. In contrast, SRSF2 is the most commonly mutated splicing factor in MDS with a more fulminant course71 (Fig. 2a). Currently, the mechanistic basis for this association of different splicing factors with distinct histological subtypes of cancer is not known. Moreover, specific mutated residues in SF3B1 appear to be associated with distinct diseases (Fig. 2d). For instance, SF3B1R625 mutations represent the most common SF3B1 mutation in uveal melanoma, yet are far less frequent in hematological malignancies64,65. Similarly, U2AF1 mutations affect both the S34 and Q157 residues in hematopoietic malignancies8,9, but only mutations affecting S34 have been identified in lung cancer56.
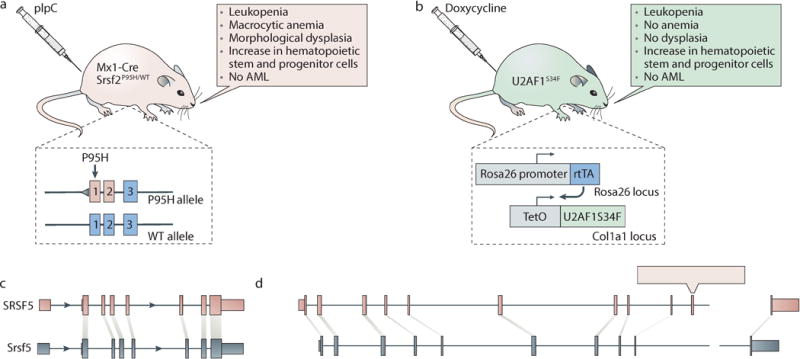
Knock-in mouse models in which mutated spliceosomal genes are expressed from their endogenous loci have been created for Srsf2 (two distinct models)72,73 and Sf3b174,75. A transgenic model of inducible U2AF1S34F expression has also been described76 (Box 2). Phenotypic analyses of a Srsf2P95H conditional knock-in model72 and the transgenic U2AF1S34F model76 revealed that the expression of these mutated proteins in post-natal hematopoietic tissue recapitulated key features of MDS, including leukopenia and increased numbers of hematopoietic stem and myeloid progenitor cells. Srsf2P95H knock-in mice developed morphological dysplasia whereas the transgenic U2AF1S34F mice did not. However, the transcriptomes of hematopoietic stem or progenitor cells (HSPCs) from each model showed that these cells had the same genome-wide alterations in exonic splicing enhancer (Srsf2P95H) and 3′ splice site (U2AF1S34F) preferences as those that were observed in patients’ cells with these mutations. Additionally, in both the Srsf2P95H and U2AF1S34F mouse models, there were some genes that were differentially spliced in mouse cells as well as patient cells, suggesting that the models recapitulate many molecular phenotypes of human disease.
Box 2. Models of spliceosomal gene mutations in cancer.
The genetic heterogeneity of primary patient samples render genetic models important tools when studying the roles of spliceosomal mutations in cancer pathogenesis. As the wild-type and mutant alleles of spliceosomal genes are consistently co-expressed at similar levels in patients, mimicking this expression pattern is likely important in order to create biologically realistic genetic models. Analyses of the hematopoietic transcriptomes of Srsf2P95H conditional knock-in72 (shown in figure panel a; light pink) and U2AF1S34F transgenic76 (shown in figure panel b; light green) murine models demonstrated that the mechanistic alterations in exon and splice site recognition induced by these mutations are conserved between human and mouse and validate these murine models for mechanistic studies of the role of splicing alterations in cancer pathogenesis. These observations are consistent with the deep conservation of SRSF2 and U2AF1 across eukaryotes. However, even though the functions of these spliceosomal proteins are conserved, the sets of downstream mis-spliced genes may differ between human and murine models due to species-specific differences in splicing. (c) Tissue-specific splicing events are more frequently conserved between human (pink) and mouse (blue) than their unregulated counterparts, as is the flanking intronic sequence14,192. This even extends to conservation between the human and fungal genomes for a splicing event that enables autoregulation of SRSF5141. (d) Nonetheless, the majority of alternative isoforms are species-specific184,193,194, such as the illustrated example of CD55 (pink, human exons; blue, mouse exons). This situation—where a subset of regulated isoforms is highly conserved, but most alternative splicing events are likely not to be—is analogous to that observed for transcription factor binding, in which detailed studies of transcription factors active in the liver revealed that the majority of binding sites are species-specific195,196. It remains to be determined what fraction of species-specific isoforms contribute to phenotypic differences between species compared with those that constitute non-functional products of noisy splicing. However, as only approximately one quarter of alternative exons are conserved between human and mouse184, mis-spliced isoforms that are relevant to human disease pathogenesis yet absent from the mouse genome are likely to exist. Given these differences between species, the parallel use of genome-engineering techniques to generate isogenic human cell lines carrying spliceosomal gene mutations in endogenous loci may prove particularly useful for detailed transcriptional studies. For example, Zhang et al.98 recently generated SRSF2P95H knock-in K562 cells to identify the changes in exon recognition and differential splicing induced by SRSF2 mutations. Combined studies of murine models, isogenic human cells, and patient cohorts will likely prove essential to identify the direct targets of mutant spliceosomal proteins with cancer relevance.
The high frequencies with which SF3B1, SRSF2, U2AF1 and ZRSR2 are subject to specific mutations in cancer suggest that spliceosomal mutations drive tumorigenesis, at least in some cellular contexts, and are not merely passenger mutations. Spliceosomal mutations are likely to occur as both initiating and secondary genetic events, a distinction that has been best studied in liquid neoplasms. The clonal architecture of MDS indicates that SF3B1 mutations are initiating genetic events77. Similar studies of secondary AML revealed that SRSF2, U2AF1 and ZRSR2 mutations also occurred early during the leukemogenic process78. In contrast, even though SF3B1 is the second most commonly mutated gene in CLL, SF3B1 mutations occur most frequently in advanced versus early disease, suggesting that they are secondary genetic events that facilitate progression11,79.
Mutations affecting 3′ splice site recognition via U2
SF3B1 is mutated at significant rates in both hematological and solid cancers, including MDS, CLL, uveal melanoma and breast cancer, rendering it the most commonly mutated spliceosomal gene (Fig. 2a). It encodes a core spliceosomal protein that binds upstream of the pre-mRNA branch site in a manner that is likely to be largely sequence independent. SF3B1 is probably required for the recognition of most 3′ splice sites17. SF3B1 mutations are concentrated in sequence encoding its HEAT repeat domains (Fig. 2b); however, the normal function of these domains is poorly characterized, rendering it difficult to predict the mechanistic consequences of SF3B1 mutations. DeBoever et al.80 recently reported that SF3B1 mutations were associated with enhanced recognition of cryptic 3′ splice sites between the branch point and normal 3′ splice site. While U2 snRNP binding to the branch point normally prevents recognition of AG dinucleotides immediately downstream of the branch point by steric occlusion, DeBoever et al.80 hypothesized that SF3B1 mutations prevent this normal steric occlusion, thereby enhancing recognition of cryptic splice sites (Fig. 3a). Darman et al.81 similarly reported that mutant SF3B1 enhanced recognition of intron-proximal cryptic 3′ splice sites, which frequently involved normally unused upstream branch points. However, the exact mechanism(s) by which mutations might alter SF3B1 interactions with pre-mRNA, components of the U2 snRNP or other proteins remains unknown. A precise understanding of how mutations alter the role of SF3B1 in RNA splicing will likely require further studies of normal SF3B1 function.
Figure 3. Current understanding of the mechanistic consequences of spliceosomal gene mutations for RNA splicing.
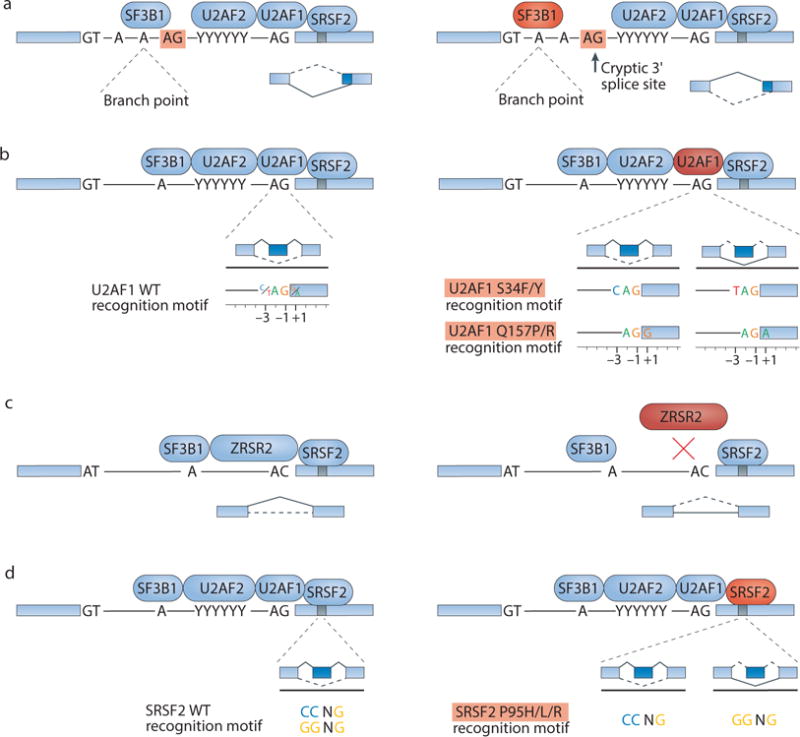
(a) Splicing factor 3B, subunit 1 (SF3B1) mutations (mutant form shown in red) alter 3′ splice site (ss) selection by permitting or enhancing recognition of cryptic upstream 3′ splice sites. It is not yet known how mutations affecting SF3B1 alter its target protein:RNA and/or protein:protein interactions to drive this cryptic splice site recognition. Sequence motifs are illustrated as genomic DNA sequence rather than pre-mRNA sequence (“T” instead of “U”). (b) U2 small nuclear RNA auxiliary factor 1 (U2AF1) mutations alter 3′ splice site consensus sequences. Wild-type U2AF1 recognizes the consensus motif yAG|r at the intron|exon boundary (y = pyrimidine, r = purine, ‘|’ = intron-exon boundary). S34F or S34Y mutations (shown in red) promote recognition of cAG|r over tAG|r, whereas Q157P or Q157R mutations (shown in red) promote recognition of yAG|g over yAG|a. (c) Zinc finger, RNA-binding motif and serine/arginine-rich 2 (ZRSR2) mutations (shown in red) cause loss of ZRSR2 function to induce splicing defects, primarily involving the aberrant retention of U12-type introns. (d) Serine/arginine-rich splicing factor 2 (SRSF2) mutations (shown in red) alter exonic splicing enhancer (ESE) preferences. Wild-type SRSF2 recognizes the consensus motif SSNG (S = C or G), whereas mutant SRSF2 preferentially recognizes the CCNG motif over the GGNG motif.
The consequences of SF3B1 mutations may be cell type-dependent and/or allele-specific, as different SF3B1 mutations may not be phenotypically equivalent. SF3B1 mutations in MDS versus CLL constitute initial versus secondary genetic insults and associate with favorable versus poor prognosis, respectively10–12,79. Different SF3B1 mutations are more enriched in MDS compared with CLL and other cancers (Fig. 2d).
Despite the close association between SF3B1 mutations and the presence of ring sideroblasts in MDS, no studies have clearly demonstrated that SF3B1 mutations induce abnormal iron metabolism. However, the recent report of Darman et al.81 that ATP-binding cassette subfamily B member 7 (ABCB7) is mis-spliced in SF3B1-mutant cells hints at possible connections between altered splicing and sideroblastic anemia. ABCB7 encodes an iron transporter that is essential for hematopoiesis and that is mutated in X-linked sideroblastic anemia with ataxia, a genetic disease that is also characterized by the presence of ring sideroblasts82,83.
U2AF1 is mutated in both liquid and solid tumors, with the highest reported rates in MDS and lung cancer (Fig. 2a). Unlike SF3B1, which is required for recognition of many or most 3′ splice sites, U2AF1 binds the 3′ splice site in a highly sequence-specific manner, and is only required for recognition of a subset of ‘AG-dependent’ 3′ splice sites84,85. U2AF1 mutations are concentrated in sequence encoding the S34 and Q157 residues, which lie within the first and second C3H zinc ‘knuckles’ of the protein (Fig. 2b). In hematological cancer, such mutations have been shown to induce sequence-specific alterations in the preferred RNA motif bound by U2AF1, which normally recognizes the motif yAG|r (y = pyrimidine; r = purine; lower-case nucleotides are preferred but not always required, while upper-case nucleotides are usually required; ‘|’ = intron-exon boundary)86–89 (Fig. 3b). Interestingly, the molecular consequences of U2AF1 mutations are allele-specific88. S34 and Q157 mutations respectively affect recognition of the −3 (pyrimidine normally preferred) and +1 (purine normally preferred) positions, where the coordinates are defined with respect to the intron-exon boundary to induce different changes in 3′ splice site recognition. At a global level, S34 mutations promote recognition of 3′ splice sites with cAG|r and aAG|r motifs over those with tAG|r, whereas Q157 mutations promote 3′ splice sites with yAG|g motifs over those with yAG|a motifs.
Many downstream targets of mutant U2AF1 have been identified using patient transcriptomes, a transgenic mouse model of U2AF1S34F and transgenic human cells bearing each of the common U2AF1 mutations affecting the S34 and Q157 residues76,86–89. These mis-spliced genes fall into biological pathways including the DNA damage response (ataxia telangiectasia- and Rad3-related (ATR) and Fanconi anemia complementation group A (FANCA)), epigenetic regulation (H2A histone family member Y (H2AFY)), and apoptosis (caspase 8 (CASP8))76,88. Intriguingly, several of the identified target genes are recurrently mutated in MDS (for example, BCL6 corepressor (BCOR)90) and other cancers (for example, CASP867,91). However, functional studies are needed to determine whether these and other mis-spliced genes contribute to the disease process.
Mutations affecting 3′ splice site recognition via U12
ZRSR2 mutations found in MDS are distributed throughout the gene, which lies on the X chromosome (Xp22.1), and frequently interrupt the coding sequence by directly or indirectly introducing in-frame stop codons (Fig. 2a). Together with the common occurrence of ZRSR2 mutations in male patients with cancer8, this pattern is consistent with loss of function. ZRSR2 mutations therefore contrast with the mutations observed in the spliceosomal genes SF3B1, SRSF2 and U2AF1, which cause missense changes at specific residues and never introduce in-frame stop codons (Fig. 2b).
Whereas biochemical assays suggested that ZRSR2 promotes recognition of both U2- and U12-type introns, the phylogenetic observation that organisms with U12-type introns have ZRSR2 and those lacking U12-type introns also lack ZRSR2 suggested that ZRSR2 is particularly important for U12-type splicing92. Madan et al.93 reported that MDS transcriptomes harboring mutations likely to inactivate ZRSR2 are characterized by frequent retention of U12-type introns, consistent with a crucial role for ZRSR2 in the minor spliceosome (Fig. 3c). ZRSR2 knockdown altered the in vitro differentiation potential of cord blood-derived CD34+ cells by promoting myeloid differentiation and impairing erythroid differentiation93, consistent with features of human MDS. However, ZRSR2 knockdown also impaired the growth of K562 cells in vitro and following subcutaneous xenografting in vivo93, indicating that ZRSR2 loss does not convey a proliferative advantage in the K562 genetic background. ZRSR2 mutations were associated with mis-splicing of genes relevant to the MAPK pathway and E2F transcription factor signaling93, but functional experiments are needed to determine whether these or other splicing changes contribute to the hematopoietic phenotypes of ZRSR2-deficient cells.
Mutations affecting exon recognition
SRSF2 mutations appear most commonly (in 40–50% of patients8) in chronic myelomonocytic leukemia (CMML), and are also enriched in subtypes of high-risk MDS, where they portend an increased risk of transformation to acute leukemia94 (Fig. 2a). SRSF2 encodes a member of the SR protein family that contributes to both constitutive and alternative splicing. SRSF2 canonically promotes exon recognition by binding to exonic splicing enhancer (ESE) motifs within pre-mRNAs through its RRM domain95–97. All recurrent SRSF2 mutations affect the P95 residue8, which is immediately downstream of the RRM domain (Fig. 2b). RNA-seq analyses of hematopoietic stem and progenitor cells from Srsf2P95H knock-in mice72 and transgenic72 and knock-in98 K562 cells expressing SRSF2P95H/L/R, and human AML and CMML patients with or without SRSF2 mutations72, revealed that SRSF2 mutations alter its normal sequence-specific RNA-binding activity. Mutant SRSF2 preferentially recognizes a C-rich CCNG motif versus a G-rich GGNG motif, whereas wild-type SRSF2 binds both motifs with similar affinity72,98,99 (Fig. 3d). These alterations in the RNA-binding activity of SRSF2 promote or repress recognition of exons containing C- or G-rich ESEs72,98.
Altered ESE recognition causes widespread splicing changes in hundreds of genes72,98. Several of these downstream mis-spliced genes are themselves recurrent mutational targets in myeloid malignancies, including enhancer of zeste homolog 2 (EZH2)100,101 and BCOR90. SRSF2 mutations promote inclusion of a ‘poison exon’ of EZH2 that introduces an in-frame stop codon to induce nonsense-mediated decay (NMD) of the EZH2 transcript and consequent global downregulation of EZH2 protein and histone H3 lysine 27 trimethylation (H3K27me3) levels72. Loss-of-function mutations in EZH2 occur in MDS100,101, and Ezh2 loss has been functionally linked to MDS development and aberrant hematopoietic stem cell self-renewal in vivo102. Therefore, decreased EZH2 levels may partially explain how SRSF2 mutations drive MDS, and also explain the previously observed mutual exclusivity of SRSF2 and EZH2 mutations in MDS71,103.
Mutations affecting other splicing factors
In addition to SF3B1, SRSF2, U2AF1 and ZRSR2, other genes encoding splicing factors are recurrently mutated in cancer. Pre-mRNA processing factor 8 (PRPF8) is subjected to mutations or hemizygous deletions in 1–5% of patients with myeloid leukemias104. Biochemical studies in yeast suggest that PRPF8 mutations may affect recognition of suboptimal 3′ splice sites104.
Genes encoding splicing factors have also been identified as recurrent targets of translocations in cancer. For example, splicing factor proline/glutamine-rich (SFPQ; also known as PSF) is reportedly recurrently fused to ABL1 in B-cell acute lymphoblastic leukemia105 and to transcription factor binding to IGHM enhancer 3 (TFE3) in papillary renal cell carcinomas106. SRSF3 is also reportedly involved in rare fusions with BCL6 in B-cell lymphomas107. Currently, it is not known if these fusions affect the function of the splicing factors involved in the chimeric protein product. In the case of SFPQ fusions, the sequence encoding the coiled-coil domain (which is important for protein dimerization) of SFPQ appears to be consistently included in the chimeric transcript108, suggesting that SFPQ fusions may contribute to cancer by promoting aberrant dimerization of SFPQ’s partner protein.
Inherited genetic variation affecting RNA splicing factors has also been implicated in cancer. A missense genetic variant in serine/arginine repetitive matrix 2 (SRRM2; also known as SRm300) was recently found to segregate with incidence of familial papillary thyroid carcinoma109. Patients carrying this SRRM2 variant exhibited mis-splicing of specific cassette exons, suggesting that the variant altered the normal function of SRRM2 in splicing. Somatic mutations may also interact with genetic variants to promote cancer. Recent work identified both somatic mutations and genetic variants affecting DEAD-box helicase 41 (DDX41) that are associated with high-penetrance familial MDS and AML110. Although the normal molecular role of DDX41 is incompletely understood, mass spectrometry data indicated that DDX41 interacts with core spliceosome components and that the likely loss-of-function DDX41 mutations perturb these interactions. Therefore, DDX41 may play a role in RNA splicing that is disrupted by MDS and AML – associated mutations, although that hypothesis remains to be tested.
Connections to other cellular processes
Splicing factor dysregulation, including spliceosomal mutations, may directly or indirectly affect many cellular processes in addition to RNA splicing. These processes include the maintenance of genome integrity, epigenetic regulation, transcription, nuclear export, and translation-dependent mRNA decay.
Depletion of SRSF1 or SRSF2 gives rise to DNA damage and genomic instability via the formation of RNA:DNA hybrids (‘R loops’)111,112, which expose the unhybridized DNA strand to DNA damage (Fig. 4a). Splicing factors are also linked to the DNA damage response. For example, BRCA1 is reportedly physically associated with SF3B1 and U2AF1 specifically in response to DNA damage113. A recent study proposed that the spliceosome is an effector of ataxia-telangiectasia mutated (ATM) signaling, wherein DNA lesions displace spliceosomes, resulting in R loop formation and ATM activation114.
Figure 4. Links between splicing factors and diverse biological processes and potential methods for therapeutic manipulation of splicing.
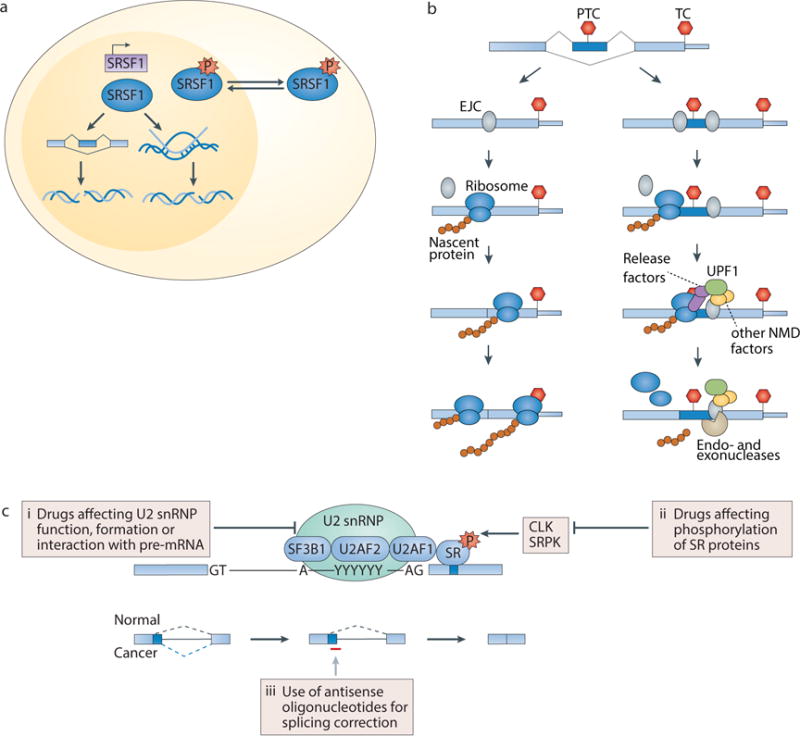
(a) Changes in the abundance, post-translational modifications and/or subcellular localization of splicing factors such as serine/arginine-rich splicing factor 1 (SRSF1) can cause DNA damage or influence the DNA damage response (DDR). Splicing factors have been linked to DNA damage and the DDR both directly (for example, insufficient levels of splicing factors can cause R loop formation and genomic instability) and indirectly (for example, through downstream changes in splicing). (b) The steps involved in nonsense-mediated decay (NMD) are shown. The exon junction complex (EJC; grey) is deposited upstream of exon-exon junctions on the processed mRNA, and is displaced by the ribosome (blue) during the pioneer (first) round of translation. Ribosome stalling at a premature termination codon (PTC) >50 nt upstream of an EJC promotes interactions between release factors (purple) and UPF1 (green), recruitment of other NMD components (orange), and RNA degradation by endo- and exonucleases (beige). In contrast, if only a normal termination codon (TC) is present, then all EJCs are displaced by the ribosome during the pioneer round of translation and NMD is not triggered. Red stop signs indicate stop codons. (c) Compounds and oligonucleotides that can disrupt or modulate normal splicing catalysis or alter splice site recognition through distinct pathways include (i) drugs affecting U2 small nuclear ribonucleoprotein (snRNP) function, formation, and/or interaction with pre-mRNA, (ii) drugs affecting post-translational modifications of serine/arginine-rich (SR) proteins and potentially other splicing factors, and (iii) oligonucleotides to manipulate specific mRNA isoforms that may be important in tumor maintenance. CLK, CDC2-like kinase; SF3B1, splicing factor 3B, subunit 1; SRPK, SR protein kinase.
Splicing is also closely linked to epigenetic regulation. Nucleosomes are preferentially positioned over exons versus introns115,116. SF3B1 is preferentially associated with nucleosomes positioned over exons, facilitating recognition of these exons116. Histone H3 lysine 36 trimethylation (H3K36me3) is further enriched over exons117,118, and modulation of H3K36me3 can influence splice site choice119. Conversely, modifying splice site recognition can influence H3K36me3 deposition120,121. The literature linking splicing and epigenetics is reviewed in more detail in Refs122,123.
Connections between splicing and epigenetics may contribute to the oncogenic activity of spliceosomal mutations. As described above, mutant SRSF2 prevents hematopoiesis in part by promoting a non-functional isoform of EZH2, resulting in global decreases in H3K27me3 levels72. Mutant U2AF1 promotes a cancer-associated isoform of the histone variant macro-H2A.176,88. Connections between SF3B1 mutations and epigenetic dysregulation have not been identified, but are plausible given the published links between splice site recognition and chromatin described above. However, further studies are needed to determine whether potential epigenetic changes caused by U2AF1 and/or SF3B1 mutations are important for cancer initiation and progression.
Splicing factor dysregulation may also affect transcription independently of epigenetic regulation. For example, SRSF2 regulates transcriptional elongation in a sequence-specific manner via the 7SK complex that governs RNA polymerase II pause release124. Similarly, hnRNP A1, hnRNP A2 and hnRNP K are linked to cancer, and hnRNP A1 and A2 have reported roles in transcription elongation whereas hnRNP K is linked to transcription termination125,126. Many splicing factors co-transcriptionally associate with the C-terminal domain of RNA polymerase II, thereby coupling transcription, splicing, cleavage and polyadenylation (reviewed in127).
Finally, molecular changes induced by splicing factor dysregulation may not be confined to the nucleus. Multiple SR proteins facilitate nuclear export of both unspliced and spliced mRNAs128,129. SRSF1 shuttles between the nucleus and cytoplasm and promotes cap-dependent translation of bound mRNAs in a eukaryotic translation initiation factor 4E (eIF4E)-dependent manner130,131. SRSF1 activates the mammalian target of rapamycin (mTOR) signaling pathway, which is required for SRSF1-mediated cell transformation40,42. Splicing itself promotes translation via the deposition of multiprotein exon-junction complexes (EJCs) near exon-exon junctions132.
NMD provides a concrete example of a cytoplasmic process that is likely affected by cancer-associated mutations affecting SF3B1, SRSF2, U2AF1 and ZRSR2, even though those proteins localize to the nucleus. NMD is a translation-dependent RNA surveillance process that degrades mRNAs containing premature termination codons (Fig. 4b; reviewed in133,134). Splicing and NMD are closely linked in human cells for several reasons. First, many NMD substrates are generated by alternative splicing, wherein premature termination codons are introduced via inclusion of alternatively spliced sequence containing an in-frame premature stop codon or exclusion of sequence resulting in a frameshift. Second, stop codons are recognized as premature by the NMD machinery if they lie >50 nucleotides upstream of a splice junction133,134. The 50 nucleotide threshold arises because the translating ribosome dislodges EJCs and prevents EJC-facilitated activation of NMD. However, if the ribosome stalls sufficiently upstream of an EJC, then NMD factors are recruited and activated133,134. Third, specific splicing factors including SRSF1, regulator of differentiation 1 (ROD1; also known as PTBP3) and PTB can enhance or repress NMD135–137.
Human cells express an abundance of mRNAs containing premature termination codons (one-third of all alternatively spliced isoforms by one estimate138), including the EZH2 poison exon that is promoted by SRSF2 mutations72. A subset of poison exons are among the most evolutionarily conserved elements in the human genome139–141. These poison exons enable splicing factors to post-transcriptionally down- or upregulate expression of specific genes, including the genes encoding many splicing factors themselves, likely explaining the extreme sequence conservation of many poison exons139–141.
Interestingly, in the earliest report of splicing factor mutations, genes involved in NMD were upregulated following overexpression of mutant U2AF18, suggesting a potential link between spliceosomal mutations and overproduction of NMD substrates. However, such high levels of NMD substrates have not been observed in subsequent studies of mutations affecting U2AF1 or other splicing factors.
The recent discovery of recurrent mutations in UPF1, which encodes a RNA helicase that is central to NMD, in pancreatic adenosquamous carcinoma provided a genetic link between NMD and cancer142. The observed mutations induced abnormal UPF1 splicing and skipping of sequence encoding core domains, potentially resulting in partial or complete loss of UPF1 function, although further work is required to determine how these mutations affect global RNA surveillance. Deficiencies in different NMD factors have been previously linked to disorders including intellectual disability143, thrombocytopenia with absent radii syndrome144 and muscular dystrophy145.
Implications for therapy
Given the crucial roles of specific alternatively spliced isoforms in cancer biology146,147, as well as the potentially increased sensitivity of cancers to global perturbation of splicing efficiency relative to normal cells148,149, pharmacological modulation of splicing may represent an important therapeutic strategy. Spliceosomal gene mutations that cause alteration or gain of function are mutually exclusive with one another and are always co-expressed with a wild-type allele, suggesting that cells bearing spliceosomal mutations may be unable to tolerate further perturbations in splicing, and could therefore be preferentially sensitive to pharmacological splicing inhibition. A number of compounds and oligonucleotides that can disrupt or modulate normal splicing catalysis or alter splice site recognition through distinct pathways have been identified (Fig. 4c).
Several compounds with antitumor activity were identified through natural product screens prior to the discovery of spliceosomal gene mutations. Synthetic analogues with higher stability, solubility and activity were subsequently developed, including E7107, meayamycin, spliceostatin A, and sudemycins (reviewed in150,151). Biochemical studies identified SF3B1 as the likely target of these drugs, consistent with the observation that mutations affecting the R1074 residue of SF3B1 confer resistance to pladienolide and E7107152,153. Unfortunately, two separate Phase I clinical trials of E7107 revealed an unexpected and unexplained side effect of visual disturbances in 5% of subjects154,155. Further efforts are needed to determine whether this was an on- or off-target effect of U2 snRNP inhibition in vivo. In the meantime, several preclinical studies are evaluating the utility and safety of sudemycins156,157 for cancer therapy.
Although the origin of splicing inhibitors’ general antitumor activities is unknown, two studies provided evidence that MYC expression renders cells sensitive to compounds that inhibit 3′ splice site recognition148,149. More recently, Lee et al.158 reported that the splicing inhibitor E7107 reduced the leukemic burden and prolonged survival of mice carrying oncogene-driven myeloid leukemias if the leukemias had Srsf2 mutations, but not if the leukemias expressed only wild-type Srsf2. Lee et al. observed similarly specific targeting of patient-derived xenograft (PDX) models of leukemias with spliceosomal mutations. These data suggest that splicing inhibitors such as E7107 are synthetically lethal with genetic lesions affecting the spliceosome.
Interventions that target post-translational modification of splicing factors might also prove effective for therapy. For example, SR proteins are phosphorylated by kinases including topoisomerase I and members of the SR protein kinase (SRPK) and CDC2-like kinase (CLK) families159–161. These kinases affect SR protein subcellular localization and splicing activity162,163, exhibit altered expression and/or activity in cancer164,165, and can potentially act as oncogenes165. Small molecules that block activity of these kinases have been identified, including TG003, an inhibitor of CLK1 and CLK4, and SRPIN340, an SRPK1 and SRPK2 inhibitor that inhibits angiogenesis166,167. In addition, indole derivatives, such as benzopyridoindoles and pyridocarbazoles, are a class of compounds that were recently discovered to modulate splicing by altering the ESE-dependent splicing activity of individual SR proteins168. Indole derivatives have been shown to modulate the splicing event that generates the cancer-associated, constitutively active ΔRon isoform of the recepteur d’origine nantais (RON, also known as MST1R) proto-oncogene and revert the invasive phenotype of cancer cells expressing ΔRon169.
Summary and future perspectives
The recent discovery of recurrent spliceosomal mutations as likely cancer drivers has underscored the pressing need to identify connections between abnormal pre-mRNA processing and tumorigenesis. Emerging evidence supports a model in which many spliceosomal mutations induce specific changes in splice site or exon recognition, frequently via altered RNA binding, leading to genome-wide splicing changes that presumably promote cancer development.
Despite these mechanistic advances, efforts to link altered splice site or exon recognition to specific pathological splicing events are nascent. Challenges including identifying and prioritizing among hundreds of downstream mis-spliced isoforms, as well as determining the biological roles of specific isoforms. Furthermore, it is unknown whether the pro-tumorigenic effects of mutated spliceosomal proteins are mediated by just a handful of mis-spliced isoforms, or instead are due to many splicing changes, which may even be functionally interdependent. Although many mis-spliced isoforms have been identified in cells bearing spliceosomal mutations, very few of these isoforms have been functionally characterized to date.
Spliceosomal mutations likely both indirectly and directly dysregulate diverse cellular processes. In principle, spliceosomal mutations could affect almost any biological process by inducing mis-splicing of key regulators (for example, the connection between SRSF2 and H3K27me3 deficiency via EZH2 mis-splicing). Spliceosomal mutations may also dysregulate processes including transcriptional elongation, the DNA damage response and NMD, in which splicing factors play key roles (Fig. 4a–b).
Although spliceosomal mutations provide the most direct link between splicing and cancer, it is also important to note that abnormal splicing is a feature of most cancers even in the absence of spliceosomal mutations5–7. Abnormal cancer-associated splicing may result from both specific and global perturbations to the splicing machinery. Specific perturbations may arise from dysregulation of single splicing factors that play pro- or anti-tumorigenic roles (Table 1), whereas global perturbations may arise from effects including potential transcriptional amplification driven by MYC149 or mutations affecting epigenetic regulators such as isocitrate dehydrogenase (IDH) or SET domain containing 2 (SETD2)5,7.
Although incomplete, our current understanding of spliceosomal mutations suggests that these mutations may create new therapeutic opportunities. Because splicing factors can act as both oncoproteins and tumor suppressors, distinct therapeutic interventions may prove necessary for treating cancers harboring different spliceosomal mutations. Possible therapeutic interventions fall into several broad categories, including restoring normal splicing and exploiting vulnerabilities to specifically target mutant cells.
Normal splicing could potentially be restored by specifically inhibiting the mutant protein, manipulating downstream splicing events or other methods. These approaches are promising, yet each requires further investigation. For example, specific inhibition or sequestration of mutant SRSF2 and U2AF1 may be possible given their altered RNA-binding preferences. However, definitive evidence that cancer cells depend on these mutated proteins, or that inhibiting the mutant allele is sufficient to restore normal splicing, is currently absent. (Mutant SRSF2 and U2AF1 likely act as oncoproteins to promote tumor formation, yet may not be required for subsequent tumor maintenance or growth.) The same caveat applies to inhibition of downstream mis-splicing. Specific mis-splicing events could potentially be corrected with antisense oligonucleotides, which have shown promise in clinical trials of disorders such as Duchenne muscular dystrophy170 and spinal muscular atrophy171,172. However, our current understanding of how spliceosomal mutations perturb cellular function is insufficient to determine which mis-splicing events to correct in cancer. Furthermore, because inhibiting a mutant oncoprotein is likely more feasible than restoring the function of a disabled wild-type protein, restoring normal splicing may not be possible in the context of spliceosomal mutations that disable tumor suppressors. For example, ZRSR2 mutations cause loss of ZRSR2 expression or function, and it is unclear whether restoring U12-type intron recognition in the absence of ZRSR2 is possible.
Conversely, it may be feasible to selectively target cells expressing mutated splicing factors. Recent work suggested that inhibiting splicing catalysis itself may provide a therapeutic index in cells bearing spliceosomal mutations158,173. Non-cell autonomous approaches to target cells with spliceosomal mutations may also be possible. Just as increased somatic mutational burdens may generate neo-epitopes and render specific subsets of cancer sensitive to cancer immunotherapies174–177, so may abnormal mRNAs generated by spliceosomal mutations result in neo-epitope production in cancers bearing these lesions. Notably, these two approaches—inhibition of splicing catalysis and immunotherapy—could potentially be efficacious in the context of spliceosomal mutations that generate oncoproteins as well as those that inactivate tumor suppressors. Recurrent mutations in SF3B1, SRSF2, U2AF1, and ZRSR2 cause very different mechanistic alterations in splicing, yet each may render cells susceptible to further perturbation of splicing catalysis or result in the generation of neo-epitopes.
Key points.
Genetic and functional data indicate that RNA splicing factors can act as oncoproteins as well as tumor suppressors.
A subset of RNA splicing factors are recurrent targets of specific point mutations in cancer. Many other splicing factors exhibit dysregulated expression in cancer.
In many cases, recurrent spliceosomal mutations alter splice site or exon recognition preferences to cause abnormal RNA splicing.
Spliceosomal mutations are sufficient to impair myeloid differentiation in murine models. In the case of SRSF2, impaired differentiation has been linked to a specific splicing change in a downstream gene (EZH2).
Spliceosomal mutations may affect cellular processes, including epigenetic regulation, the DNA damage response and nonsense-mediated decay, in addition to regulation of RNA splicing.
Small molecules that disrupt splicing catalysis and/or targeted correction of specific splicing changes may provide novel therapeutic opportunities for cancers bearing spliceosomal mutations.
Acknowledgments
H.D. is supported by a grant from the US Department of Defense Breast Cancer Research Program (W81XWH-14-1-0044). E.K. is supported by the Worldwide Cancer Research Fund. R.K.B. and O.A.-W. are supported by grants from the Edward P. Evans Foundation, the Department of Defense Bone Marrow Failure Research Program (BM150092) and National Institutes of Health/National Heart, Lung and Blood Institute (NIH/NHLBI) (R01 HL128239). O.A.-W. is supported by an NIH K08 Clinical Investigator Award (1K08CA160647-01), a US Department of Defense Postdoctoral Fellow Award in Bone Marrow Failure Research (W81XWH-12-1-0041), the Starr Cancer Consortium (I8-A8-075), the Josie Robertson Investigator Program, a Damon Runyon Clinical Investigator Award with support from the Evans Foundation, the Mr William H. Goodwin and Mrs Alice Goodwin Commonwealth Foundation for Cancer Research, and the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center. R.K.B. is supported by the Ellison Medical Foundation (AG-NS-1030-13) and NIH/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01 DK103854).
Glossary
- Major spliceosome
A ribonucleoprotein complex consisting of five small nuclear RNAs (termed U1, U2, U4, U5 and U6), each in complex with a set of proteins to form snRNPs, that are together responsible for excision of the majority of introns
- Minor spliceosome
A ribonucleoprotein complex that catalyzes splicing of a small subset of ‘U12-type’ introns. The introns recognized by the minor spliceosome are typically defined by different sequence elements than those that define U2-type introns, which are recognized by the major spliceosome
- Small nuclear ribonucleoprotein complexes
(snRNPs). These complexes assemble on pre-mRNA to catalyze splicing
- U2AF complex
A heterodimeric protein complex consisting of U2AF1 and U2AF2. U2AF1 and U2AF2 bind the polypyrimidine tract and AG dinucleotide of the 3′ splice site to facilitate splice site recognition. Only a subset of ‘AG-dependent’ 3′ splice sites require U2AF1 binding for efficient splice site recognition
- Constitutive splice sites
Splice sites that are always recognized and used by the spliceosome. Similarly, constitutive exons are always included in the mature mRNA
- Alternative splice sites
Splice sites that are variably recognized and used by the spliceosome. Similarly, alternative exons (also known as cassette or skipped exons) are sometimes, but not always, included in the mature mRNA. Recognition of alternative splice sites is frequently cell type-specific and may rely upon the binding of additional trans-acting factors
- Serine/arginine-rich proteins
(SR proteins). A family of splicing factors that frequently promote splicing, although their action is context-dependent. Many of these proteins bind pre-mRNA in a sequence-specific manner to activate splicing. Some of the family are implicated in other cellular processes, including mRNA export and translation
- Heterogeneous nuclear ribonucleoproteins
(hnRNPs). Many members of this protein family are splicing factors, although they also participate in other diverse RNA metabolic processes. These proteins frequently repress splicing, although their action is context-dependent
- Acute myeloid leukemia
A type of cancer characterized by the rapid growth of abnormal white blood cells that accumulate in the bone marrow and interfere with the production of normal blood cells
- Myelodysplastic syndromes
(MDS). A heterogeneous group of clonal disorders of hematopoiesis characterized by an impaired ability to generate mature blood cells as well as aberrant cell morphologies (termed ‘dysplasia’).
- Chronic lymphocytic leukemia
(CLL). A type of cancer characterized by accumulation of aberrant mature-appearing B lymphocytes.
- SF3B1
This gene encodes a key component of the U2 snRNP that binds upstream of the branch point to facilitate 3′ splice site recognition. SF3B1 is likely required for the splicing of most introns and is the most commonly mutated splicing factor in cancer.
- SRSF2
This gene encodes an SR protein that binds specific exonic splicing enhancer motifs to promote recognition and inclusion of exons containing these motifs
- ZRSR2
A gene encoding a component of the minor spliceosome that contacts the 3′ splice site of specific U12-type introns to promote their excision
- Synthetic lethality
The situation in which two cellular perturbations (for example, two distinct mutations, or a mutation and a particular drug) result in cell death when combined whereas each perturbation alone does not
- Secondary AML
(sAML) Acute myeloid leukemia that develops following a previous chronic myeloid malignancy such as a myelodysplastic syndrome
- Cryptic 3′ splice sites
Potential 3′ splice sites that are not normally recognized by the spliceosome. By chance, introns and exons contain many AG dinucleotides that are not used as splice sites. Perturbations such as spliceosomal mutations can cause such ‘decoy splice sites’ to be incorrectly recognized as authentic splice sites
- Stop codon
UAA, UAG, or UGA codons, signaling the end of translation. Also known as a termination codons
- Chronic myelomonocytic leukemia
A clonal disorder with features of both myelodysplastic and myeloproliferative syndromes in which there are too many monocytes in the blood
- Exonic splicing enhancer
A typically short sequence motif in pre-mRNA that is bound by a splicing factor to promote exon recognition and subsequent inclusion of the exon in the mature mRNA. Many SR proteins bind exonic splicing enhancers to activate splicing
- Poison exon
A cassette exon containing an in-frame premature stop codon. A premature stop codon lies upstream of the normal stop codon, resulting in premature termination of translation of the mRNA when included in a transcript. Poison exons can induce nonsense-mediated decay of the mRNA or production of a truncated protein
- Nonsense-mediated decay
An RNA surveillance process that recognizes and degrades mRNAs containing premature stop codons, as well as other abnormal RNAs and a subset of normal coding transcripts. Splicing is closely linked to NMD, as exon-exon junctions are important components of NMD activation in human cells
- RNA polymerase II pause release
The process by which RNA polymerase II that is paused (not actively transcribing) after the initiation of transcription is released, enabling transcriptional elongation
- Frameshift
The disruption of an open reading frame by the insertion or deletion of nucleotide sequence whose length is not a multiple of three
- Expressed sequence tag
(EST). Portions of cDNA sequences
- Unannotated splicing
Splicing events that have not been previously reported by published studies or genomic databases such as Ensembl, UCSC, Vega and RefSeq
- ψ value
The percentage of all mRNAs transcribed from a gene that correspond to a particular isoform or contain a particular alternatively spliced sequence relative to all transcripts of the parent gene. For example, the ψ value for a cassette exon is the fraction of all mRNAs that contain the cassette exon. The ψ value is independent of gene expression and falls within the range 0–100%
Biographies
Heidi Dvinge: Dr. Dvinge is a Department of Defense Postdoctoral Fellow in Dr. Robert Bradley’s lab at Fred Hutchinson Cancer Research Center. Dr. Dvinge studies abnormalities in RNA splicing that distinguish cancerous from normal cells.
Eunhee Kim: Dr. Kim is a research associate in Dr. Omar Abdel-Wahab’s lab at Memorial Sloan Kettering Cancer Center and is funded by the Worldwide Cancer Research Fund. Her research focuses on understanding the role of mutations in genes encoding core splicing proteins in cancer.
Omar Abdel-Wahab: Dr. Abdel-Wahab is an Assistant Member at Memorial Sloan Kettering Cancer Center whose lab focuses on functional genetic studies of hematological malignancies. The lab has a specific interest in understanding alterations in epigenetic modifiers and other transcriptional modifiers, such as spliceosomal mutations, in the pathogenesis and therapy of leukemia.
Robert K. Bradley: Dr. Bradley is an Associate Member at Fred Hutchinson Cancer Research Center whose lab focuses on genetic and genomic studies of RNA processing. The lab has a specific interest in RNA-mediated disease mechanisms, such as abnormal RNA splicing and surveillance in neoplastic and genetic disease.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discovery. 2013;3:1228–1237. doi: 10.1158/2159-8290.CD-13-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes & Development. 2010;24:2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–1335. doi: 10.1016/j.cell.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 4.Jung H, et al. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nature Genetics. 2015;47:1242–1248. doi: 10.1038/ng.3414. [DOI] [PubMed] [Google Scholar]
- 5.Dvinge H, Bradley RK. Widespread intron retention diversifies most cancer transcriptomes. Genome Medicine. 2015;7:45. doi: 10.1186/s13073-015-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danan-Gotthold M, et al. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Research. 2015;43:5130–5144. doi: 10.1093/nar/gkv210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon JM, et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Research. 2014;24:241–250. doi: 10.1101/gr.158253.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. A landmark study demonstrating frequent mutations in genes encoding spliceosomal proteins in myeloid malignancies. [DOI] [PubMed] [Google Scholar]
- 9.Graubert TA, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nature Genetics. 2012;44:53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papaemmanuil E, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. The N Engl J Med. 2011;365:2497–2506. doi: 10.1056/NEJMoa1109016. A key study revealing frequent mutations in the gene encoding the spliceosomal protein SF3B1 in CLL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quesada V, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nature Genetics. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 13.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics. 2008;40 doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 14.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. A landmark study describing the use of RNA-seq to quantify splicing across human tissues. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Turunen JJ, Niemelä EH, Verma B, Frilander MJ. The significant other: splicing by the minor spliceosome. Wiley Interdisciplinary Reviews. RNA. 2013;4:61–76. doi: 10.1002/wrna.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matera AG, Wang Z. A day in the life of the spliceosome. Nature Reviews Molecular Cell Biology. 2014;15:108–121. doi: 10.1038/nrm3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fica SM, et al. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013;503:229–234. doi: 10.1038/nature12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graveley BR, Hertel KJ, Maniatis T. A systematic analysis of the factors that determine the strength of pre-mRNA splicing enhancers. EMBO J. 1998;17:6747–6756. doi: 10.1093/emboj/17.22.6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu XD, Ares M., Jr Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15:689–701. doi: 10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh R, Valcarcel J. Building specificity with nonspecific RNA-binding proteins. Nat Struct Mol Biol. 2005;12:645–653. doi: 10.1038/nsmb961. [DOI] [PubMed] [Google Scholar]
- 23.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma. 2013;122:191–207. doi: 10.1007/s00412-013-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krecic AM, Swanson MS. hnRNP complexes: composition, structure, and function. Curr Opin Cell Biol. 1999;11:363–371. doi: 10.1016/S0955-0674(99)80051-9. [DOI] [PubMed] [Google Scholar]
- 26.Zahler AM, Lane WS, Stolk JA, Roth MB. SR proteins: a conserved family of pre-mRNA splicing factors. Genes & Development. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 27.Kohtz JD, et al. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 28.Krecic AM, Swanson MS. hnRNP complexes: composition, structure, and function. Current Opinion in Cell Biology. 1999;11:363–371. doi: 10.1016/S0955-0674(99)80051-9. [DOI] [PubMed] [Google Scholar]
- 29.Pandit S, et al. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013;50:223–235. doi: 10.1016/j.molcel.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han J, et al. SR proteins induce alternative exon skipping through their activities on the flanking constitutive exons. Mol Cell Biol. 2011;31:793–802. doi: 10.1128/MCB.01117-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xue Y, et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Llorian M, et al. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat Struct Mol Biol. 2010;17:1114–1123. doi: 10.1038/nsmb.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huelga SC, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012;1:167–178. doi: 10.1016/j.celrep.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dasgupta T, Ladd AN. The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip Rev RNA. 2012;3:104–121. doi: 10.1002/wrna.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konieczny P, Stepniak-Konieczna E, Sobczak K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Research. 2014;42:10873–10887. doi: 10.1093/nar/gku767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ule J, et al. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 37.Saltzman AL, Pan Q, Blencowe BJ. Regulation of alternative splicing by the core spliceosomal machinery. Genes & Development. 2011;25:373–384. doi: 10.1101/gad.2004811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jurica MS, Moore MJ. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 39.Barash Y, et al. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 40.Karni R, et al. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14:185–193. doi: 10.1038/nsmb1209. This paper demonstrated that modest overexpression of the splicing factor SRSF1 is pro-tumorigenic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anczuków O, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19:220–228. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karni R, Hippo Y, Lowe SW, Krainer AR. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci USA. 2008;105:15323–15327. doi: 10.1073/pnas.0801376105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia R, Li C, McCoy JP, Deng CXX, Zheng ZMM. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. International Journal of Biological Sciences. 2010;6:806–826. doi: 10.7150/ijbs.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Y, et al. Downregulation of splicing factor SRSF3 induces p53β, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene. 2013;32:2792–2798. doi: 10.1038/onc.2012.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen MA, Wilkinson JE, Krainer AR. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol. 2014;21:189–197. doi: 10.1038/nsmb.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen-Eliav M, et al. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol. 2013;229:630–639. doi: 10.1002/path.4129. [DOI] [PubMed] [Google Scholar]
- 47.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clower CV, et al. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci USA. 2010;107:1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Babic I, et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metabolism. 2013;17:1000–1008. doi: 10.1016/j.cmet.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Golan-Gerstl R, et al. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71:4464–4472. doi: 10.1158/0008-5472.CAN-10-4410. [DOI] [PubMed] [Google Scholar]
- 51.Sweetser DA, et al. Delineation of the minimal commonly deleted segment and identification of candidate tumor-suppressor genes in del(9q) acute myeloid leukemia. Genes, Chromosomes, and Cancer. 2005;44:279–291. doi: 10.1002/gcc.20236. [DOI] [PubMed] [Google Scholar]
- 52.Gallardo M, et al. hnRNP K Is a Haploinsufficient Tumor Suppressor that Regulates Proliferation and Differentiation Programs in Hematologic Malignancies. Cancer Cell. 2015;28:486–499. doi: 10.1016/j.ccell.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moumen A, Masterson P, O’Connor MJ, Jackson SP. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell. 2005;123:1065–1078. doi: 10.1016/j.cell.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 54.Zong FY, et al. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014;10:e1004289. doi: 10.1371/journal.pgen.1004289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Angeloni D. Molecular analysis of deletions in human chromosome 3p21 and the role of resident cancer genes in disease. Briefings in Functional Genomics & Proteomics. 2007;6:19–39. doi: 10.1093/bfgp/elm007. [DOI] [PubMed] [Google Scholar]
- 56.Imielinski M, et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oh JJ, et al. 3p21.3 tumor suppressor gene H37/Luca15/RBM5 inhibits growth of human lung cancer cells through cell cycle arrest and apoptosis. Cancer Research. 2006;66:3419–3427. doi: 10.1158/0008-5472.CAN-05-1667. [DOI] [PubMed] [Google Scholar]
- 58.Rintala‐Maki ND, Goard CA. Expression of RBM5‐related factors in primary breast tissue. Journal of Cellular Biochemistry. 2007;100:1440–1458. doi: 10.1002/jcb.21134. [DOI] [PubMed] [Google Scholar]
- 59.Hernandez J, et al. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol. 2016;13:466–472. doi: 10.1080/15476286.2016.1144004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bechara EG, Sebestyén E, Bernardis I, Eyras E, Valcárcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Molecular Cell. 2013;52:720–733. doi: 10.1016/j.molcel.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, et al. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell. 2014;26:374–389. doi: 10.1016/j.ccr.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shapiro IM, et al. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harbour JW, et al. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nature Genetics. 2013;45:133–135. doi: 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martin M, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nature Genetics. 2013;45:933–936. doi: 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biankin AV, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ellis MJ, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maguire SL, et al. SF3B1 mutations constitute a novel therapeutic target in breast cancer. The Journal of Pathology. 2015;235:571–580. doi: 10.1002/path.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Papaemmanuil E, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–3627. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim E, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell. 2015;27:617–630. doi: 10.1016/j.ccell.2015.04.006. This study demonstrated that SRSF2 mutations are sufficient to drive myelodysplasia, are distinct from loss-of-function of SRSF2 and change the RNA-binding affinity of the protein to alter exon recognition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kon A, et al. SRSF2 P95H Mutation Causes Impaired Stem Cell Repopulation and Hematopoietic Differentiation in Mice (Abstract 1649) Blood. 2015;126 [Google Scholar]
- 74.Obeng EA, et al. Mutant Splicing Factor 3b Subunit 1 (SF3B1) Causes Dysregulated Erythropoiesis and a Stem Cell Disadvantage (Abstract 828) Blood. 2014;124 [Google Scholar]
- 75.Mupo A, et al. Sf3b1 K700E Mutation Impairs Pre-mRNA Splicing and Definitive Hematopoiesis in a Conditional Knock-in Mouse Model (Abstract 140) Blood. 2015;126 [Google Scholar]
- 76.Shirai CL, et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell. 2015;27:631–643. doi: 10.1016/j.ccell.2015.04.008. This study presented one of the first in vivo models of spliceosomal gene mutations in cancer, which revealed the biological effects of the U2AF1-S34F mutation on hematopoiesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mian SA, et al. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat Commun. 2015;6:10004. doi: 10.1038/ncomms10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lindsley RC, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–1376. doi: 10.1182/blood-2014-11-610543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rossi D, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118:6904–6908. doi: 10.1182/blood-2011-08-373159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DeBoever C, et al. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Computational Biology. 2015;11:e1004105. doi: 10.1371/journal.pcbi.1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Darman RB, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Reports. 2015;13:1033–1045. doi: 10.1016/j.celrep.2015.09.053. This study revealed that SF3B1 mutations promote recognition of cryptic 3′ splice sites in cancer samples and genetically modified cell lines. [DOI] [PubMed] [Google Scholar]
- 82.Allikmets R, et al. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A) Hum Mol Genet. 1999;8:743–749. doi: 10.1093/hmg/8.5.743. [DOI] [PubMed] [Google Scholar]
- 83.Pondarre C, et al. Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood. 2007;109:3567–3569. doi: 10.1182/blood-2006-04-015768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu S, Romfo CM, Nilsen TW, Green MR. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature. 1999;402:832–835. doi: 10.1038/45590. [DOI] [PubMed] [Google Scholar]
- 85.Reed R. The organization of 3′ splice-site sequences in mammalian introns. Genes & Development. 1989;3:2113–2123. doi: 10.1101/gad.3.12b.2113. [DOI] [PubMed] [Google Scholar]
- 86.Przychodzen B, et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. doi: 10.1182/blood-2013-01-480970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brooks AN, et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PloS One. 2014;9:e87361. doi: 10.1371/journal.pone.0087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ilagan JO, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Research. 2015;25:14–26. doi: 10.1101/gr.181016.114. This study demonstrated that U2AF1 mutations cause alteration, not loss of function, and that mutations in the first versus second zinc fingers of U2AF1 induce different changes in 3′ splice site preference. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Okeyo-Owuor T, et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia. 2015;29:909–917. doi: 10.1038/leu.2014.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Damm F, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122:3169–3177. doi: 10.1182/blood-2012-11-469619. [DOI] [PubMed] [Google Scholar]
- 91.Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shen H, Zheng X, Luecke S, Green MR. The U2AF35-related protein Urp contacts the 3′ splice site to promote U12-type intron splicing and the second step of U2-type intron splicing. Genes & Development. 2010;24:2389–2394. doi: 10.1101/gad.1974810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Madan V, et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nature Communications. 2015;6:6042. doi: 10.1038/ncomms7042. This work demonstrated that U12-type introns are poorly recognized in patients carrying presumed loss-of-function ZRSR2 mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bejar R, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. Journal of Clinical Oncology. 2012;30:3376–3382. doi: 10.1200/JCO.2011.40.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Graveley BR, Maniatis T. Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol Cell. 1998;1:765–771. doi: 10.1016/s1097-2765(00)80076-3. [DOI] [PubMed] [Google Scholar]
- 96.Liu HX, Chew SL, Cartegni L, Zhang MQ, Krainer AR. Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Molecular and Cellular Biology. 2000;20:1063–1071. doi: 10.1128/mcb.20.3.1063-1071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schaal TD, Maniatis T. Multiple distinct splicing enhancers in the protein-coding sequences of a constitutively spliced pre-mRNA. Molecular and Cellular Biology. 1999;19:261–273. doi: 10.1128/mcb.19.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang J, et al. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci USA. 2015;112:E4726–4734. doi: 10.1073/pnas.1514105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Daubner GM, Clery A, Jayne S, Stevenin J, Allain FH. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012;31:162–174. doi: 10.1038/emboj.2011.367. This study described nuclear magnetic resonance solution structures of the RNA recognition motif domain of SRSF2 in complex with RNA that explained the ability of SRSF2 to bind both G- and C-rich motifs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ernst T, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nature Genetics. 2010;42:722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 101.Nikoloski G, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nature Genetics. 2010;42:665–667. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 102.Muto T, et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. The Journal of Experimental Medicine. 2013;210:2627–2639. doi: 10.1084/jem.20131144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Haferlach T, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kurtovic-Kozaric A, et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia. 2015;29:126–136. doi: 10.1038/leu.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Duhoux FP, et al. The t(1;9)(p34;q34) fusing ABL1 with SFPQ, a pre-mRNA processing gene, is recurrent in acute lymphoblastic leukemias. Leukemia Research. 2011;35:e114–117. doi: 10.1016/j.leukres.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 106.Mathur M, Samuels HH. Role of PSF-TFE3 oncoprotein in the development of papillary renal cell carcinomas. Oncogene. 2007;26:277–283. doi: 10.1038/sj.onc.1209783. [DOI] [PubMed] [Google Scholar]
- 107.Chen W, Itoyama T, Chaganti RS. Splicing factor SRP20 is a novel partner of BCL6 in a t(3;6)(q27;p21) translocation in transformed follicular lymphoma. Genes, Chromosomes & Cancer. 2001;32:281–284. doi: 10.1002/gcc.1191. [DOI] [PubMed] [Google Scholar]
- 108.Lee M, et al. The structure of human SFPQ reveals a coiled-coil mediated polymer essential for functional aggregation in gene regulation. Nucleic Acids Research. 2015;43:3826–3840. doi: 10.1093/nar/gkv156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tomsic J, et al. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci Rep. 2015;5:10566. doi: 10.1038/srep10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Polprasert C, et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell. 2015;27:658–670. doi: 10.1016/j.ccell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005;122:365–378. doi: 10.1016/j.cell.2005.06.008. This study demonstrated that loss of SRSF1 results in R loop formation and DNA damage. [DOI] [PubMed] [Google Scholar]
- 112.Xiao R, et al. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Molecular and Cellular Biology. 2007;27:5393–5402. doi: 10.1128/MCB.00288-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Savage KI, et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol Cell. 2014;54:445–459. doi: 10.1016/j.molcel.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tresini M, et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature. 2015;523:53–58. doi: 10.1038/nature14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tilgner H, et al. Nucleosome positioning as a determinant of exon recognition. Nat Struct Mol Biol. 2009;16:996–1001. doi: 10.1038/nsmb.1658. [DOI] [PubMed] [Google Scholar]
- 116.Kfir N, et al. SF3B1 association with chromatin determines splicing outcomes. Cell Rep. 2015;11:618–629. doi: 10.1016/j.celrep.2015.03.048. [DOI] [PubMed] [Google Scholar]
- 117.Kolasinska-Zwierz P, et al. Differential chromatin marking of introns and expressed exons by H3K36me3. Nature Genetics. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Spies N, Nielsen CB, Padgett RA, Burge CB. Biased chromatin signatures around polyadenylation sites and exons. Mol Cell. 2009;36:245–254. doi: 10.1016/j.molcel.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Luco RF, et al. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. This paper demonstrated that histone modifications can influence alternative splicing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.de Almeida SFF, et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat Struct Mol Biol. 2011;18:977–983. doi: 10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- 121.Kim S, Kim H, Fong N, Erickson B, Bentley DL. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc Natl Acad Sci USA. 2011;108:13564–13569. doi: 10.1073/pnas.1109475108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Khan DH, Jahan S, Davie JR. Pre-mRNA splicing: role of epigenetics and implications in disease. Advances in Biological Regulation. 2012;52:377–388. doi: 10.1016/j.jbior.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 124.Ji X, et al. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell. 2013;153:855–868. doi: 10.1016/j.cell.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lemieux B, et al. A Function for the hnRNP A1/A2 Proteins in Transcription Elongation. PLoS One. 2015;10:e0126654. doi: 10.1371/journal.pone.0126654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mikula M, Bomsztyk K, Goryca K, Chojnowski K, Ostrowski J. Heterogeneous nuclear ribonucleoprotein (HnRNP) K genome-wide binding survey reveals its role in regulating 3′-end RNA processing and transcription termination at the early growth response 1 (EGR1) gene through XRN2 exonuclease. J Biol Chem. 2013;288:24788–24798. doi: 10.1074/jbc.M113.496679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hsin JP, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes & Development. 2012;26:2119–2137. doi: 10.1101/gad.200303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang Y, Gattoni R, Stevenin J, Steitz JA. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell. 2003;11:837–843. doi: 10.1016/s1097-2765(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 129.Muller-McNicoll M, et al. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes & Development. 2016;30:553–566. doi: 10.1101/gad.276477.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sanford JR, Gray NK, Beckmann K, Caceres JF. A novel role for shuttling SR proteins in mRNA translation. Genes & Development. 2004;18:755–768. doi: 10.1101/gad.286404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Michlewski G, Sanford JR, Caceres JF. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol Cell. 2008;30:179–189. doi: 10.1016/j.molcel.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 132.Nott A, Le Hir H, Moore MJ. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes & Development. 2004;18:210–222. doi: 10.1101/gad.1163204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chang YFF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annual Review of Biochemistry. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 134.Popp MW, Maquat LE. Organizing principles of mammalian nonsense-mediated mRNA decay. Annual Review of Genetics. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang Z, Krainer AR. Involvement of SR proteins in mRNA surveillance. Mol Cell. 2004;16:597–607. doi: 10.1016/j.molcel.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 136.Brazao TF, et al. A new function of ROD1 in nonsense-mediated mRNA decay. FEBS Lett. 2012;586:1101–1110. doi: 10.1016/j.febslet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 137.Ge Z, Quek BL, Beemon KL, Hogg JR. Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. Elife. 2016;5 doi: 10.7554/eLife.11155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA. 2003;100:189–192. doi: 10.1073/pnas.0136770100. This study demonstrated that approximately one-third of alternative isoforms of human genes contain premature termination codons that likely trigger NMD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 2007;446:926–929. doi: 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- 140.Ni JZ, et al. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes & Development. 2007;21:708–718. doi: 10.1101/gad.1525507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lareau LF, Brenner SE. Regulation of splicing factors by alternative splicing and NMD is conserved between kingdoms yet evolutionarily flexible. Molecular Biology and Evolution. 2015;32:1072–1079. doi: 10.1093/molbev/msv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu C, et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nature Medicine. 2014;20:596–598. doi: 10.1038/nm.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tarpey PS, et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nature Genetics. 2007;39:1127–1133. doi: 10.1038/ng2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Albers CA, et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nature Genetics. 2012;44:435–439. doi: 10.1038/ng.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Feng Q, et al. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. eLife. 2015;4 doi: 10.7554/eLife.04996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nature Reviews Cancer. 2008;8:880–887. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 148.Hubert CG, et al. Genome-wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes & Development. 2013;27:1032–1045. doi: 10.1101/gad.212548.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hsu TY, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525:384–388. doi: 10.1038/nature14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bonnal S, Vigevani L, Valcárcel J. The spliceosome as a target of novel antitumour drugs. Nature Reviews Drug Discovery. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 151.Webb TR, Joyner AS, Potter PM. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discovery Today. 2013;18:43–49. doi: 10.1016/j.drudis.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yokoi A, et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. The FEBS Journal. 2011;278:4870–4880. doi: 10.1111/j.1742-4658.2011.08387.x. [DOI] [PubMed] [Google Scholar]
- 153.Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nature Chemical Biology. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- 154.Eskens FA, et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clinical Cancer Research. 2013;19:6296–6304. doi: 10.1158/1078-0432.CCR-13-0485. [DOI] [PubMed] [Google Scholar]
- 155.Hong DS, et al. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) spliceosome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs. 2013;32:436–444. doi: 10.1007/s10637-013-0046-5. [DOI] [PubMed] [Google Scholar]
- 156.Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chemical Biology. 2011;6:582–589. doi: 10.1021/cb100356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Xargay-Torrent S, et al. The splicing modulator sudemycin induces a specific antitumor response and cooperates with ibrutinib in chronic lymphocytic leukemia. Oncotarget. 2015;6:22734–22749. doi: 10.18632/oncotarget.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lee SC-W, et al. Therapeutic targeting of spliceosomal mutant myeloid leukemias through modulation of splicing catalysis (Abstract 4) Blood. 2015;126 [Google Scholar]
- 159.Rossi F, et al. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature. 1996;381:80–82. doi: 10.1038/381080a0. [DOI] [PubMed] [Google Scholar]
- 160.Gui JF, Lane WS, Fu XD. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- 161.Colwill K, et al. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. The EMBO Journal. 1996;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- 162.Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. The Journal of Biological Chemistry. 2008;283:1223–1227. doi: 10.1074/jbc.R700034200. [DOI] [PubMed] [Google Scholar]
- 163.Yeakley JM, et al. Phosphorylation regulates in vivo interaction and molecular targeting of serine/arginine-rich pre-mRNA splicing factors. The Journal of Cell Biology. 1999;145:447–455. doi: 10.1083/jcb.145.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gout S, et al. Abnormal expression of the pre-mRNA splicing regulators SRSF1, SRSF2, SRPK1 and SRPK2 in non small cell lung carcinoma. PLoS One. 2012;7 doi: 10.1371/journal.pone.0046539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yoshida T, et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Research. 2015;75:1516–1526. doi: 10.1158/0008-5472.CAN-14-2443. [DOI] [PubMed] [Google Scholar]
- 166.Dawid GN, et al. Regulation of Vascular Endothelial Growth Factor (VEGF) Splicing from Pro-angiogenic to Anti-angiogenic Isoforms A Novel Therapeutic Strategy For Angiogenesis. Journal of Biological Chemistry. 2010;19:5532–5540. doi: 10.1074/jbc.M109.074930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Elianna MA, et al. WT1 Mutants Reveal SRPK1 to Be a Downstream Angiogenesis Target by Altering VEGF Splicing. Cancer Cell. 2010;20:768–780. doi: 10.1016/j.ccr.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Soret J, et al. Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors. Proc Natl Acad Sci USA. 2005;102:8764–8769. doi: 10.1073/pnas.0409829102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ghigna C, et al. Pro-metastatic splicing of Ron proto-oncogene mRNA can be reversed: Therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biology. 2010;7:495–503. doi: 10.4161/rna.7.4.12744. [DOI] [PubMed] [Google Scholar]
- 170.Aartsma‐Rus A, Fokkema I, Verschuuren J. Theoretic applicability of antisense‐mediated exon skipping for Duchenne muscular dystrophy mutations. Human Mutation. 2009;30:293–299. doi: 10.1002/humu.20918. [DOI] [PubMed] [Google Scholar]
- 171.Hua Y, Sahashi K, Hung G, Rigo F. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes & Development. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Passini MA, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Science Translational Medicine. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhou Q, et al. A chemical genetics approach for the functional assessment of novel cancer genes. Cancer Research. 2015;75:1949–1958. doi: 10.1158/0008-5472.CAN-14-2930. [DOI] [PubMed] [Google Scholar]
- 174.Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rizvi NA, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Le DT, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gubin MM, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Alamancos GP, Agirre E, Eyras E. Methods to study splicing from high-throughput RNA Sequencing data. Methods Mol Biol. 2014;1126:357–397. doi: 10.1007/978-1-62703-980-2_26. [DOI] [PubMed] [Google Scholar]
- 179.Engstrom PG, et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nature Methods. 2013;10:1185–1191. doi: 10.1038/nmeth.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006;7(Suppl 1):S12.1–14. doi: 10.1186/gb-2006-7-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cunningham F, et al. Ensembl 2015. Nucleic Acids Research. 2015;43:9. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rosenbloom KR, et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Research. 2015;43:81. doi: 10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nature Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Barbosa-Morais NL, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338:1587–1593. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- 185.Engström PGG, et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nature Methods. 2013;10:1185–1191. doi: 10.1038/nmeth.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Steijger T, et al. Assessment of transcript reconstruction methods for RNA-seq. Nature Methods. 2013;10:1177–1184. doi: 10.1038/nmeth.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocrine-related Cancer. 2011;18:96. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Antonarakis ES, et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N Engl J Med. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Xing Y, Lee CJ. Protein modularity of alternatively spliced exons is associated with tissue-specific regulation of alternative splicing. PLoS Genetics. 2005;1:e34. doi: 10.1371/journal.pgen.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yeo GW, Van Nostrand E, Holste D, Poggio T, Burge CB. Identification and analysis of alternative splicing events conserved in human and mouse. Proc Natl Acad Sci USA. 2005;102:2850–2855. doi: 10.1073/pnas.0409742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. 2012;338:1593–1599. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Odom DT, et al. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nature Genetics. 2007;39:730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schmidt D, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Moran-Jones K, Grindlay J, Jones M, Smith R, Norman JC. hnRNP A2 regulates alternative mRNA splicing of TP53INP2 to control invasive cell migration. Cancer Research. 2009;69:9219–9227. doi: 10.1158/0008-5472.CAN-09-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Golan-Gerstl R, et al. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Research. 2011;71:4464–4472. doi: 10.1158/0008-5472.CAN-10-4410. [DOI] [PubMed] [Google Scholar]
- 199.LeFave CV, Squatrito M, Vorlova S. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. The EMBO Journal. 2011;30:4084–4097. doi: 10.1038/emboj.2011.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Xu Y, et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes & Development. 2014;28:1191–1203. doi: 10.1101/gad.241968.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Adler AS, et al. An integrative analysis of colon cancer identifies an essential function for PRPF6 in tumor growth. Genes & Development. 2014;28:1068–1084. doi: 10.1101/gad.237206.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Izaguirre DI, et al. PTBP1-dependent regulation of USP5 alternative RNA splicing plays a role in glioblastoma tumorigenesis. Molecular Carcinogenesis. 2012;51:895–906. doi: 10.1002/mc.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Fushimi K, et al. Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc Natl Acad Sci USA. 2008;105:15708–15713. doi: 10.1073/pnas.0805569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bonnal S, et al. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Molecular Cell. 2008;32:81–95. doi: 10.1016/j.molcel.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 205.Zhou X, et al. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nature Communications. 2014;5:4581. doi: 10.1038/ncomms5581. [DOI] [PubMed] [Google Scholar]
- 206.Wu SJJ, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. 2012;120:3106–3111. doi: 10.1182/blood-2012-02-412296. [DOI] [PubMed] [Google Scholar]
- 207.Damm F, et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012;119:3211–3218. doi: 10.1182/blood-2011-12-400994. [DOI] [PubMed] [Google Scholar]