

Abstract
Background
This study was designed to explore the molecular mechanism underlying the effect of cellular miRNAs and EBV miRNA upon the expression of targets such as PTEN, and their involvement in the pathogenesis of Burkitt lymphoma.
Material/Methods
In this study, we examined several differentially expressed cellular miRNAs in EBV-positive versus EBV-negative Burkett lymphoma tissue samples, and confirmed PTEN as targets of cellular miR-142 by using a bioinformatics tool, luciferase reporter system, oligo transfection, real-time PCR, and Western blot analysis.
Results
We further confirmed the binding site of miR-142 in the 3′UTR of the target genes, and established the negative regulatory relationship between miRNA and mRNAs with luciferase activity assay. To verify the regulatory relationship between the miRNAs and PTEN, we evaluated the expression of PTEN in the tissue samples, and found that PTEN was downregulated in EBV- positive Burkett lymphoma. Additionally, lymphoma cells were transfected with EBV-BART-6-3p and miR-142 and we found that EBV-BART-6-3p and miR-142 synergistically reduced expression of IL-6R and PTEN. Furthermore, we also examined viability of the cells in each treatment group, and showed that EBV-BART-6-3p and miR-142 synergistically promoted proliferation of the cells.
Conclusions
These findings improve our knowledge about the role of miR-142/EBV-BART-6-3p and their target, PTEN, in the development of Burkett lymphoma; they could be novel therapeutic targets for the treatment of EBV-positive Burkett lymphoma.
MeSH Keywords: Burkitt Lymphoma, Epstein-Barr Virus Infections, MicroRNAs, PTEN Phosphohydrolase
Background
Burkett lymphoma (BL) is a key model for understanding multi-stage tumorigenesis [1]. It was the first tumor for which the association with an infectious agent was demonstrated [2]. The discovery of its association with the Epstein-Barr virus (EBV) in the 1960s became a cornerstone of human tumor virology [3]. Later, in the 1980s, the identification of c-MYC/Ig fusion at the site of t [4,5] translocation has set a path that has led to the findings of molecular oncogenic changes in many other tumor contexts [6,7]. The World Health Organization (WHO) classification has listed BL that originated from lymphoid and hematopoietic tissues as an aggressive B-cell non-Hodgkin lymphoma with 3 subsets: immunodeficiency-associated (ID-BL), sporadic (sBL), and endemic (eBL) [8].
MicroRNAs (miRNAs), a group of noncoding small RNAs consisting of 19 to 25 nucleotides, are highly conserved, and tissue-specific, playing important roles in posttranscriptional gene regulation (Kim and Lim 2014). Additionally, a synergistic effect was shown by several miRNAs targeting the same mRNA (Nam et al. 2014). miRNAs are crucial in the development of cell differentiation, apoptosis, and proliferation and are closely related to tumorigenesis in human disease (Huang et al. 2014). There are 44 different mature miRNAs generated by EBV, which are categorized into BamHI-A rightward transcript (BART) miRNAs and BamHI fragment H rightward open reading frame 1 (BHRF1) miRNAs [9–12]. The EBV lytic cycle has been reported to be regulated by viral miRNAs by several studies. The levels of BRLF1 and BZLF1 increased remarkably in Mutu and C666-1 cells transfected with a miR-BART6-3p antagomir [13]. The expression of early genes mediated by EVB is suppressed by miR-BART6-3p via silencing of Dicer. Substantial down-regulation of BRLF1 andBZLF1 in these cells with Dicer silencing were observed [13]. Some other studies reported that the accumulation of lytic proteins (early antigen diffuse component [Ea-D]), BMRF1, and BZLF1, led to over-expression of miR-BHRF1-1 in SUNE1, a nasopharyngeal carcinoma (NPC) cell line, and consequently the EBV copy number increased [14].
Ambrosio et al. reported that EBV-BART-6-3P functions as an oncogene (or oncomiR) by promoting the proliferation of BL cells via targeting PTEN, and play an active role in Burkitt lymphomagenesis [15]. Simultaneously, they identified some cellular miRNAs that are differentially expressed between the EBV-positive and EBV-negative BL samples, while no further study was carried out to explore the roles of those cellular miRNAs in the development of BL. In the present study, we examined and compared the expression of those cellular miRNAs in the samples we collected, and found miR-142 was aberrantly upregulated in the EBV-positive BL samples. Interestingly, we further identified PTEN as virtual targets of miR-142 by performing computational analysis, and the EBV-coded miRNA and cellular miRNA work synergistically on the biological behaviors of the EBV-positive BL cells.
Material and Methods
Cases collection
We collected 34 BL cases at the Department of Hematology, The First People’s Hospital of Jining. A pathologist reviewed the cases and diagnoses were confirmed according to the WHO criteria. Among the 34 BL cases, 18 were EBV-positive and 16 were EBV-negative. Our study was approved by the Ethics Committee at The First People’s Hospital of Jining, and written informed consent was obtained from all study participants.
Reverse transcriptase (RT) quantitative polymerase chain reaction (qPCR)
RNAzol reagent (Invitrogen, Carlsbad, CA) was used to extract total RNA in accordance with the manufacturer’s protocol. Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen, Carlsbad, CA), oligo (dT) primers (Macrogen, Seoul, South Korea) and 3 μg total RNA were used to synthesize cDNA. The cDNA was then amplified using quantitative PCR performed with SYBR green quantitative PCR (qPCR) kit (TaKaRa, Tokyo, Japan) on an ABI 7900 real-time PCR system (Applied Biosystems, Foster City, USA). MiR-181, -197, -574, -501, -142, -7, and -609 and mRNA of PTEN were measured. The cycler protocol was 10 min at 95°C, 40 cycles of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C. Dissociation curves were checked routinely to confirm specific amplification of the PCR product. The incubating temperature of reaction mixtures ramped from 60°C to 95°C at a heating rate of 0.1°C/s, and maintained at 95°C for 60 s. Fluorescence measure was performed continuously. The 2−ΔΔCt method was used to calculate relative gene expression with U6 as an internal standard.
Western blotting
Whole cell extracts were prepared with a cell lysis reagent (0.1% SDS, 1% Triton X-100, 130 mM NaCl, 50 nM Tris–HCl pH 8.0) containing protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s instructions. Cell lysates were separated by 10% SDS-PAGE gel and transferred to Hybond ECL nitrocellulose membrane (Millipore, Billerica, MA). The blots were then incubated by anti-PTEN antibody and anti-β-actin (all diluted at 1:1000, purchased from SCBT, Santa Cruz, CA). Secondary antibody conjugated to HRP and the ECL Western Blotting Kit (Applygen, Beijing, China) were used for detection according to the manufacturer’s protocol.
Cell culture
Ramos cell line (Cell Bank of the Chinese Academy of Sciences, Shanghai, China) was used for in vitro studies. All cells were routinely cultured in RPMI 1640 medium supplemented with 100 μg/ml streptomycin, 100 U/ml penicillin, 2 mM glutamine, and 10% fetal bovine serum (FBS) (all from Lonza, Switzerland).
Oligonucleotide transfection
Ramos cells were incubated in a 6-well plate (Greiner, Bahlingen, Germany) at 1×105 cells/well in 2 ml RPMI 1640 containing antibiotics (Wisent, Quebec City, Canada) and 10% fetal bovine serum (Gibco, Grand Island, NY). Mimics of miR-142 and EBV-BART-6-3p were purchased from Shanghai GenePharma (Shanghai, China). After reaching 80% confluence, the oligos were transfected into Ramos cells with FuGENE HD (Roche, Mannheim, Germany) in accordance with the manufacturer’s protocol. Cells transfected with 25 nM of a stability-enhanced non-targeting small RNA oligonucleotide were used as a negative control (NC).
Cell proliferation assay
Ramos cells were incubated in serum-free RPMI for 48 h, and then 10 μl of MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium bromide) (Millipore, Billerica, MA, USA) were added, followed by incubation for 4 h. The formazan product was dissolved by addition of 100 μl acidic isopropanol, and the absorbance was determined at 570 nm (reference wavelength 630 nm).
Luciferase reporter assay
We amplified the full-length 3′ UTRs of PTEN and subcloned them into the multiple restrictive sites between the poly (A) site of the psiCHECK-2 plasmid (Promega, Madison, WI) and the firefly luciferase-coding sequence. An EZ change site-directed mutagenesis kit (Enzynomics, Daejeon, South Korea) was used to introduce mutations into the seed sequences of PTEN. We seeded Ramos cells in a plate of 96 wells (5×103 cells/well) to investigate the influence of miR-142 and miR-BART-6-3p on the expression of PTEN. After 24 h, the cells were cotransfected with a psiCHECK reporter vector containing PTEN and miR-142 or EBV-BART-6-3p. At 48 h after transfection, the cells were assayed for luciferase activity using the Dual-Glo luciferase reporter assay system (Promega). Firefly luciferase activity was used to normalize renilla luciferase activity for each sample.
Statistical analysis
The t test was used to analyze data. Curve fit and analysis were conducted by GraphPad Prism software (GraphPad Software, San Diego, CA). P value <0.05 was considered to indicate statistical significance. All results are shown as mean ± standard deviations (SD).
Results
MiR-142 was differentially expressed in EBV-positive cells
Several cellular microRNAs were reported to be differentially expressed in EBV-infected cells in previous studies. To study the underlying molecular mechanism of EBV infection involved in the tumorigenesis of BL, we explored the expression of these microRNAs (including miR-181, miR-197, miR-574, miR-501, miR-501, miR-142, miR-7, and miR-609) among EBV-positive cells compared with EBV-negative cells. As shown in Figure 1, significant overexpression of miR-197, miR-510, and miR-142 and relatively evident downregulation of miR-181 were detected among EBV-positive samples compared with EBV-negative samples.
Figure 1.
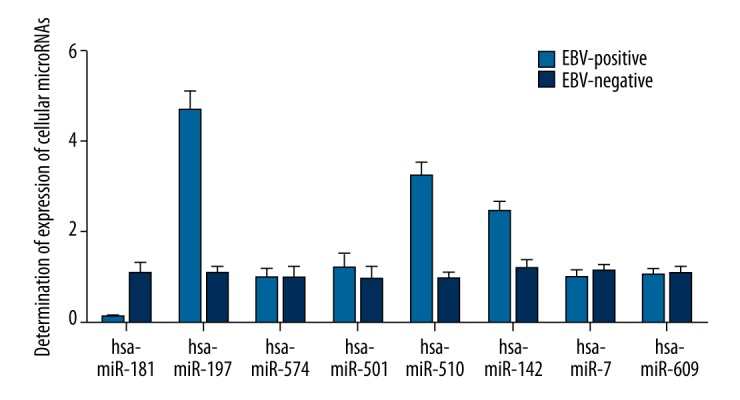
Expression of several miRNAs (miR-181, miR-197, miR-510, miR-501, miR-574 miR-7, miR-609, and miR-142) was measured using real-time PCR, and miR-181, miR-197, miR-510, and miR-142 were found to be differentially expressed.
PTEN were targets of miR-142 and EBV-BART-6-3p
To identify the targets of the differentially expressed miRNAs in our study, we searched an online miRNA database (www.mirdb.org) for genes with seed sequence in the 3′UTR that matched the miRNAs. We found the putative target binding sites of miR-142 within the 3′UTR of PTEN mRNAs. As shown in Figure 2, miR-142 and EBV-BART-6-3p matched with 3′UTR of PTEN.
Figure 2.

PTEN is a shared target of miR-142 and EBV-BART-6-3p. (A) miR-142 binds to PTEN gene on its putative binding site on 3′UTR (2427-2434). (B) EBV miRNA EBV-BART-6-3p binds to PTEN gene on a different site.
Overexpression of cellular/EBV miRNA inhibited the expression of PTEN
To verify the relationship between the cellular/EBV miRNA and PTEN, we conducted luciferase reporter assay with constructs containing wild-type and mutant PTEN 3′UTR segments in Ramos cells overexpressing miR-142 or miR-BART6-3p, respectively. As shown in Figure 3, relative luciferase activity of wild-type PTEN 3′UTR was significantly downregulated compared with mutant IL-6R 3′UTR and the negative controls in miR-142 overexpressing Ramos cells. However, in the Ramos cells overexpressing miR-BART6-3p, both the wild-type and mutant PTEN 3′UTR treatment groups showed obvious underexpression compared with the negative controls (Figure 3), which indicated that 2427–2434 bp in the 3′UTR of PTEN is the binding site of miR-142, which excluded the possibility that 2444–2450 bp is the binding site of EBV-BART-6-3p.
Figure 3.

Luciferase assay was used to evaluate the regulatory association between miR-142/EBV-BART-6-3p and PTEN. (A) Relative luciferase activity of miR-142 overexpressing cells transfected with constructs containing wild-type PTEN 3′UTR was clearly lower than in cells transfected with mutant PTEN 3′UTR and the negative controls. (B) Relative luciferase activity of EBV-BART6-3p-overexpressing cells transfected with constructs containing wild-type/mutant PTEN 3′UTR was clearly lower than in the negative controls.
PTEN was downregulated in EBV-positive BL cells
To verify the regulatory relationship between miRNAs and PTEN, we also examined the mRNA and protein expression levels of PTEN in the EBV-positive BL cells where EBV-BART6-3p and miR-142 were overexpressed using Western blot analysis and real-time PCR. As shown in Figure 4A, the relative density of Western blot of PTEN in EBV-positive cells was clearly lower than in the EBV-negative cells. Similarly, as shown in Figure 4B, PTEN mRNA expression in EBV-positive cells was clearly inhibited compared with EBV-negative cells, indicating the downregulation of PTEN protein expression in EBV-positive cells
Figure 4.

Expression of PTEN was evaluated in EBV-positive and EBV-negative BL samples. (A) mRNA expression level of PTEN is notably reduced in EBV-positive cell samples compared with EBV-negative cell samples. (B) Protein expression level of PTEN is notably reduced in EBV-positive cell samples compared with EBV-negative cell samples.
Synergistic effect of EBV-BART6-3p and miR-142 on the expression of PTEN
To clarify the relationship between cellular miRNA, EBV miRNA, and PTEN, we explored the mRNA and protein expression level of PTEN when cell groups were treated with cellular miRNA mimics, EBV miRNA mimics, and both cellular miRNA and EBV miRNA, as compared with the negative controls. As shown in Figure 5, the mRNA and protein expression of PTEN was slightly decreased in the Ramos cells individually treated with miR-142 and EBV-BART-6-3p compared with the negative controls, while in the Ramos cells co-transfected with miR-142 and EBV-BART-6-3p, the expression of PTEN was dramatically reduced, indicating the synergistic effect of EBV-BART6-3p and miR-142 on the expression of PTEN.
Figure 5.

mRNA and protein expression of PTEN were determined in Ramos cells transfected with miR-142 or miR-BART6-3p mimics. (A) Expression level of PTEN protein showed a slight decrease in cells transfected with miR-142 mimics or miR-BART6-3p mimics individually when compared with the negative controls, while cells treated with both miR-142 mimics and miR-BART6-3p mimics exhibited a dramatic decrease as compared with the negative controls. (B) Expression level of PTEN mRNA showed a slight decrease in cells transfected with miR-142 mimics or miR-BART6-3p mimics individually when compared with the negative controls, while cells treated with both miR-142 mimics and miR-BART6-3p mimics exhibited a dramatic decrease as compared with the negative controls.
Synergistic effect EBV-BART6-3p and miR-142 upon the viability of Ramos cells
Because PTEN was reported to accelerate the process of cell apoptosis, we also examined cell viability of sample groups treated with miR-142 mimics, EBV-BART6-3p mimics, and miR-142 mimics together with miR-BART6-3p mimics compared with the negative controls. As shown in Figure 6, cells treated with both cellular and EBV miRNAs showed a significantly higher trend of viability, while cells treated with miR-142 mimics and miR-BART6-3p mimics showed equally low viability, all compared with the negative controls, clearly indicating the synergistic effect of miR-142 and EBV-BART6-3p upon the proliferation of Ramos cells via targeting PTEN.
Figure 6.
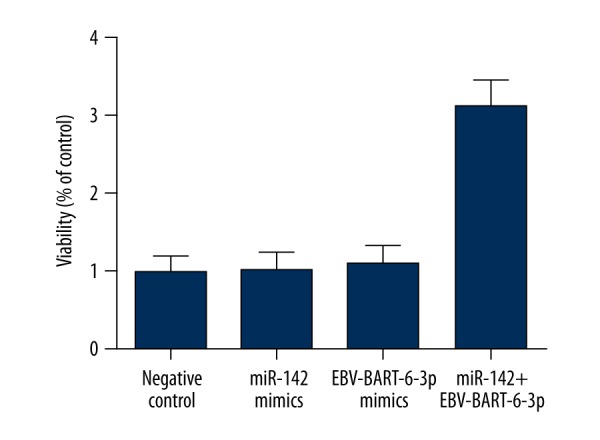
Viability in Ramos cells transfected with miR-142 mimics or miR-BART6-3p mimics individually was slightly higher than in the controls, while the cells treated with both miR-142 mimics and miR-BART6-3p mimics exhibited dramatically increased viability as compared with the negative controls.
Discussion
Epstein-Barr virus (EBV) is associated with a variety of malignancies, such as Burkitt lymphoma, gastric carcinoma, nasal natural killer/T-cell lymphoma (NNL), and Hodgkin’s disease [16], but the underlying molecular mechanism remains largely unknown. In this study, we identified 1 cellular miRNA (miR-142) that functions together with EBV-BART-6-3p as oncogenes to suppress the expression of PTEN, a known tumor suppressor, and inhibit the proliferation of BL cells.
miR-142 was initially identified as a hematopoietic-specific miRNA [17] expressed in numerous hematopoietic cell lineages [18]. Recently, it was reported that miR-142 is abnormally expressed in human T-cell lymphotropic virus type-1 (HTLV-1) adult T-cell leukemia [19]. Another study showed that miR-142 reduces cyclic adenosine monophosphate (cAMP) generation in conventional CD4+T cells through modulating the mRNA expression level of adenylyl cyclase 9 [20], which is a major enzyme catalyzing the generation of cAMP from ATP; as a result, cAMP activates protein kinase A (PKA) by directing binding. In this way, cAMP not only inhibits normal T-cell proliferation but also negatively regulate T-leukemia cells through the PKA pathway [21,22]. The data indicated that underexpression of miR-142 might be responsible for the abnormally enhanced proliferation of T-leukemic cells and leukemogenesis by decreasing cAMP levels. In addition, the miR-142 gene has been found to be located within the breakpoint junction of a t (9: 14) translocation [23–25], suggesting a potential association between the dysregulation of miR-142 and leukemogenesis. Lv et al. showed that miR-142 functions as an oncogene in tumorigenesis of T-cell acute lymphoblastic leukemia (T-ALL) as evidenced as its ability to promote the proliferation of leukemic cell growth and decrease the sensitivity to the treatment with glucocorticoid (GC) [26]. In this study, we examined several differentially expressed cellular miRNAs, including miR-181, miR-197, miR-574, miR-501, miR-501, miR-142, miR-7, and miR-609, in EBV-positive versus EBV-negative BL tissue samples, and identified the differential expression of miR-142 in the BL samples compared with controls. In addition, we confirmed PTEN as shared targets of cellular miR-142 and EBV miRNA by using bioinformatics tools, and we further confirmed the binding site of miR-142 in the 3′UTR of the target genes, and established the negative regulatory relationship between miRNAs (miR-142 and EBV-BART-6-3p) and mRNA (PTEN) with luciferase activity assay. We also determined the mRNA and protein expression levels of PTEN in EBV-positive and EBV-negative BL samples, and we found that the expression level of PTEN in EBV-positive cells was clearly lower than in the EBV-negative cells.
Phosphatase and tensin homolog (PTEN) is a protein encoded by the PTEN gene. It is one of the most frequently mutated genes in human cancer [27,28] with identified cancer-related mutations scattered over the entire PTEN gene [29]. PTEN is characterized by the presence of a tensin-like domain and a catalytic domain resembling the dual-specificity protein tyrosine phosphatases [30]. It negatively regulates intracellular levels of phosphatidylinositol-3,4,5-trisphosphate in cells, the primary signaling pathway for cell survival and proliferation, and functions as a tumor suppressor [30]. Most of the PTEN mutations are identified in the C-terminus of the protein, suggesting that the elements in this region may play a critical role in tumor suppression [31]. The C-terminal sequence of PTEN is critical for its nuclear localization [32], control of anchorage-independent proliferation [33], and the migratory and invasive ability of the cells [34]. In this study, we transfected the Ramos cells with miR-142 mimics and EBV-BART-6-3p, and determined the expression of PTEN, finding that the mRNA and protein expression of PTEN was slightly decreased in the Ramos cells individually treated with miR-142 and EBV-BART-6-3p compared with the negative controls, while in the Ramos cells co-transfected with miR-142 and EBV-BART-6-3p the expression of PTEN was dramatically reduced, indicating the synergistic effect of EBV-BART6-3p and miR-142 on the expression of PTEN. Moreover, we investigated the viability of the Ramos cells transfected with miR-142 mimics and EBV-BART-6-3p, individually or in combination, and we found that cells treated with both cellular and EBV miRNAs showed a significantly higher trend of viability, while cells treated with miR-142 mimics and miR-BART6-3p mimics both showed low viability, all compared with the negative controls, clearly indicating the synergistic effect of miR-142 and EBV-BART6-3p upon the proliferation of the Ramos cells via targeting PTEN.
Conclusions
In the present study we demonstrated that overexpressed miR-142 in EBV-positive BL is critical in the control of cell proliferation of BL in collaboration with EBV-BART-6-3p and functions as a pro-oncogenic gene. Both the EBV-coded miRNA and cellular miR-142 regulate the initiation and progression of BL, at least in part, by targeting PTEN.
Footnotes
Source of support: Departmental sources
Conflict of interest
There are no conflicts of interest.
References
- 1.Burkitt D. A sarcoma involving the jaws in African children. Br J Surg. 1985;46:218–23. doi: 10.1002/bjs.18004619704. [DOI] [PubMed] [Google Scholar]
- 2.Magrath I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br J Haematol. 2012;156:744–56. doi: 10.1111/j.1365-2141.2011.09013.x. [DOI] [PubMed] [Google Scholar]
- 3.Epstein MA, Achong BG, Pope JH. Virus in cultured lymphoblasts from a New Guinea Burkitt lymphoma. Br Med J. 1967;2:290–91. doi: 10.1136/bmj.2.5547.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dave SS, Fu K, Wright GW, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006;354:2431–42. doi: 10.1056/NEJMoa055759. [DOI] [PubMed] [Google Scholar]
- 5.Qiu J, Cosmopoulos K, Pegtel M, et al. A novel persistence associated EBV miRNA expression profile is disrupted in neoplasia. PLoS Pathog. 2011;7:e1002193. doi: 10.1371/journal.ppat.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalla-Favera R, Bregni M, et al. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA. 1982;79:7824–27. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Care A, Cianetti L, Giampaolo A, et al. Translocation of c-myc into the immunoglobulin heavy-chain locus in human acute B-cell leukemia. A molecular analysis. EMBO J. 1986;5:905–11. doi: 10.1002/j.1460-2075.1986.tb04302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leoncini L, Raphael M, Stein H, et al. International Agency for Research on Cancer, editor. Burkitt lymphoma. Lyon, France: IARC Press; 2008. [Google Scholar]
- 9.Chen SJ, Chen GH, Chen YH, et al. Characterization of Epstein-Barr virus miRNAome in nasopharyngeal carcinoma by deep sequencing. PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0012745. pii: e12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai X, Schafer A, Lu S, et al. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006;2:e23. doi: 10.1371/journal.ppat.0020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu JY, Pfuhl T, Motsch N, et al. Identification of novel Epstein-Barr virus microRNA genes from nasopharyngeal carcinomas. J Virol. 2009;83:3333–41. doi: 10.1128/JVI.01689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim do N, Chae HS, Oh ST, et al. Expression of viral microRNAs in Epstein-Barr virus-associated gastric carcinoma. J Virol. 2007;81:1033–36. doi: 10.1128/JVI.02271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iizasa H, Wulff BE, Alla NR, et al. Editing of Epstein-Barr virus-encoded BART6 microRNAs Controls Their Dicer Targeting and Consequently Affects Viral Latency. J Biol Chem. 2010;285:33358–70. doi: 10.1074/jbc.M110.138362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Chen X, Li L, et al. EBV encoded miR-BHRF1-1 potentiates viral lytic replication by downregulating host p53 in nasopharyngeal carcinoma. Int J Biochem Cell Biol. 2012;44:275–79. doi: 10.1016/j.biocel.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Ambrosio MR, Navari M, Di Lisio L, et al. The Epstein Barr-encoded BART-6-3p microRNA affects regulation of cell growth and immuno response in Burkitt lymphoma. Infect Agent Cancer. 2014;9:12. doi: 10.1186/1750-9378-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nature Reviews Cancer. 2004;4:757–68. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 17.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 18.Merkerova M, Belickova M, Bruchova H. Differential expression of microRNAs in hematopoietic cell lineages. Eur J Haematol. 2008;81:304–10. doi: 10.1111/j.1600-0609.2008.01111.x. [DOI] [PubMed] [Google Scholar]
- 19.Bellon M, Lepelletier Y, Hermine O, Nicot C. Deregulation of microRNA involved in hematopoiesis and the immune response in HTLV-I adult T-cell leukemia. Blood. 2009;113:4914–17. doi: 10.1182/blood-2008-11-189845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang B, Zhao J, Lei Z, et al. miR-142-3p restricts cAMP production in CD4+CD25– T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep. 2009;10:180–85. doi: 10.1038/embor.2008.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamir A, Granot Y, Isakov N. Inhibition of T lymphocyte activation by cAMP is associated with down-regulation of two parallel mitogen-activated protein kinase pathways, the extracellular signal-related kinase and c-Jun N-terminal kinase. J Immunol. 1996;157:1514–22. [PubMed] [Google Scholar]
- 22.Lerner A, Kim DH, Lee R. The cAMP signaling pathway as a therapeutic target in lymphoid malignancies. Leuk Lymphoma. 2000;37(1–2):39–51. doi: 10.3109/10428190009057627. [DOI] [PubMed] [Google Scholar]
- 23.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–38. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 24.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 25.Gauwerky CE, Huebner K, Isobe M, et al. Activation of MYC in a masked t(8;17) translocation results in an aggressive B-cell leukemia. Proc Natl Acad Sci USA. 1986;86:8867–71. doi: 10.1073/pnas.86.22.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lv M, Zhang X, Jia H, et al. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-alpha and cAMP/PKA pathways. Leukemia. 2012;26:769–77. doi: 10.1038/leu.2011.273. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Yen C, Liaw D, Podsypanina K, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–47. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 28.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 29.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 30.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–78. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 31.Shen WH, Balajee AS, Wang J, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 32.Terrien E, Chaffotte A, Lafage M, et al. Interference with the PTEN-MAST2 interaction by a viral protein leads to cellular relocalization of PTEN. Sci Signal. 2012;5:ra58. doi: 10.1126/scisignal.2002941. [DOI] [PubMed] [Google Scholar]
- 33.Georgescu MM, Kirsch KH, Akagi T, et al. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci USA. 1999;96:10182–87. doi: 10.1073/pnas.96.18.10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leslie NR, Yang X, Downes CP, Weijer CJ. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr Biol. 2007;17:115–25. doi: 10.1016/j.cub.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]