

Abstract
A phase 1 pharmacokinetic (PK) and pharmacodynamic (PD) study was conducted to demonstrate similarity of a proposed pegfilgrastim biosimilar to its reference product. In a single‐dose, randomized, assessor‐blinded, 2‐way crossover, active‐controlled PK/PD study, 66 healthy adults received the proposed pegfilgrastim biosimilar and US‐licensed pegfilgrastim reference product. Primary end points were pegfilgrastim AUCt and Cmax (PK), and absolute neutrophil count AUECt and Emax (PD). Safety and immunogenicity were also measured. Fifty‐six subjects completed both arms of the study. Mean pegfilgrastim concentration–time profile for both products was similar, with the 90% confidence intervals (CI) of the relative mean ratio for the primary end points falling within the predefined acceptance criteria of 80%–125% (91.7%–116.1% and 86.7%–110.2% for AUCt and Cmax, respectively). PD similarity was also demonstrated by the 95%CI of the relative mean ratio of the primary end point parameters within the predefined acceptance margins of 80%–125% (96.0%–101.6% and 92.6%–100.1% for AUECt and Emax, respectively). No statistically meaningful PK/PD differences were observed. No clinically meaningful safety or immunological differences were observed with the proposed pegfilgrastim biosimilar that were not previously identified with the reference product. The proposed pegfilgrastim biosimilar product is highly similar to the reference product with regard to PK/PD.
Keywords: biosimilars, filgrastim, pegfilgrastim, neutropenia, myelosuppressive chemotherapy, highly similar, supportive care, oncology, biologics
Human granulocyte colony‐stimulating factor (G‐CSF) is an endogenous hematopoietic growth factor that promotes the growth, proliferation, differentiation, and maturation of neutrophil precursors. It induces neutrophil terminal differentiation and enhances the function of mature neutrophils by increasing phagocytic activity and antibody‐dependent cell‐mediated cytotoxicity.1, 2 In the clinical setting, hematopoietic growth factors are used to attenuate the effects of myelosuppressive chemotherapy treatment and prevent myelosuppression, most commonly in the form of neutropenia. Prior to the advent of hematopoietic growth factors, chemotherapy‐induced neutropenia resulted in the disruption of full chemotherapy doses on the appropriate schedule and often compromised clinical outcome.3 Specifically, myelosuppressive chemotherapy regimens were often restricted by dose‐limiting toxicities to avoid occurrence of febrile neutropenia and to avoid delays in subsequent treatment cycles.
Pegylation consists of a reaction between a 20‐kDa monomethoxy polyethylene glycol propionaldehyde and the N‐terminal amino group of G‐CSF to form a Schiff base; this base, on reduction, forms a secondary amine bond between the protein and polyethylene glycol to form pegfilgrastim. Pegfilgrastim functions as a long‐acting form of filgrastim that requires only once‐per‐cycle administration for the management of chemotherapy‐induced neutropenia. In both experimental animals and healthy human volunteers, pegfilgrastim has decreased renal clearance and increased plasma half‐life compared with unpegylated filgrastim, thus sustaining the duration of the pharmacological effect.4 Pegfilgrastim was first introduced into clinical practice by Amgen, Inc., under the proprietary name Neulasta® (International Non‐Proprietary Name: pegfilgrastim).
The 2009 Biologics Price Competition and Innovation (BPCI) Act established an abbreviated approval pathway for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with a biological product already licensed by the Food and Drug Administration (FDA).5 The Apotex pegfilgrastim biosimilar development program was undertaken in accordance with FDA guidance,6, 7, 8 with the aim of establishing biosimilarity to the US‐licensed pegfilgrastim reference product and to provide a biosimilar treatment option for patients in the United States.
Apotex, Inc., and Intas Pharmaceuticals Limited have codeveloped a proposed pegfilgrastim biosimilar product as a biosimilar to the reference product, with the same proposed indication—to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a clinically significant incidence of febrile neutropenia. The analytical similarity of the proposed pegfilgrastim biosimilar and the reference product with respect to their physicochemical profile was established using a wide range of rigorous analytical techniques. The nonclinical primary pharmacodynamics (PD) studies and assessment of repeat‐dose toxicity, toxicokinetics, and local tolerance studies also support a conclusion that the proposed pegfilgrastim biosimilar is biosimilar to the reference product (unpublished results).
As expected for a biosimilar product, the focus of its clinical development program was to confirm the efficacy and safety of the proposed biosimilar with that of the reference product in head‐to‐head studies, not to establish its therapeutic effects in the same disease conditions and target populations a priori. These have already been established for pegfilgrastim through the extensive clinical studies completed with the reference product as part of the regulatory authorities' licensure of that original product. Thus, this article is intended to present the evidence of clinical similarity of the proposed pegfilgrastim biosimilar and the reference product with respect to clinical pharmacology (pharmacokinetics [PK] and PD), safety, and immunogenicity in targeted confirmatory studies.
Methods
Study Design
This was a phase 1 single‐dose, randomized, assessor‐blinded, 2‐way crossover, active‐controlled PK and PD study of the proposed biosimilar and the reference product in healthy adult male and female volunteer subjects. Each subject was randomly assigned to receive a single fixed 6‐mg dose of pegfilgrastim, administered by injection subcutaneously in the upper arm at time zero (0). The study duration included 2 study periods of 28 days each with an 8‐week washout period between administration of treatments (approximately 85 days total study duration). Subjects received pegfilgrastim either in the form of Apotex's proposed pegfilgrastim biosimilar (6 mg/0.6 mL prefilled syringe) or the reference product (Neulasta; 6 mg/0.6 mL prefilled syringe) in accordance with the randomization scheme and the alternate source of pegfilgrastim for the second study period.
Study Population
Healthy, nonsmoking male or female volunteers aged 18–55 years were recruited. All volunteers were required to have a body mass index within 18.5–29.9 kg/m2 and a body weight of 60–100 kg. The randomized volunteers included 66 healthy subjects (49 men and 17 women) from 1 study site (Apotex, Inc., BioClinical Development, Clinical Operations) in Toronto, Ontario, Canada, who had signed informed consent and had passed the screening processes for eligibility. The study was approved by Health Canada and Canadian Research Ethics Board and was conducted in compliance with Good Clinical Practice guidelines. The first subject was screened on March 4, 2013, and the clinical conduct of the study was completed on July 8, 2013.
Volunteers also had to meet the following inclusion criteria: acceptable alcohol and/or drug screen at check‐in of each period; acceptable health, blood pressure, pulse rate, and temperature at check‐in; and female subjects of childbearing potential to be either sexually inactive (abstinent) for 60 days prior to the first dose and throughout the study or use an acceptable method of birth control as described in the protocol. Oral, injectable, or topical contraceptives and contraceptive implants were permitted, as they are acceptable methods of contraception. Volunteers with clinically significant blood chemistry or significant abnormal electrocardiogram (ECG) results were excluded from the study. Additional exclusion criteria included a positive test for human immunodeficiency virus or hepatitis; a history or presence of significant asthma, cancer, chronic bronchitis, seizure, diabetes, migraine, hypertension, cardiovascular, pulmonary, neurological, or chronic psychiatric conditions, hepatic, renal, hematopoietic, or gastrointestinal or ongoing infectious diseases, or any other significant abnormality as evidenced by a medical history and physical examination; sickle cell disorder; and requiring other medication at the time of the study. Also exclusion criteria were history of drug or alcohol abuse within the last 6 months; any known enzyme‐inducing or ‐inhibiting drug taken within 30 days before the study; history of anaphylaxis, idiopathic urticarial, undiagnosed wheezing; infection within 2 weeks prior to dosing; use of lithium within 2 weeks of the beginning of the study or a plan to use lithium during the study or within 2 weeks after the end of the study; participation in an investigational drug study within a minimum of 30 days prior to dosing of this study; blood donation of 50 to 499 mL of whole blood within 30 days or more than 499 mL of whole blood within 56 days prior to drug administration; and women who were pregnant or breast‐feeding at any point during the study. Subjects were required to abstain from caffeine/xanthine‐containing foods or beverages 48 hours prior to dosing, and until 360 hours (day 15) after dosing.
Study End Points
Primary End Points
The primary PK end point parameters were peak concentration (Cmax) and area under the curve (AUCt). The primary PD end point parameters were absolute neutrophil count (ANC), area under the effect curve from time zero measured up to the last sampling time (AUECt), and the maximum effect observed over the sampling interval (Emax).
Secondary End Points
The secondary PK end point parameters were area under the plasma concentration–time curve from time zero to infinity (AUCinf), the sampling time at which Cmax occurred (Tmax), the apparent elimination half‐life of the drug (Thalf), the apparent volume into which the drug is distributed (Vd), and the rate at which the drug is cleared from the body (Cl). The secondary PD end point parameters were Tmax (the sampling time at which Emax occurred) for ANC, AUECt, and Emax for absolute CD34+ cell count.
Bioanalytical Methods
The total volume of blood taken for each subject during the study, including diagnostic and immunogenicity testing, was 453 mL.
PK Samples
In each study period, venous blood samples for the determination of pegfilgrastim plasma concentrations were collected at 0 hours (ie, between 5 and 45 minutes prior to dosing) and 1, 2, 3, 4, 6, 8, 10, 12, 14, 16, 18, 20, 24 (day 1), 28, 32, 36, 40, 48 (day 2), 60, 72 (day 3), 84, 96 (day 4), 108, 120 (day 5), 144 (day 6), 168 (day 7), 192 (day 8), 216 (day 9), 240 (day 10), 264 (day 11), and 288 (day 12) hours after subcutaneous administration of the study drugs. Plasma pegfilgrastim levels were quantified using a validated enzyme‐linked immunosorbent assay method. The method was validated in accordance with the principles of current Good Laboratory Practices and in accordance with the FDA Draft Guidance on Bioanalytical Method Validation (2001),9 as well as the European Medicines Agency (EMA) Guideline on Bioanalytical Method Validation (2011).10
PD Samples
In each study period, venous blood samples for the determination of ANC were collected at 0 hours (ie, between 5 and 45 minutes prior to dosing) and 1, 2, 4, 8, 12, 18, 24 (day 1), 28, 32, 36, 40, 48 (day 2), 60, 72 (day 3), 84, 96 (day 4), 108, 120 (day 5), 144 (day 6), 168 (day 7), 216 (day 9), 264 (day 11), 312 (day 13), and 360 (day 15) hours after the subcutaneous injection of the study drug(s) in each period. And for absolute CD34+ cell count, blood samples were collected at 0 hours (ie, between 5 and 45 minutes prior to dosing) and 24 (day 1), 48 (day 2), 72 (day 3), 84, 96 (day 4), 108, 120 (day 5), 144 (day 6), 168 (day 7), 192 (day 8), 240 (day 10), and 288 (day 12) hours after the subcutaneous injection of the study drug(s) in each period. ANC was determined by a Beckman Coulter LH780, SN AN1062 Cell Counter, and blood samples were stored at 4°C and analyzed within 24 hours of collection.
In each study period, venous samples for the determination of CD34+ count were collected as described above. A fluorescence‐activated cell sorting Canto II‐ flow cytometry analyzer was used to determine the CD34+ count using fluorescence sensitivity, with the 3‐color International Society of Hematotherapy and Graft Engineering method for the enumeration of CD34+ hematopoietic stem cells.
Safety
Safety was assessed based on observed adverse events (AEs), clinical laboratory tests, physical examinations, assessment of immunogenicity (ie, antibody formation), and results from vital sign assessments and ECGs.
Immunogenicity
Immunogenicity sampling times occurred at 0 and 672 hours (day 28) in each study period. Immunogenicity assessment to detect antidrug antibodies was performed using Meso‐Scale Discovery technology. The method was validated in accordance with the FDA Guidance for Industry on Bioanalytical Method Validation (2001)9 and Bioanalytical Method Validation (2013),11 the FDA Draft Guidance on Assay Development for Immunogenicity Testing Therapeutic Proteins (2009),12 EMA Guideline on Immunogenicity Assessment of Biotechnology‐Derived Therapeutic Proteins (2007),13 and the EMA Guideline on Bioanalytical Method Validation (2011).10
Immunogenicity assessment was based on a multitiered approach consisting of screening, confirmatory and neutralizing assay components. In tier 1, all samples were screened to detect the presence of binding antibodies to pegylated filgrastim. Samples with a mean replicate relative light units (RLU) response greater than or equal to the plate cut point were reported as “potentially positive” and analyzed to confirm specificity of the response in tier 2. If the mean replicate RLU response was less than the cut point, the sample was reported as “negative.” Drug‐treated samples analyzed in the tier 2 specificity/confirmatory assay that showed inhibition greater than or equal to the specificity cut point in the presence of pegylated filgrastim were confirmed positive for the presence of anti–pegylated filgrastim antibodies. Samples with percent inhibition less than the specificity cut point were reported as “negative.” In tier 3 analysis, samples that were confirmed positive for antibodies during tier 2 analysis were titered until a negative (below the plate cut point) response was obtained. The reportable titer result was determined as the lowest concentration of the diluted sample that was detected at or above the plate cut point and was reported as the reciprocal of that dilution. During the course of analysis of 190 samples from the study, no samples were confirmed to be positive.
Statistical Analyses
The results were analyzed using SAS statistical analysis software, version 8.2 (SAS Institute, Inc., Cary, North Carolina). The statistical analysis was performed in accordance with the FDA guidance on Statistical Approaches to Establishing Bioequivalence (January 2001) for the 2‐way crossover study design.14 For the protocol‐defined PK analysis, analysis of variance (ANOVA) was performed on the log‐transformed AUCt, AUCinf, Cmax, Cl, and Vd parameters and on the raw data for Tmax, apparent first‐order terminal elimination rate constant (Kel), and Thalf parameters using the SAS GLM procedure. The two one‐sided hypothesis15 was tested at the α = 0.05 level of significance for the AUCt and Cmax parameters by constructing the 90% confidence interval (CI) for the ratio between the test and reference means.
For each of the ANC and CD34+ cell counts, ANOVA was performed on the logarithmic transformation of the AUECt and Emax and on the raw data for Tmax. The two one‐sided hypothesis15 at the α = 0.025 level of significance was tested for AUECt and Emax by constructing the 95%CI for the ratio between the test and reference formulation means for the PD end points.
Acceptance Criteria
Bioequivalence with regard to the PK profile was concluded between the proposed pegfilgrastim biosimilar and the reference product if the 90%CIs of the primary end point parameters were contained within the 80%–125% equivalence margins. In addition, the 90%CIs of the secondary end point parameters were also calculated and assessed against these equivalence margins as supportive evidence of PK similarity.
Bioequivalence with regard to PD was concluded between the proposed pegfilgrastim biosimilar and the reference product if the 95%CIs of the primary end point parameters were contained within the 80%–125% equivalence margins. In addition, the 95%CIs of the secondary end point parameters were also calculated and assessed against these equivalence margins as supportive evidence of PD biosimilarity.
Results
Subject Disposition
Sixty‐six healthy volunteer subjects (49 men and 17 women) were enrolled and randomized into the study. Of these 66 subjects, 56 subjects (84.85%) completed both periods of the study. In period 1, 33 subjects were exposed to the proposed pegfilgrastim biosimilar, and 33 subjects were exposed to the reference product. In period 2, 27 subjects were exposed to the proposed pegfilgrastim biosimilar, and 30 subjects were exposed to the reference product.
Ten subjects in total (15.15%) discontinued the study. The main reasons for discontinuation were noncompliance with the study requirements, as 4 subjects did not attend the clinic for scheduled visits (1 subject [1.52%] in the proposed pegfilgrastim biosimilar group vs 3 subjects [4.55%] in the reference product group). Other discontinuations included 1 withdrawal by subject (1.52%) for personal reasons in the proposed pegfilgrastim biosimilar group, 3 subjects (4.55%) who experienced adverse events, and 1 subject (1.52%) in the proposed pegfilgrastim biosimilar group and 2 subjects (3.03%) in the reference product group with abnormal white blood cell count, with 1 subject because of a right wrist fracture and 1 subject because of hypersensitivity classified as a serious adverse event. Prior to period 2 dosing and hence prior to PK/PD statistical analyses, 2 subjects (3.03%) were withdrawn for PK/PD reasons (1 subject in the proposed pegfilgrastim biosimilar group [1.52%] vs 1 subject in the reference product group [1.52%]). Discontinued subjects were not replaced. Because these subjects did not complete both study periods, in accordance with the protocol, they were not included in the PK population.
A summary of the disposition of the subjects is presented in Table 1 (for additional demographic information, please see the online SDC). In addition, approximately 60% of subjects were white. The median age (range) was 41 years (20 to 55 years). The median body weight was 77.8 kg and ranged between 61 and 96.6 kg.
Table 1.
Summary of Subject Disposition, Overall n = 66 (Safety Analysis Set)
| Proposed | Pegfilgrastim | ||
|---|---|---|---|
| Pegfilgrastim | Reference | Overall, | |
| Description | Biosimilar, n (%) | Product, n (%) | n (%) |
| Subjects receiving medication (population safety analysis set) | 60 (90.91) | 63 (95.45) | 66 (100) |
| Subjects completing study (PK/PD population) | 56 (84.85) | 56 (84.85) | 56 (84.85) |
| Reasons for discontinuation | |||
| Discontinued for any reason | 4 (6.06) | 6 (9.09) | 10 (15.15) |
| Dismissed because of adverse event | 1 (1.52) | 2 (3.03) | 3 (4.55) |
| Dismissed because of noncompliance with study drug | 1 (1.52) | 3 (4.55) | 4 (6.06) |
| Withdrawal by subject | 1 (1.52) | 0 | 1 (1.52) |
| Withdrawn for PK/PD reasonsa | 1 (1.52) | 1 (1.52) | 2 (3.03) |
Subjects were withdrawn prior to the completion of the clinical phase and hence prior to PK/PD analysis.
Pharmacokinetics
The mean pegfilgrastim concentration–time profile (linear plot) obtained after the single subcutaneous administration of the proposed pegfilgrastim biosimilar and the reference product is presented in Figure 1. Mean pegfilgrastim concentrations increased as expected after single‐dose subcutaneous administration, with peak levels occurring approximately 24 to 25 hours postdose and concentrations returning to near baseline levels and/or below limit of quantification (BLQ) by 288 hours (12 days) postdose.
Figure 1.
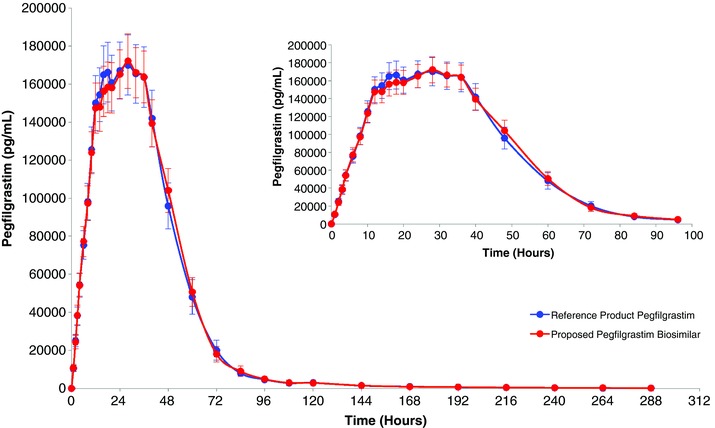
Mean plasma (average concentration)–time profile of pegylated filgrastim (linear plot) following a fixed single subcutaneous injection of 6 mg of the proposed pegfilgrastim biosimilar or the reference product pegfilgrastim in healthy volunteers. Insert displays 0–96 hours.
As seen in Table 2, the results demonstrate that the 90%CIs of the relative mean ratios for the primary pharmacokinetic end points of the study, AUCt and Cmax, were within the predefined acceptance criteria of 80%–125% (ie, 91.7%–116.1% and 86.7%–110.2% for AUCt and Cmax, respectively), with corresponding relative mean ratios near 100% (103.2% and 97.7%, respectively). For these primary PK end points, statistical significance was not detected for the treatment, period, or sequence effects. The nonsignificant period and sequence effects indicate no carryover of the drug from the first dosing period to the second period and that the order of dosing is not relevant, thereby supporting the adequacy of the washout period and the control of the study conditions. In addition, the secondary PK end points further support the PK conclusions of this study, as the relative mean ratios (with the 90%CIs) for the AUCinf, Cl, and Vd parameters were contained within the 80%–125% (ie, 100.8% [88.3%–115.0%], 96.9% [86.1%–109.0%], and 101.6% [89.0%–116.0%], respectively) and the relative mean ratios (with 90%CIs) for the untransformed parameters Tmax and Thalf are also contained with the limits of 80%–125% (ie, 105.2% [95.9%–114.5%], and 103.2% [96.1%–110.3%], respectively).
Table 2.
Summary of Pegfilgrastim PK Parameters Following a Fixed Single Subcutaneous Injection of 6 mg of Proposed Pegfilgrastim Biosimilar or Pegfilgrastim Reference Product to Healthy Subjects (PK Population)a
| Proposed Pegfilgrastim | Pegfilgrastim Reference | |||
|---|---|---|---|---|
| Biosimilar, Arithmetic | Product, Arithmetic | Relative Meanb | ||
| PK Parameter | Mean (SD), n = 56 | Mean (SD), n = 56 | Ratio, % | 90%CI |
| AUCt (pg·h/mL) | 8 165 681 | 8 125 513 | 103.2 | 91.7–116.1 |
| (5 261 409) | (6 005 813) | |||
| Cmax (pg/mL) | 190 076 | 194 909 | 97.7 | 86.7–110.2 |
| (113 710) | (129 021) | |||
| AUCinf c (pg·h/mL) | 8 108 814 | 8 410 220 | 100.8 | 88.3–115.0 |
| (5 443 322) | (6 067 565) | |||
| Tmax (h) | 25.82 | 24.18 | 105.2 | 95.9–114.5 |
| (8.00) | (9.20) | |||
| Thalf (h)c | 58.03 | 55.09 | 103.2 | 96.1–110.3 |
| (22.46) | (16.41) | |||
| Vd (mL)c | 105 461 | 97 139 | 101.6 | 89.0–116.0 |
| (103 629) | (93 230) | |||
| Cl (mL/h) | 1185 | 1206 | 96.9 | 86.1–109.0 |
| (1072) | (858) |
The drug content of the batches of the proposed pegfilgrastim biosimilar and reference product employed in this study differed by greater than 5% (ie, 8%); any differences in PK parameter between the 2 products could be significantly obscured by this difference in drug content. To avoid bias by ensuring that the concentration data for pegfilgrastim were accurately reflective of the drug content of the test and reference products, prior to conducting PK and statistical analyses, the pegfilgrastim concentration data for the proposed biosimilar and reference product were corrected for protein content and purity of the corresponding batch used.
Based on the least‐squares estimates of the geometric means of AUCt, AUCinf, Cmax, Cl, and Vd and on arithmetic means for Tmax and Thalf parameters.
N = 50 for proposed pegfilgrastim biosimilar and n = 53 for reference product; Thalf, AUCinf, and Vd parameters were not determined if the log‐linear terminal phase was not clearly defined.
Pharmacodynamics
As shown in Figure 2A, ANC increased as expected after single‐dose subcutaneous administration, with peak levels occurring approximately 64 and 61 hours postdose for the proposed pegfilgrastim biosimilar and the reference product, respectively, and returning to baseline levels by approximately 360 hours postdose. As demonstrated by the PD results in Table 3, the 95%CI of the relative mean of each of the primary PD end point parameters for ANC, AUECt, and Emax, was fully contained within the predefined acceptance criterion of 80%–125% (ie, 96.0%–101.6% and 92.6%–100.1% for AUECt and Emax, respectively), with corresponding relative mean ratios near 100% (98.8% and 96.3%, respectively). These data support the pharmacodynamic similarity of the proposed pegfilgrastim biosimilar and the reference product.
Figure 2.
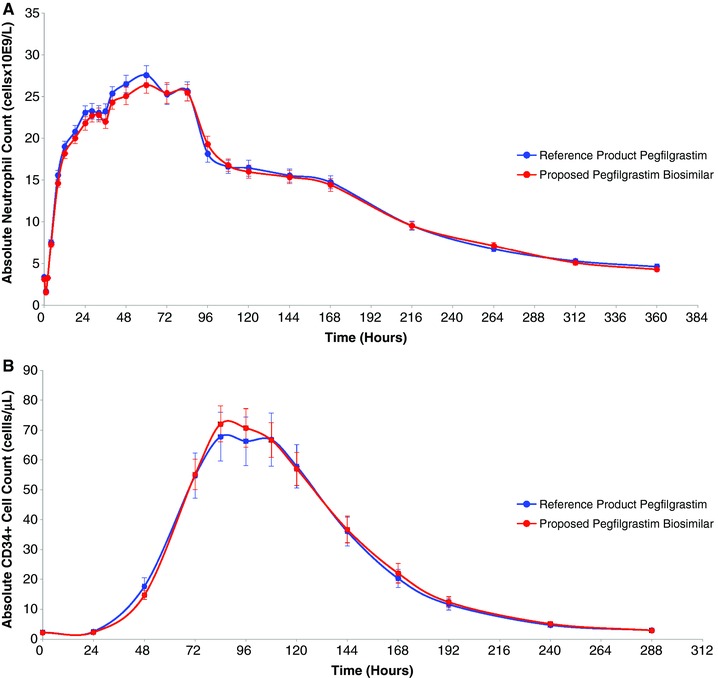
Average cell count–time profile of absolute (A) neutrophil counts and (B) CD34+ cell counts (linear plot) following a fixed single subcutaneous injection of 6 mg of the proposed pegfilgrastim biosimilar or the reference product pegfilgrastim in healthy volunteers.
Table 3.
Summary of ANC and CD34+ PD End Point Parameters Following a Fixed Single Subcutaneous Injection of 6 mg of Proposed Pegfilgrastim Biosimilar or Pegfilgrastim Reference Product to Healthy Subjects (PD Population)
| Proposed Pegfilgrastim | Pegfilgrastim Reference | |||
|---|---|---|---|---|
| Biosimilar, Arithmetic | Product, Arithmetic | Relative Meana | ||
| End Points | Mean (SD), n = 56 | Mean (SD), n = 56 | Ratio, % | 95%CI |
| ANC PD Parameter | ||||
| AUECt (cells × 109·h/L) | 4749.85 | 4817.55 | 98.8 | 96.0–101.6 |
| (1247.09) | (1314.54) | |||
| Emax (cells × 109/L) | 29.75 | 30.94 | 96.3 | 92.6–100.1 |
| (7.99) | (8.72) | |||
| Tmax (h) | 63.43 | 60.86 | 103.8 | 96.1–111.4 |
| (16.54) | (18.94) | |||
| CD34+ Count PD Parameter | ||||
| AUECt (cells·h/μL) | 7153.34 | 6991.64 | 105.9 | 99.5–112.7 |
| (5187.76) | (6798.59) | |||
| Emax (cells/μL) | 78.82 | 76.03 | 106.8 | 98.7–115.5 |
| (50.02) | (67.34) | |||
| Tmax (h) | 94.07 | 96.64 | 97.6 | 94.0–101.3 |
| (12.06) | (14.55) | |||
Based on the least‐squares estimates of the geometric means of AUECt and Emax and based on the least‐squares estimates of the arithmetic means for Tmax.
This pharmacodynamic similarity is further evidenced by the results for the additional PD marker, absolute CD34+ cell count, illustrated in Figure 2B as the average cell count–time profile. The 95%CIs for the relative mean of the absolute CD34+ cell count end point parameters AUECt and Emax were well contained within the acceptance margins of 80%–125%. The corresponding data are presented in Table 3.
Safety
No AEs were observed in the phase 1 clinical trial that were not previously observed with use of the reference product. All events were mild to moderate in severity and were similar in treatment groups, with slightly more moderate events for the reference product compared with the proposed pegfilgrastim biosimilar. The most frequent AEs, defined as those occurring in at least 5% of exposures to either study drug, are shown organized by system organ class (online SDC). Common AEs were distributed among several of the major system organ classes. Overall, the distribution of common AEs was relatively similar after exposure to the proposed pegfilgrastim biosimilar and the reference product and relatively similar in the 2 study periods. The 3 most common AEs were increased white blood cell count, reported in 66 subjects (100%); followed by bone pain, reported in 55 subjects (83.3%); and headache, reported in 44 subjects (66.7%). The white cell count AEs were mild in severity without any apparent difference between the treatment groups. Frequency of bone pain was slightly higher in period 1 (87.88%) for the proposed pegfilgrastim biosimilar compared with the reference product (75.76%), with the incidence of reported bone pain decreasing in period 2 for both treatments and with a slightly higher incidence reported in the reference product group (73.33%) than in the proposed pegfilgrastim biosimilar (66.67%). The related bone pain AEs were mild to moderate in severity without any apparent difference in severity between the treatment groups.
There were no deaths in the study, and 1 subject experienced a serious adverse event (SAE) that was reported in period 1. A 54‐year‐old female subject taking the reference product experienced a hypersensitivity reaction that required hospital admission but resolved without sequelae. The SAE was moderate in severity and considered probably related to the study drug. No clinically significant effects were seen with regard to vital signs, biochemistry, or hematology in the study.
Discussion
The BPCI Act, enacted as part of the Patient Protection and Affordable Care Act in 2010, gave the FDA the authority to implement an abbreviated pathway for the licensure of biological products that are demonstrated to be biosimilar to or interchangeable with a “standalone” reference biologic product already licensed by the FDA. The term biosimilarity is defined in section 351(k) of the Public Health Service (PHS) Act to mean that the biological product is “highly similar to the reference product notwithstanding minor differences in clinically inactive components and that there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product.”5
Various factors were considered in the overall design of the clinical pharmacology study in regard to the assessment and demonstration of similarity in both efficacy and safety for the proposed pegfilgrastim biosimilar and the reference product. Such factors included the reported pharmacokinetic, pharmacodynamic, efficacy, safety, and immunogenicity profile of pegfilgrastim medicinal products in conjunction with the relevant regulatory guidance for biosimilars.6, 8, 14, 16, 17, 18, 19 With regard to PK/PD, the exposure‐response information is important for the confirmation of biosimilar safety, purity, and potency of any biological product, as well as for the determination of any potential clinically meaningful differences between 2 products. The phase 1 study was conducted in a healthy volunteer population that is appropriate both for the PK/PD assessment of the proposed pegfilgrastim biosimilar and the pegfilgrastim class of medicinal products in general. In both healthy and patient populations, the mechanism of action of pegfilgrastim medicinal products is the same, whereby pegfilgrastim elicits its effects on hematopoietic cells by binding to specific cell surface receptors stimulating proliferation and differentiation of committed progenitor cells of the granulocyte‐neutrophil lineage into functionally mature neutrophils.20 Because the bone marrow in a healthy subject population is functionally unimpaired (in comparison with patients undergoing myelosuppressive chemotherapy), the bone marrow of this subject population is expected to be more responsive to stimulation with granulocyte colony‐stimulating factors.21 Moreover, factors that can affect the PK of the drug are more easily controlled in healthy subjects, thereby allowing more accurate and precise determination of PK parameters. The primary PD marker, ANC, is a relevant parameter used to demonstrate the PD similarity between the proposed biosimilar and the reference product for the approved indication. The underlying rationale for measuring ANC, is the role of ANC in defining neutropenia to assess the duration of neutropenia along with other affiliated relevant efficacy end points.22 The importance of ANC measures to define neutropenia is evident in the clinical studies supporting the original approval of the reference product.22 The secondary PD marker, absolute CD34+ cell count, was also assessed as supportive evidence of the PD similarity of the proposed pegfilgrastim biosimilar and the reference product.
The single‐dose crossover design is considered appropriate to access equivalence between the proposed pegfilgrastim biosimilar and the reference product. The 2‐period, 2‐treatment crossover design helps to minimize the impact of intersubject variability and therefore lowers the required sample size. As detailed below, it is also the most appropriate and sensitive design for the assessment and determination of PK and PD similarity between the 2 products in consideration of the pharmacological and safety properties of pegfilgrastim in healthy subjects. Pharmacologically, although pegfilgrastim elicits its effects by the same mechanism of action as filgrastim, as a consequence of its pegylation, pegfilgrastim is primarily eliminated via neutrophil‐mediated clearance such that the serum clearance of pegfilgrastim is related to the number of neutrophils, resulting in a relatively long half‐life for pegfilgrastim in both healthy subjects and, to a greater degree, in cancer patients because of a lower number of neutrophils.23, 24 Pharmacokinetically, although the terminal elimination half‐life for pegfilgrastim after subcutaneous dose administration is longer than that of filgrastim,22 this half‐life is still considered short enough (ie, shorter than 5 days) to allow for a crossover study design in healthy subjects employing a sufficiently long washout period (ie, at least 8 weeks for >10 half‐lives of the drug) between the first and second treatment administration. This study design is also considered appropriate from a PD standpoint because the PD response to pegfilgrastim, as measured by ANC, is a direct and readily available measure24 correlating well with drug exposure. The overall favorable safety profile and the low incidence of immunogenicity with pegfilgrastim22 further supported this crossover study design. A fixed single dose of 6 mg was selected and employed in this study, as it is the only FDA‐approved dose for pegfilgrastim25 and was expected to be sufficient to provide an adequate PK and PD response while also being safe in a healthy volunteer subject population.
As seen from the data, the proposed pegfilgrastim biosimilar and reference product demonstrated bioequivalence for all PK end point parameters. In addition, there was no indication of any clinically relevant differences between the PK data reported in this study and the published PK data for the reference product.24 In this study, both products were also shown to be biosimilar for all PD parameters, and no unexpected safety concerns (including immunogenicity) were seen with the proposed pegfilgrastim biosimilar.
In addition to this clinical study, through state‐of‐the‐art analytical studies and nonclinical pharmacodynamic repeat‐dose toxicity/toxicokinetics and local tolerance studies, it has been demonstrated that the proposed pegfilgrastim biosimilar is biosimilar (ie, analytically highly similar) to the reference product (unpublished results). The pharmacokinetic and pharmacodynamic results from this phase 1 study contribute to demonstration of PK/PD similarity of the proposed pegfilgrastim biosimilar and the reference product, thereby supporting the lack of any expected clinically meaningful differences between these 2 pegfilgrastim medicinal products in a clinical setting in a patient population. This is consistent with what was observed in efficacy and safety (including immunogenicity) in a phase 3 clinical study comparing the proposed pegfilgrastim biosimilar head to head with the reference product in breast cancer patients receiving chemotherapy (unpublished results).
The totality of the evidence supports the biosimilarity between Apotex's proposed pegfilgrastim biosimilar and the US‐licensed reference product. FDA approval of the proposed pegfilgrastim biosimilar may provide clinicians with additional treatment options for treating neutropenia in patients undergoing myelosuppressive chemotherapy.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Declaration of Conflicting Interests
Authors of this publication do not have any research support from sources other than Apotex, Inc. and do not have any financial involvement or declare any other competing interests.
Funding
Research support came from Apotex, Inc.
References
- 1. Welte K, Platzer E, Lu L, et al. Purification and biochemical characterization of human pluripotent hematopoietic colony‐stimulating factor. Proc Natl Acad Sci U S A. 1985;82:1526–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Souza LM, Boone TC, Gabrilove J, et al. Recombinant human granulocyte colony‐stimulating factor: effects on normal and leukemic myeloid cells. Science. 1986;232:61–65. [DOI] [PubMed] [Google Scholar]
- 3. Leonard RC, Miles D, Thomas R, Nussey F. Impact of neutropenia on delivering planned adjuvant chemotherapy: UK audit of primary breast cancer patients. Br J Cancer. 2003;89:2062–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Molineux G, Kinstler O, Briddell B, et al. A new form of filgrastim with sustained duration in vivo and enhanced ability to mobilize PBPC in both mice and humans. Exp Hematol. 1999;27:1724–1734. [DOI] [PubMed] [Google Scholar]
- 5. Office of the Legislative Counsel . 2013. Section 351(k) of the Public Health Service Act. US House of Representatives; 2013. http://legcounsel.house.gov/Comps/PHSA_CMD.pdf. Accessed April 8, 2015.
- 6. FDA . 2012. Draft guidance for industry: scientific considerations for demonstrating biosimilarity to a reference product. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291128.pdf. Accessed April 8, 2015.
- 7. FDA . 2015. Guidance for industry: quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product. http://www.fda.gov/ucm/groups/fdagov‐public/@fdagov‐drugs‐gen/documents/document/ucm291134.pdf. Accessed February 26, 2016.
- 8. FDA . 2014. Draft guidance for industry: clinical pharmacology data to support a demonstration of biosimilarity to a reference product. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/cm397017.pdf. Accessed April 8, 2015.
- 9. FDA . 2001. Guidance for industry: bioanalytical method validation. http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf. Accessed April 8, 2015.
- 10. EMA Committee for Medicinal Products for Human Use (CHMP) . 2011. Guideline on bioanalytical method validation. EMEA/CHMP/EWP/192217/2009 Rev.1 Corr.*. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf. Accessed April 8, 2015.
- 11. FDA . 2013. Draft guidance for industry: bioanalytical method validation. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm368107.pdf. Accessed April 8, 2015.
- 12. FDA . 2009. Guidance for industry: assay development for immunogenicity testing of therapeutic proteins. http://www.fda.gov/downloads/Drugs/Guidances/UCM192750.pdf. Accessed April 8, 2015.
- 13. EMA CHMP. 2007. Guideline on immunogenicity assessment of biotechnology‐derived therapeutic proteins. EMEA/CHMP/BMWP/14327/2006. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf. Accessed April 8, 2015.
- 14. FDA . 2001. Guidance for industry: statistical approaches to establishing bioequivalence. http://www.fda.gov/downloads/Drugs/.../Guidances/ucm070244.pdf. Accessed February 25, 2016.
- 15. Schuirmann DJ. A comparison of the two one‐sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15:657–680. [DOI] [PubMed] [Google Scholar]
- 16. EMA CHMP. 2006. Guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: non‐clinical and clinical issues. EMEA/CHMP/42832/2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf. Accessed April 8, 2015.
- 17. EMA CHMP. 2006. Annex to guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: non‐clinical and clinical issues. EMEA/CHMP/BMWP/31329/2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003955.pdf. Accessed April 8, 2015.
- 18. EMA CHMP. 2010. Guideline on the investigation of bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1/Corr**. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed April 8, 2015.
- 19. FDA . Draft guidance for industry: bioavailability and bioequivalence studies submitted in NDAs or INDs—general considerations. 2014. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm389370.pdf. Accessed April 8, 2015.
- 20. Curran MP, Goa KL. Pegfilgrastim. Drugs. 2002;62:1207–1213. [DOI] [PubMed] [Google Scholar]
- 21. Gascon P, Fuhr U, Sorgel F, et al. Development of a new G‐CSF product based on biosimilarity assessment. Ann Oncol. 2010;21:1419–1429. [DOI] [PubMed] [Google Scholar]
- 22. Molineux G. Pegfilgrastim: using pegylation technology to improve neutropenia support in cancer patients. Anticancer Drugs. 2003;14(4):259–264. [DOI] [PubMed] [Google Scholar]
- 23. Zamboni WC. Pharmacokinetics of pegfilgrastim. Pharmacotherapy. 2003;23(8 pt 2):9S–14S. [DOI] [PubMed] [Google Scholar]
- 24. Yang BB, Kido A. Pharmacokinetics and pharmacodynamics of pegfilgrastim. Clin Pharmacokinet. 2011;50(5):295–306. [DOI] [PubMed] [Google Scholar]
- 25. FDA . 2002. Product approval information. http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2002/pegfamg013102L.htm. Accessed February 25, 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.