

Abstract
It is well established that the formation of transthyretin (TTR) amyloid fibrils is linked to the destabilization and dissociation of its tetrameric structure into insoluble aggregates. Isotope labeling is used for the study of TTR by NMR, neutron diffraction, and mass spectrometry (MS). Here MS, thioflavin T fluorescence, and crystallographic data demonstrate that while the X‐ray structures of unlabeled and deuterium‐labeled TTR are essentially identical, subunit exchange kinetics and amyloid formation are accelerated for the deuterated protein. However, a slower subunit exchange is noted in deuterated solvent, reflecting the poorer solubility of non‐polar protein side chains in such an environment. These observations are important for the interpretation of kinetic studies involving deuteration. The destabilizing effects of TTR deuteration are rather similar in character to those observed for aggressive mutations of TTR such as L55P (associated with familial amyloid polyneuropathy).
Keywords: amyloid proteins, deuteration, isotope effects, native mass spectrometry, transthyretin
Transthyretin (TTR) is a 55 kDa homotetrameric protein responsible for transporting thyroxine and retinol, and is also implicated in amyloidosis. Aggregation of the wild‐type (WT) TTR causes senile systemic amyloidosis (SSA), predominantly affecting the heart. Mutated TTR causes familial amyloid polyneuropathy (FAP) and/or cardiomyopathy (FAC), depending on the identity of the mutation the patient has inherited. TTR deposits occur throughout the peripheral and autonomic nervous system, and accumulate in various organ systems, such as gastrointestinal tract, heart, and kidney.1 Over the years, numerous experimental techniques have been applied to the study of TTR and the misfolding that ultimately leads to the deposition of insoluble amyloid fibrils. X‐ray crystallography has been extensively used for structural studies of this protein and there are currently over 200 structures of TTR and many of its variants including complexes formed with various ligands.2 Unfortunately none of these data fully explain the mechanism of TTR amyloid fibril formation. There are increasing efforts in exploiting other techniques such as NMR and neutron crystallography as alternative methods to study the assembly processes.3, 4, 5 A number of these techniques require deuteration of the protein, or some other form of isotope labeling. While it is known from neutron scattering and NMR work that isotope labeling can usually be reliably (and isomorphously) used in structural studies (crystallography, SANS, dynamics),6, 7, 8 there has been some discussion about the effect that it may have on protein assembly and folding processes.9 In this work, both hydrogenated (HTTR) and deuterated (DTTR) forms of TTR have been studied. High‐resolution structures of these analogues have been compared in detail and the stability of the tetramer has been assessed by measuring the exchange kinetics between labeled and unlabeled tetramers using MS, and by SDS‐PAGE analysis as a function of pH. The impact of deuteration on amyloid fibril formation has also been studied using thioflavin T (ThT) fluorescence.
The X‐ray crystal structures of HTTR and DTTR were determined to a resolution of 1.30 Å and 1.42 Å respectively (Figure 1 A, Table S1 in the Supporting Information). Alignment of the backbone atoms of both chains A and B for the two structures showed that they are essentially identical, with an overall root‐mean‐square deviation (RMSD) of 0.11 Å. These results further validate the increasingly widespread use of perdeuteration in neutron crystallography.10 It should be noted that while the two structures look identical at the resolution obtained, atomic resolution data (≤1 Å)11 may help to further clarify the small structural effects associated with H/D isotopic replacement.12
Figure 1.
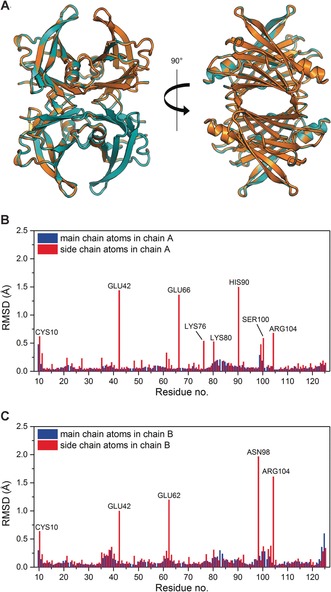
A) Superposition of the X‐ray crystal structures of HTTR (orange color) and DTTR (teal color) show close similarity of the two variants, RMSD=0.11 Å over 908 main chain atoms. Image was generated and rendered with Pymol. Panels (B) and (C) show the calculated RMSD of the main chain atoms (blue) and side chain atoms (red) between HTTR and DTTR for residues in chain A and chain B, respectively. The residues Cys10, Glu42, Glu66, Lys76, Lys80, His90, Ser100, and Arg104 in chain A, and the residues Cys10, Glu42, Glu62, Asn98, and Arg104 in chain B are disordered and have poor density in the crystal structure analysis.
The amyloid formation kinetics of HTTR and DTTR were assessed under fibrillating conditions by measuring ThT fluorescence over 80 hours (Figure 2 A). Fibril formation from DTTR was evident within an hour, as shown by a three‐fold increase in ThT fluorescence by comparison with HTTR (Figure 2 A inset). In general, DTTR showed an approximately two‐fold higher fibril accumulation than HTTR. Dissociation of the tetrameric state is thought to be the rate‐limiting step for TTR amyloidogenesis.1 The tetramer stability of HTTR and DTTR was therefore examined as a function of pH (Figure 2 B,C). The samples were incubated in buffers whose pH ranged from 2 to 11 for 72 hours at 37 °C and then analyzed by PAGE (see the Supporting Information). DTTR presented a higher propensity for tetramer dissociation than HTTR. In particular, DTTR monomers were present between pH 6.5 and 11, whereas HTTR was tetrameric in this pH range. At pH 5.5 and 6, the proportion of monomers formed was greater for DTTR than for HTTR. These results indicate that the perdeuteration of TTR destabilizes the quaternary state and promotes the subsequent dissociation into monomers.
Figure 2.
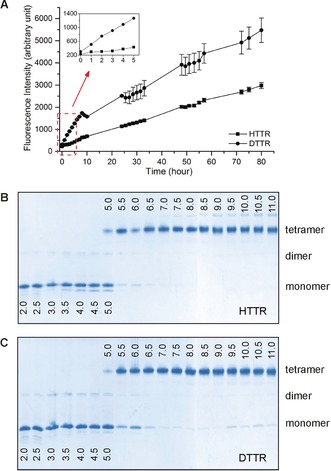
A) ThT fluorescence intensities measured during the fibrillation of HTTR and DTTR (λ ex=440 nm, λ em=480 nm). Each data point is the average of triplicate measurements. The error bars represent the standard deviations. B) HTTR and C) DTTR tetramer stability as a function of pH. The pH value is indicated on each lane. Three major bands were detected, corresponding to TTR tetramer (56 kDa), dimer (28 kDa), and monomer (14 kDa).
The isotope effect following protein perdeuteration was further elucidated using native MS13 to monitor and compare the tetramer subunit exchange as a function of time for equimolar mixtures of HTTR and DTTR, and also for equimolar mixtures of HTTR and [13C, 15N]‐labeled TTR (CNTTR). The latter study was used as a reference, based on the assumption that 13C‐ and 15N‐labeling of proteins is not generally thought to introduce significant isotope effects, as noted in previous work where a similar labeling strategy was used to investigate subunit exchange between WT and the L55P mutant of TTR.14 Given these types of mixture, the various possible TTR tetramers resulting from the combination of pure HTTR (4H) and pure DTTR (4D) samples are summarized in Figure S3. These different homo‐ and hetero‐ tetramers have distinct m/z signals in the MS measurements and therefore their relative abundance can be established as a function of time (see the Supporting Information). In the first experiment, 4H and 4D samples were mixed, and MS spectra recorded over a period of 8 days (Figure 3 A,B). Thirty eight minutes after initiating the reaction, only 4H and 4D signals were detected (Figure 3 A). 16 hours after the reaction onset, the MS data also indicated the presence of 2H2D and 1H3D hetero‐tetramers (Figure S4 A). The peaks that could be assigned to the 3H1D hetero‐tetramer were not resolvable from the other signals, as previously reported.14 In the second experiment, 4H and pure CNTTR (4CN) samples were prepared and monitored over 10 days (Figure 3 C and 3D). In common with the first experiment, only 4H and 4CN homo‐tetramers were present at the beginning of the reaction (Figure 3 C), and 4H, 4CN, 2H2CN, and 1H3CN were detected after 16 hours (Figure S4 B). The relative abundance of TTR tetrameric species throughout the two experiments is shown in Figure 4 A,B. These plots and the associated half‐life (t 1/2) measurements (Table S4), indicate that the HTTR/DTTR subunit exchange in the tetramer occurs more rapidly than it does for the HTTR/CNTTR. The t 1/2 of 4D and 4CN were about 19 hours and 57 hours, respectively (Table S4). Thus, the exchange involving the d‐subunits was about 3 times faster than that for the CN‐subunits. This increase in subunit exchange implies that deuteration causes a destabilization of the TTR tetramer.
Figure 3.
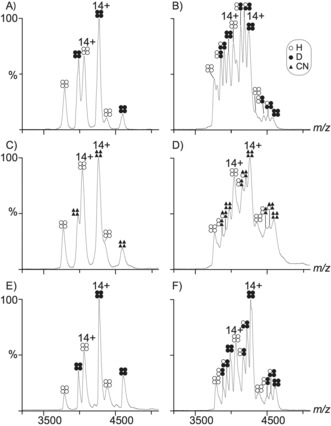
Native MS spectra of mixed unlabeled and isotope‐labeled TTR. A) 38 minutes after mixing HTTR and DTTR. In the spectrum six peaks are present matching the masses of the unlabeled (4H) and labeled (4D) TTR homo‐tetramers. B) TTR spectrum after 8 days of the HTTR and DTTR mixing. C) TTR spectrum 7 minutes after mixing 4H and 4CN. D) The same reaction was monitored after 10 days. H‐ammonium acetate was used for the samples shown in A–D. E) 7 minutes after mixing HTTR and DTTR. F) TTR spectrum after 8.8 days of mixing HTTR and DTTR. d‐ammonium acetate dissolved in D2O was used for the samples in (E) and (F).
Figure 4.
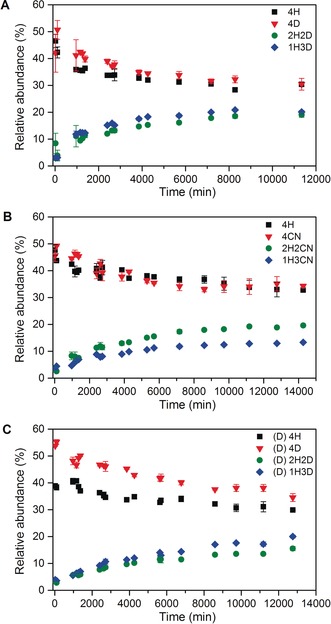
The changes in relative abundance of each species during the subunit exchange between A) HTTR and DTTR, B) HTTR and CNTTR, both in H‐ammonium acetate, and C) HTTR and DTTR in d‐ammonium acetate. Each data point is the average of 2–4 replicate measurements. The error bars represent the standard deviations.
These observations are in accordance with previous studies where deuterated proteins have been found to be less stable to heat denaturation.15, 16, 17 Hattori et al.18 have shown that the oligomerization propensity of d‐phycocyanin is lower than that of H‐phycocyanin. The destabilizing effects of protein perdeuteration may be considered in terms of the different properties of C−H and C−D bonds. In particular, the vibrational amplitudes and frequencies of the various C−D bonds involved are smaller than those of the C‐H interactions, indicating that the C−D bond lengths are shorter and stronger than the C‐H counterparts. For this reason, the deuterated side chains present a reduced size and interact less favorably with each other. Moreover, smaller side chains have lower steric hindrance for non‐polar amino acid side chains. This leads to weak intermolecular and intramolecular hydrophobic interactions.17, 19
In addition to the experiments described above, an investigation was carried out to study the effect of deuterated solvent on TTR subunit exchange (Figures 3 E,F, and 4 C). This was essentially a duplication of the first MS experiment, apart from the fact that a D2O‐based solvent was used instead of a H2O‐based one (see the Supporting Information). Here the first emergence of the 2H2D and 1H3D hetero‐tetramers occurred after 23 hours instead of 16 hours (Figure S4 C); the t 1/2 of 4D in the d‐solvent was about 65 hours (Table S4). This exchange was 3.4 times slower than that for the H‐solvent (i.e. about 19 hours), suggesting that the d‐solvent enhances TTR tetramer stability. Deuterated buffers are considered as inefficient solvents for non‐polar amino acid side chains and therefore (i) drive the burial of non‐polar surface groups and (ii) make more compact van der Waals packing in folded proteins.20 Stronger hydrophobic interactions amongst the TTR tetramers may stabilize the complex and slow down the subunit exchange. Previous studies have already demonstrated that D2O makes proteins more compact and less flexible,18, 21 and stabilizes viral RNA.22 In the work of Jasnin et al.,20 the authors have shown that macromolecules are less flexible in D2O than in H2O.
There is relatively little information available on the effect of deuterium substitution on macromolecules.9, 15, 16, 17, 20 While this study shows consistent and reproducible effects of protein and solvent deuteration on TTR tetramer stability, isotope effects such as these are generally rather subtle and unpredictable. It should be noted that the macromolecular isotope effect described here for DTTR is relatively small by comparison with the effects of serious aggressive mutations such as TTR‐L55P (associated with FAP) where a very much faster subunit exchange has been reported.14 In the overall context of this work, where the faster subunit exchange, decreased tetramer stability, and enhanced fibril formation of the deuterated TTR show similar trends to that observed for L55P, it is interesting to speculate that the use of deuteration may provide a useful probe of the factors underlying amyloidoses associated with TTR and other proteins. Undoubtedly, details of the solvent structure and amino acid protonation states at the monomer‐monomer and dimer‐dimer interfaces are likely to be crucial for uncovering the mechanism of amyloidosis. Neutron crystallographic studies of these aspects of the L55P and other pathologically related TTR mutations are currently in progress.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work used the MS platform of the Grenoble Instruct Center (ISBG: UMS 3518 CNRS‐CEA‐UJF‐EMBL) with the support from FRISBI (grant number ANR‐10‐INSB‐05‐02) and GRAL (grant number ANR‐10‐LABX‐49‐01) within the Grenoble Partnership for Structural Biology (PSB). V.T.F. acknowledges support from the EPSRC under grant numbers GR/R99393/01 and EP/C015452/1 which funded the creation of the Deuteration Laboratory (d‐Lab) in the Life Sciences group of the ILL. We also acknowledge Dr. Luca Signor for his help on the mass spectrometry under denaturing conditions.
A. W. Yee, M. Moulin, N. Breteau, M. Haertlein, E. P. Mitchell, J. B. Cooper, E. Boeri Erba, V. T. Forsyth, Angew. Chem. Int. Ed. 2016, 55, 9292.
Contributor Information
Dr. Elisabetta Boeri Erba, Email: elisabetta.boeri-erba@ibs.fr.
Prof. V. Trevor Forsyth, Email: tforsyth@ill.fr
References
- 1. Johnson S. M., Wiseman R. L., Sekijima Y., Green N. S., Adamski-werner S. L., Kelly J. W., Acc. Chem. Res. 2005, 38, 911–921. [DOI] [PubMed] [Google Scholar]
- 2. Palaninathan S. K., Curr. Med. Chem. 2012, 19, 2324–2342. [DOI] [PubMed] [Google Scholar]
- 3. Fitzpatrick A. W. P., Debelouchina G. T., Bayro M. J., Clare D. K., Caporini M. A., Bajaj V. S., Jaroniec C. P., Wang L., Ladizhansky V., Müller S. A., et al., Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yokoyama T., Mizuguchi M., Nabeshima Y., Kusaka K., Yamada T., Hosoya T., Ohhara T., Kurihara K., Tomoyori K., Tanaka I., et al., J. Struct. Biol. 2012, 177, 283–290. [DOI] [PubMed] [Google Scholar]
- 5. Haupt M., Blakeley M. P., Fisher S. J., Mason S. A., Cooper J. B., Mitchell E. P., Forsyth V. T., IUCrJ 2014, 1, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Artero J. B., Härtlein M., McSweeney S., Timmins P., Acta Crystallogr. Sect. D 2005, 61, 1541–1549. [DOI] [PubMed] [Google Scholar]
- 7. Haertlein M., Moulin M., Devos J. M., Laux V., Dunne O., Forsyth V. T., Methods Enzymol. 2016, 566, 113–157. [DOI] [PubMed] [Google Scholar]
- 8. Cooper S. J., Raftery J., Helliwell J. R., Brockwell D., Attwood D., Barber J., Chem. Commun. 1998, 1063–1064. [Google Scholar]
- 9. White J., Heß D., Raynes J., Laux V., Haertlein M., Forsyth T., Jeyasingham A., Eur. Biophys. J. 2015, 44, 367–371. [DOI] [PubMed] [Google Scholar]
- 10. Blakeley M. P., Langan P., Niimura N., Podjarny A., Curr. Opin. Struct. Biol. 2008, 18, 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cuypers M. G., Mason S. A., Blakeley M. P., Mitchell E. P., Haertlein M., Forsyth V. T., Angew. Chem. Int. Ed. 2013, 52, 1022–1025; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1056–1059. [Google Scholar]
- 12. Fisher S. J., Helliwell J. R., Acta Crystallogr. Sect. A 2008, 64, 359–367. [DOI] [PubMed] [Google Scholar]
- 13. Boeri Erba E., Petosa C., Protein Sci. 2015, 24, 1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keetch C. A., Bromley E. H. C., McCammon M. G., Wang N., Christodoulou J., Robinson C. V., J. Biol. Chem. 2005, 280, 41667–41674. [DOI] [PubMed] [Google Scholar]
- 15. Piszczek G., Lee J. C., Tjandra N., Lee C. R., Seok Y. J., Levine R. L., Peterkofsky A., Arch. Biochem. Biophys. 2011, 507, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meilleur F., Contzen J., Myles D. A. A., Jung C., Biochemistry 2004, 43, 8744–8753. [DOI] [PubMed] [Google Scholar]
- 17. Brockwell D., Yu L., Cooper S., McCleland S., Cooper a, Attwood D., Gaskell S. J., Barber J., Protein Sci. 2001, 10, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hattori A., Crespi H. L., Katz J. J., Biochemistry 1965, 4, 1225–1238. [Google Scholar]
- 19. Hattori A., Crespi H. L., Katz J. J., Biochemistry 1965, 4, 1213–1225. [DOI] [PubMed] [Google Scholar]
- 20. Jasnin M., Tehei M., Moulin M., Haertlein M., Zaccai G., Eur. Biophys. J. 2008, 37, 613–617. [DOI] [PubMed] [Google Scholar]
- 21. Efimova Y., Haemers S., Wierczinski B., Norde W., van Well A., Biopolymers 2006, 85, 264–273. [DOI] [PubMed] [Google Scholar]
- 22. Verheyden B., Andries K., Rombaut B., Vaccine 2001, 19, 1899–1905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary