

Abstract
Palmitoylethanolamide (PEA) has been suggested to have useful analgesic properties and to be devoid of unwanted effects. Here, we have examined critically this contention, and discussed available data concerning the pharmacokinetics of PEA and its formulation. Sixteen clinical trials, six case reports/pilot studies and a meta‐analysis of PEA as an analgesic have been published in the literature. For treatment times up to 49 days, the current clinical data argue against serious adverse drug reactions (ADRs) at an incidence of 1/200 or greater. For treatment lasting more than 60 days, the number of patients is insufficient to rule out a frequency of ADRs of less than 1/100. The six published randomized clinical trials are of variable quality. Presentation of data without information on data spread and nonreporting of data at times other than the final measurement were among issues that were identified. Further, there are no head‐to‐head clinical comparisons of unmicronized vs. micronized formulations of PEA, and so evidence for superiority of one formulation over the other is currently lacking. Nevertheless, the available clinical data support the contention that PEA has analgesic actions and motivate further study of this compound, particularly with respect to head‐to‐head comparisons of unmicronized vs. micronized formulations of PEA and comparisons with currently recommended treatments.
Keywords: adverse drug reactions, clinical trials, inflammation, pain, pharmacokinetics, Palmitoylethanolamide
Introduction
Palmitoylethanolamide (PEA, N‐(2‐hydroxyethyl) hexadecamide, palmidrol; structure shown in Figure 1) belongs to the family of N‐acylethanolamines (NAEs), endogenous biologically active lipids including the endogenous cannabinoid receptor ligand anandamide and the satiety factor oleoylethanolamide. PEA was identified in the 1950s as being an active anti‐inflammatory agent in chicken egg yolk 1, 2. In mammals, PEA is produced on demand from the lipid bilayer and is ubiquitous, with tissue concentrations in the mid to high pmol/g range being found in rodents 3. Preclinical and clinical studies suggest PEA may potentially be useful in a wide range of therapeutic areas, including eczema, pain and neurodegeneration and at the same time to be essentially devoid of unwanted effects in humans (see e.g. 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 for examples, and 20 for a review of the clinical data accrued up to 2012 with respect to pain). PEA is currently marketed for veterinary use (skin conditions, Redonyl™, [Innovet]) and as a nutraceutical in humans (Normast™, Pelvilen™ [Epitech]), PeaPure™ [JP Russel Science Ltd]) in some European countries (e.g. Italy, Spain; it is sold as a food supplement in other countries, such as the Netherlands). It also is a constituent of a cream (Physiogel AI™, Stiefel) marketed for dry skin.
Figure 1.
Structure of PEA. The compound is sometimes referred to as NAE 16:0, where 16 and 0 refer to the number of carbon atoms and double bonds, respectively, in the acyl side chain. The related compounds anandamide and oleoylethanolamide are NAE 20:4 and NAE 18:1, respectively, using this nomenclature
Most reviews on the subject of PEA and its clinical potential have presented it in a fairly cursory manner, with the exception of a very recent meta‐analysis 21. In addition, the pharmacokinetic properties of PEA have not been considered to any extent. In the present review, we have focused on these issues.
Preclinical pharmacology of PEA
The pharmacological properties of PEA with respect to pain, inflammation and mechanism(s) of action in preclinical models have been well reviewed elsewhere 20, 22, 23 and will only be mentioned briefly here. PEA shows efficacy in a variety of pain models including carrageenan‐ and prostaglandin‐induced hyperalgesia 6, 15, 18, the formalin test of persistent pain 8, 9, visceral hyperalgesia produced by instillation of nerve growth factor into the bladder 7, 12, and the sciatic nerve ligature model of neuropathic pain 14, whereas the acute thermal pain response is not affected 8. The proposed mechanism(s) of action of PEA involve effects upon mast cells 6, CB2‐like cannabinoid receptors 9, 12, ATP‐sensitive K+‐channels 18, TRP channels 24, and NFkB 15, although the most robust evidence is for an action of PEA upon the nuclear receptor peroxisome proliferator‐activated receptor α (PPARα) 13. These are by no means the only actions of PEA: it can also, for example, interact as an agonist with GPR119, an orphan receptor involved in glucagon‐like peptide‐1 secretion 25, 26, and will, at least in theory, affect endocannabinoid signalling by acting as a competing substrate for the endocannabinoid homologue anandamide (N‐arachidonoylethanolamine). Some of these actions are shared by the endogenous NAEs N‐oleoylethanolamine and N‐stearoylethanolamine 13, 25, 27, but clinical data to our knowledge is lacking with respect to these compounds.
Pharmacokinetic considerations
There is very little data available in the open literature concerning the pharmacokinetic properties of PEA. To our knowledge, the bioavailability (F) and apparent volume of distribution (V d) of PEA have not been reported. In view of this, we have attempted to provide some ‘ball‐park’ estimates using data from a recent study investigating the plasma concentration of PEA following oral treatment of nine male Wistar rats (body weight 150–250 g) with 100 mg kg−1 of PEA in a corn oil suspension 28. The focus of that study was to find pro‐drugs for PEA, and so the authors were content to report the area under the curve for the measurement period (AUC0‐8h) and the approximate t max value. The plasma concentrations, in nM, reported in Table 2 of 28 are shown graphically in Figure 2. PEA is also relatively short‐lived in human plasma: Petrosino et al. 29 reported in graphical form plasma PEA levels 0, 2, 4 and 6 h after oral administration of 300 mg micronized PEA to 10 healthy volunteers. There was a significant increase (from ~10 to ~23 pmol ml−1 plasma) at the 2 h time point, returning to baseline at the higher time points.
Table 2.
Case reports and pilot studies investigating PEA in patients with pain
| Type of study | No. of cases | Type of pain | PEA dosage | Formulation | Treatment length | Outcome (all VAS scale unless marked with * or †) | Unwanted effects | Ref |
|---|---|---|---|---|---|---|---|---|
| Pilot study, open‐label | 4 | Chronic pelvic pain associated with endometriosis | 400 mg daily (combined with polydatin) | M | 90 days | Reduction of pain intensity | None reported | 59 |
| Case report collection | 7 | Chronic idiopathic axonal neuropathy and pain | 1200 mg daily to 2000 mg | M | Different amongst patients ranging from weeks to months | Reduction in pain intensity in all patients* | None reported | 60 |
| Case report | 1 | Chronic regional pain syndrome type 1 | 1200 mg daily (combined with topical ketamine cream) | M | 2 months | Reduction of pain intensity† | None reported | 61 |
| Case report | 1 | Multiple sclerosis and central neuropathic pain | Up to 1200 mg daily (combined with acupuncture) | UM or M | 9 months, intermittant | Reduction of pain intensity* | None identified‡ | 62 |
| Case report | 1 | Pudendal neuralgia | Up to 900 mg daily | NS | 1 year | ‘Improvement of neuralgia and associated symptoms’† | NS | 63 |
Abbreviations: M, Micronized; NS, not stated in the article; UM, Ultramicronized; VAS, Visual analogue scale.
NRS used.
Other or unidentified evaluation method.
Patient developed a cough early on in the study. The cough continued after PEA was stopped, and so the compound was reinstated.
Figure 2.
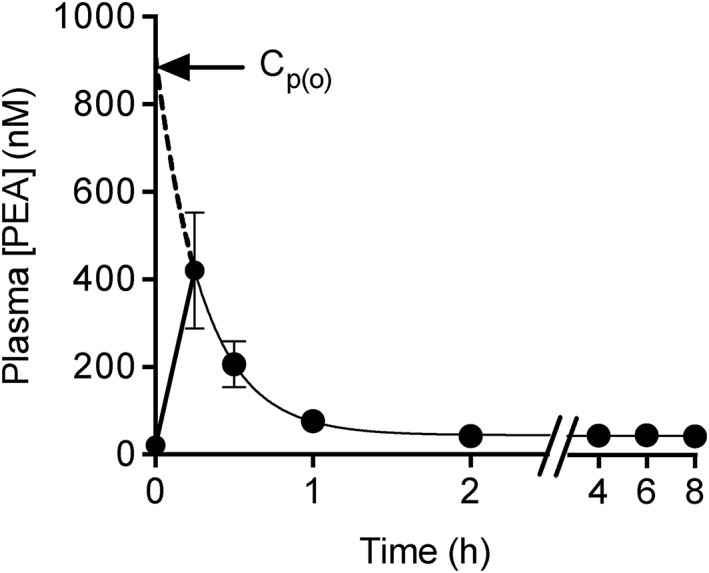
Plasma concentrations of PEA following oral dosing of 100 mg kg−1 to male Wistar rats (body weight 150–250 g). The data are taken from Table 2 of 28 and are shown as means ± SEM, n = 9. The time points from 0.25–8 h were fitted to a one‐phase decay model using the least squares method (GraphPad Prism 6.0 h for the Macintosh). The model returns the extrapolated plasma concentration at t = 0 (Cp(o), 913 ± 16 nM), the value to which the curve asymptotes (44 ± 1 nM, i.e. the data ≥2 h), the rate constant (3.4 ± 0.06 h−1) and hence the t 1/2 value (0.21 h). Needless to say, the large data spread at the first time point renders the values approximate
Assuming a simple one compartment model with first‐order absorption and distribution, a plasma elimination half‐time of ~12 min in the rat can be calculated using the time points between 15 min and 8 h of the data of 28, with an extrapolated (and very approximate) concentration at t = 0 (‘Cp(o)’) of 910 nM (arrowed in Figure 2), corresponding to 0.27 mg l−1. The AUC1‐8h of 6525 ± 1372 ng PEA min ml−1 reported in 28 corresponds to a value of 37 ± 10 × 10−6 of given dose h−1, assuming a total blood volume of 6.25 ml/100 g, of which 55% is plasma. Our interpretation of the data in 28 is that most of the PEA is outside of the blood following oral administration (for further analysis determining approximate V d values for a given bioavailability, see Appendix S1).
The tissue distribution of PEA has also been studied: Grillo et al. 30 reported that in a small sample (n = 3–4 per group), administration of PEA (10 mg kg−1) emulsified in corn oil increased levels of this lipid in the heart and brain of DBA/2 mice 24 and/or 48 h after subcutaneous injections. Artamonov et al. 31 investigated the distribution of N‐[9,10‐3H] PEA in male 150–200 g Wistar rats 20 min after oral administration (dose ~100 mCi, 3.3 × 10−5 mol/100 g of body weight, corresponding to approximately 100 mg kg−1). They found that approximately 0.95% of the administered PEA was found in the brain, with a very heterogeneous distribution: NAE levels of 10 400, 65, 110, 7.4 and 2.2 pmol mg−1 of tissue were recovered in the hypothalamus, white matter, brain stem, cerebellum and brain cortex, respectively (means of three experiments). The corresponding values for pituitary gland and adrenal organs were 2050 and 85 pmol mg−1 of tissue, respectively. Very little of the total tritium recovered in the hypothalamus was in lipids other than NAE (e.g. free fatty acids), whereas 28 and 34% of the label was metabolized in the pituitary and cerebellum, respectively 31. The very heterogeneous distribution in the brain is surprising for a lipophilic compound, and would suggest preferential retention by the hypothalamus. One explanation for such retention would be a selective expression of a PEA binding moiety in the hypothalamus. Interestingly, PPARα can be ruled out as such a target, because its expression in the hypothalamus is low 32.
Clinical studies with PEA
The clinical studies identified by our search (see Appendix S2 for details) are summarized in Tables 1 and 2. We found 21 clinical studies, of which 16 were clinical trials enrolling a range of 20 to 636 patients and five were case/pilot studies. In the clinical trials, PEA was used for periods ranging from 14 days to 120 days, and the doses ranged from 300 mg to 1200 mg daily. The administration form of PEA was in most cases oral tablets except some occasional use of sublingual formulations (sachets), and the commonest form of evaluation was the visual analogue scale (VAS), where the patient makes a subjective assessment of her/his pain level on a 10 cm line where the left side represents no pain, and the right side represents the worst imaginable pain 33, 34. With one exception (35, possibly a ‘floor effect’), all available clinical trials reported significantly reduced pain intensity and an almost complete absence of unwanted effects, the latter confirming early field studies of PEA in healthy individuals 4.
Table 1.
Clinical trials investigating the effect of PEA in pain. Trials are listed in descending order with respect to the number of participants
| Type of study | No. of patients | Type of pain | PEA dosage | Formulation | Treatment length | Outcome (all VAS scale unless marked with ¶ or **) | Unwanted effects | Conflict of interest | Sponsor | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| Double blind randomized controlled multi‐centre, placebo | 636 (1/3 placebo) | Low back pain (lumbosciatica) | 1 or 2 × 300 mg daily | UM or M Normast®/Epitech group | 21 days | 600 mg better than 300 mg, both doses significantly better than placebo at t = 21 days | None reported | Unknown | Unknown | 41, † |
| (Observational) prospective cohort | 610 (564 completions) | Chronic pain of different etiopathogenesis | 1200 mg daily for 3 weeks followed by 600 mg daily for 4 weeks | UM or M Normast®/Epitech group | 49 days | Significant decrease in pain intensity in all patients (P = 0.0001)¶ | None reported | Declare no conflict of interest | NS | 49 |
| Non‐randomized, non‐controlled, PEA as add‐on compared to only standard treatment * | 118 (64 received PEA) | Low back pain (lumbosciatica) | 600 mg daily | Formulation unknown Manufacturer unknown | 30 days | Significant changes for both groups, a slightly larger decrease in pain intensity with PEA compared to standard treatment.* No significant change in ODI | None reported | Declare no conflict of interest | Angelini, Spain | 50 |
| Double blind, randomized, controlled, placebo | 111 (1/3 placebo) | Lumbosciatic pain | 1 or 2 × 300 mg daily | UM or M Normast®/Epitech group | 21 days | Significant reduction of pain intensity with PEA regardless of simultaneous treatment with other drugs compared to placebo at days 21 | None reported | NS | NS | 43, † |
| Observational | 80 | Fibromyalgia | Starting with 600 mg daily for 1 month following 300 mg daily for month 2–3 | UM or M PEA–m®, PEA‐um®, Epitech Group | 6 months (PEA 3 months) | Addition of PEA to the treatment regimen significantly reduced VAS pain scores further | None reported | Declare no conflict of interest | None | 51 |
| Randomized, double‐blind, 3 parallel‐group, placebo‐controlled | 61 (1/3 placebo, 1/3 celecoxib) | Chronic pelvic pain | 800 mg daily (combined with transpolydatin) | M Manufacturer unknown | 3 months | Significantly reduced pain intensity with PEA compared to placebo, although Celecoxib was more effective than PEA | None reported | NS | NS | 44 |
| Prospective cohort | 47 | Endometriotic pain | 800 mg daily (combined with transpolydatin) | M Tablets Manufacturer unknown | 90 days | Significant decrease in pain intensity | NS | NS | NS | 52 |
| Randomized, controlled | 30 (1/2 acupuncure) | Radiculopathy | 600 mg daily | UM or M Normast®/Epitech group | 120 days | Significant decrease in chronic pain intensity with PEA compared to acupuncture treatment only** | Unknown | Unknown | 6 | 53, ‡ |
| Open‐label | 30 | Diabetic or traumatic neuropathic pain | 1200 mg daily | UM Sachets, Tablets Manufacturer unknown | 40 days | Significant reduction of pain intensity. VAS, health questionnaire five dimensions for quality of life (EQ‐ED50) and NP Symptom Inventory (NPSI) used | NS | Declare no conflict of interest | Associazone Neuropatie Chroniche Piemonte ONLUS | 54 |
| Randomized, split mouth, single blinded | 30 | Postoperative pain due to lower third molar surgery | 600 mg daily | UM or M Normast®/Epitech group | 15 days | Significant decrease in postoperative pain with PEA treatment | One case of drowsiness and one case of palpitations | Unknown | Unknown | 37 |
| Open‐label | 30 | Neuropathic pain, different types | 600 mg daily (combined with pregabalin) | Unknown | 45 days | Significant reduction of pain intensity | None reported | Unknown | Unknown | 55, ‡ |
| Open‐label | 30 | Peripheral diabetic neuropathy | 600 mg daily | M Normast®/Epitech group | 60 days | Significant reduction in pain intensity [Total Symptom Score TSS]** | None reported | Declare no conflict of interest | NS | 56 |
| Controlled trial | 26 | Carpal tunnel syndrome | 600 mg or 1200 mg daily | UM or M Normast®/Epitech group | 30 days | Significant improvement of CTS induced median nerve latency time. Also improvement of subjective discomfort, and Tinel's sign** | None reported | NS | NS | 57 |
| Triple‐blind, randomized, controlled | 24 (1/2 ibuprofen) | Temporomandibular joint inflammatory pain | 900 mg daily for 7 days and then 600 mg daily for 7 days more | UM or M Normast®/Epitech group | 14 days | Significantly larger reduction in pain intensity compared to ibuprofen treatment on day 14 | None reported | NS | None | 42 |
| Open‐label | 20 | Chemotherapy‐induced neuropathy | 600 mg daily | NS | 60 days | Significant reduction in pain intensity** | Unknown | NS | NS | 58 |
| Double blind, randomized and TENS § ‐alone controlled | 20 (1/2 TENS alone) | Vestibulodynia | 800 mg daily (combined with transpolydatin and TENS) | NS | 60 days | No significant improvement with PEA treatment | Three cases of mild transient gastrointestinal symptoms | NS | NS | 35 |
Abbreviations: M, Micronized; NM, Not micronized; NS, not stated in the article; NRS, Numerical rating scale; UM, Ultramicronized; VAS, Visual analogue scale.
NSAIDs, analgesics, muscle relaxants, corticosteroids; the exact treatment differed for patients / treatment centres.
Article in Spanish.
Article in Italian.
Transcutaneous electrical nerve stimulation. The six blinded RCTs that we identified are references 41, 43, 44, 37, 42 and 35.
NRS used.
Other or unidentified evaluation method. ODI, Oswestry Disablity Index (measures quality of life in patients with low back pain).
A meta‐analysis into the clinical utility of micronized and ultra‐micronized PEA on pain intensity in patients suffering from chronic and/or neuropathic pain has recently been published 21. The authors of 21, of whom two were employees of Epitech (the makers of Normast and other PEA preparations), obtained raw data from corresponding authors of 12 studies (six published in journals, two published abstracts and four manuscripts either in preparation or submitted for publication) that met the inclusion criteria (including availability of raw data and comparable methods for assessing pain intensity). The authors concluded on the basis of their analyses that PEA was an effective treatment for pain with no registered serious adverse effects. Their analysis was based upon 12 studies that met their inclusion criteria (three placebo‐controlled double blind studies, two open‐label randomized vs. standard therapy and seven open‐label studies without a comparator) in patients with a variety of aetiologies. Several outcomes were presented, of which a key finding was the difference in the number of patients achieving ≤3 in the NRS/VAS scores (55/263 [20.9%] for the controls, 760/1138 [66.7% of the PEA treatment groups) [21]. The fact that approximately half of the included patients came from the open‐label studies (703/30 PEA/control vs. 266/485 PEA/control for the double blind studies) is perhaps a weakness of the study, although a Cox survival analysis (reduction in pain intensity to ≤3 on an NRS/VAS scale as endpoint) favoured both PEA over control and the double blind over the open‐label studies (other factors with modest, but significant effects in this analysis were gender and age (<65 vs. ≥65); pain aetiology did not contribute significantly to the analysis). Whilst the strength of the article is that it has access to raw data, this is mitigated by a lack of discussion as to the quality of the key studies. Additionally, the authors did not discuss the issue of publication bias 36, whereby studies with less satisfactory outcomes would either not have been visible in their searches or alternatively might been excluded due to unavailability of the raw data. We cannot address this issue here, but we have investigated the strengths and weaknesses of the key randomized controlled trials (RCTs), and further considered how to interpret the clearly promising data with respect to adverse effects.
Tolerability of PEA
As noted by other authors 20, 21, PEA appears to be well tolerated indeed. The only adverse event (not necessarily drug‐related) that has been reported was for a patient treated with 300 mg Normast™ following impacted third molar extraction 37. The patient, who was not taking any other drugs, reported palpitations lasting 2–3 h on the third day of Normast™ treatment. This occurred 1 h after Normast™ consumption, and the patient did not continue with the trial after this event. This low rate of adverse events is remarkable indeed: after all, patients treated with placebo in double blind studies report adverse events. For example, in a recent multicentre, randomized double‐blind study in patients with uncontrolled moderate to severe back pain, 35% of the placebo‐treated patients reported treatment‐emergent adverse events (primarily nausea, constipation, vomiting, dizziness, headache and somnolence) 38. As we do not have access to the study protocols, we cannot say whether the lack of adverse events found with PEA in the studies reflects a true low rate, or whether mild/moderate adverse events were not documented or reported.
The likelihood of observing an adverse drug reaction (ADR) is dependent upon the number of patients observed, the frequency threshold of the ADR, and whether it occurs early on or after prolonged treatment. Frequencies of ADRs are divided into ‘very common’ (≥1/10), ‘common’ (≥1/100 and <1/10), ‘uncommon’ (≥1/1000 and <1/100), ‘rare’ (≥1/10 000 and <1/1000), and ‘very rare’ (<1/10 000). As a general rule of thumb, the 95% likelihood of observing an ADR at a frequency threshold of 1/n in a study requires 3n patients 39. In other words, at least 300 patients would be needed for a 95% likelihood of observing a single ADR at a frequency of occurrence of 1/100 39. For two and three ADRs to be observed at this frequency, the number increases to 480 and 650, respectively 39. If we consider only the data in Table 1, and disregard for simplicity differences in dosaging, then a total of 1590 patients were treated with PEA. However, the number of patients drops off rapidly with increasing treatment time (shown visually in Figure 3; note that the y‐axis on the graph is logarithmic, not linear). For treatment times ≤49 days, the rule of thumb described above suggests that ADRs occurring this early on in treatment would be likely to have been seen for an incidence of 1/200 or greater. But remember, these numbers refer to a 95% likelihood of observing a single ADR 39, and of recognizing it as such. Nonetheless, the current clinical data argue against ‘very common’ or ‘common’ serious ADRs being found with PEA following these treatment times, whereas there is insufficient data to give information in the ‘uncommon’ or ‘rare’ categories.
Figure 3.
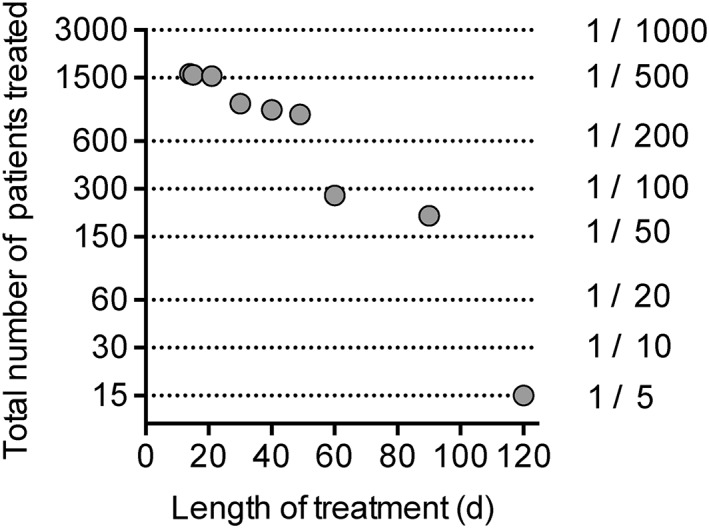
Number of patients treated with PEA in the studies summarized in Table 1 as a function of the length of treatment. The dotted lines represent the number of patients needed for a 95% likelihood of observing a single ADR at the frequency of occurrence shown 39
Treatment of chronic pain is not likely to be short term, and for ≥60 days of treatment, the number of patients is insufficient to rule out a frequency of ADRs of less than 1/100. That does not, of course, mean that such ADRs will occur, merely that there is insufficient data to judge whether or not they do occur.
Efficacy of PEA
The studies are summarized in Tables 1 and 2. The total number of participants is high in two trials (n ≈ 600) whilst the others are more modest in size, ranging from 20 to 118 participants in all. Some of the trials compare PEA to placebo, others investigate PEA as an add‐on to standard treatments. Many of the PEA clinical trials have limitations in terms of design: case reports (Table 2) have little value in terms of external validity, and open labelled trials (Table 1) do not take into account placebo effects, which are a major issue in pain studies 40. The strongest indicator of efficacy is the RCT and we identified six blinded RCTs.
The efficacy of PEA in the six blinded RCTs is summarized in more detail, together with our assessment of their strengths and weaknesses, in Table 3. The largest of the studies, investigating the effects of PEA on lumbosciatica 41 was included in the meta‐analysis of 21. The differences between days 0 and 21 for the VAS scores can be used to calculate a treatment effect size, assuming that the VAS scores are normally distributed (this was not stated explicitly in the article), and leaving aside the issue that VAS is an ordinal measure. From their data and using an online calculator (http://www.psychometrica.de/effect_size.html; last accessed 14 June 2016), we estimate Cohen's d values of 0.43 (95% CI 0.23–0.62) and 1.35 (95% CI 1.14–1.56) for 300 and 2 × 300 mg PEA, respectively. The latter value is a large effect size.
Table 3.
Efficacy and strengths/weaknesses of the six blinded RCT investigating the effects of PEA in pain
| Efficacy | Strengths (+) and Weaknesses (−) |
|---|---|
|
Guida et al.
41
lumbosciatic algias with 300 or 600 mg Normast
Significant reduction of VAS scores on day 21 from 6.6 ± 1.7 (means ± SD) to 4.6 ± 1.7 for placebo; 6.5 ± 1.9 → 3.6 ± 1.8 for 300 mg PEA and 7.1 ± 1.8 → 2.1 ± 1.7 for 600 mg PEA. Similar result found for Roland‐Morris disability questionnaire (measures back pain) Very few dropouts in the PEA groups |
+ Well‐powered study (636 patients) |
| + Clear inclusion and exclusion criteria | |
| + Intention to treat analysis; repeat measurements of VAS scores and drug safety | |
| − Efficacy data only reported for end of study data (Day 21). Authors did not report their findings on Days 7 and 14 of treatment. | |
| Canteri et al. 43 follow‐up study to 41 , significant reduction on day 21 for PEA groups compared to placebo | Smaller, confirmatory study (35–38 patients per group) − Efficacy data reported graphically (without error bars) for end of study data alone (day 21) |
|
Bacci et al.
37
Patients undertaking bilateral lower third molar extractions; randomized, split‐mouth, single‐blind study. Placebo or Normast 600 mg from 6 days before surgery to 9 days after surgery
VAS scores on day 3 were 3.8 ± 3.1 cm vs 5.5 ± 2.4 cm; day 7: 1.0 ± 1.8 vs. 1.5 ± 2.2 cm (Normast vs. placebo) |
Small study (30 patients, 26 completed protocol) − hard to assess its clinical relevance compared with, say, the standard NSAID treatment, where studies of pain relief have usually focussed on h after dosing 64 as opposed to days, as here |
|
Cobellis et al.
44
women with pelvic pain, PEA (2 × 400 mg) + transpolydatin (40 × 2 mg/day) vs. placebo and vs. celecoxib (200 × 2 mg/kg/day × 7 days)
reported VAS scores for dysmenorrhea, deep dyspareunia and non‐menstrual chronic pelvic pain. Found efficacy of PEA + transpolydatin at 3 months (example for pelvic pain score): placebo reduced from ~7.3 → ~4.8 cm; PEA + transpolydatin reduced from ~7.5 → ~2.2 cm (celecoxib reduced from ~7.8 → ~1.4 cm). |
+ Used non‐parametric statistics for VAS scores since they were not normally distributed. |
| + Long‐term treatment | |
| + Comparator drug (celecoxib) | |
| − Small study (20–21 patients/group) | |
| − Data presented graphically rather than in a table | |
|
Marini et al.
42
patients with temporomandibular joint inflammatory pain, Normast (300 + 600 mg per day) vs. ibuprofen (600 mg ×3 per day) for two weeks.
Similar reduction in VAS days 1–8. Thereafter ibuprofen plateaued out (at ~3.7 cm) whereas PEA group continued down to 0.8 cm on day 14. |
− Small study (24 patients) The patient population appears to be remarkably homogeneous in terms of their pain scores, with, for example, baseline and final VAS (in mm) of 70 ± 0.22 and 7.7 ± 0.19 (means ± SE) for the PEA group. The variation of the VAS is usually an order of magnitude higher. |
|
Murina et al.
35
60 days of treatment with PEA (2 × 400 mg per day) + polydatin (2 × 40 mg per day) vs. placebo in patients with vestibulodynia. All patients received transcutaneous electrical nerve stimulation (TENS).
Large response in placebo (TENS) group (from 6.2 ± 1.1 → 2.3 ± 1.5 cm). Greater effect of TENS in patients with recent onset of the disorder who were given PEA + polydatin |
+ Long treatment time |
| − Small study (20 patients) | |
| − Large response in placebo (TENS) group (from 6.2 ± 1.1 → 2.3 ± 1.5 cm) per sig reduce chance of seeing additional PEA effect | |
| − Unclear whether the significant finding was part of the original study design or a post‐hoc subgroup analysis |
None of the RCTs discussed above were flagged in our ClinicalTrials.gov search, so issues such as primary outcome changes and/or unmotivated subgroup analysis, issues which mar many RCTs 65, 66 have not been examined. However, it is reasonable to assume that reductions in VAS scores are a primary outcome.
In terms of the strengths/weaknesses of the studies, there are several issues that emerge, the small size of most of the other studies being the most obvious. Key issues are the nonreporting of time points other than the final time point 41, lack of (or surprisingly small values 42), information as to the variation in VAS scores among the patients; data presented graphically rather than in tables 43, 44; floor effects in the comparator group and possible post‐hoc subgroup analyses 35; and evaluation time points that are difficult to compare with current treatments 37. Two of the studies had NSAID comparator groups; in one, the patients fared better with celecoxib than with PEA + transpolydatin 44, whilst in the other, the patients fared equally well with PEA and ibuprofen over the first eight days, after which the effect of ibuprofen plateaued out, whilst those patients treated with PEA continued to improve 42. All in all, the data point to efficacy of PEA over placebo (assuming no publication bias), but more information is needed to be able to gauge this efficacy vs. current treatment regimes.
Formulation of PEA
PEA is a poorly water‐soluble substance and as such the dissolution rate is often the rate‐limiting step for oral absorption and bioavailability. Dissolution rate is influenced by, among other factors, particle size and therefore drug substances are usually micronized in order to achieve a more rapid dissolution.
In the clinical trials discussed here, ultramicronized or micronized PEA was used except in three studies where the quality of PEA was unknown or not stated (Tables 1–3). Focus has been placed on the importance of micronization of PEA, in particular the advantages (or lack thereof) of micronized PEA over unmicronized PEA (see 45 for a flavour of this particular debate; note the conflict of interest statement at the end of that article). In brief, the process of micronization results in smaller particles and hence a larger total surface area. This allows the gastrointestinal milieu more access to free surfaces on the drug particle and hence a faster dissolution can be achieved. This may lead to a better adsorption of the drug molecules 46. There is a report in rodents that orally administered micronized and ultramicronized PEA are more efficacious than unmicronized PEA in the carrageenan model of inflammatory pain 47. However, in that study the formulations of PEA were dissolved in carboxymethylcellulose prior to oral or intraperitoneal administration, i.e. already in solution, which would be expected to bypass the contribution of the micronization. Head‐to‐head comparisons of the different formulations of PEA in humans are lacking, and thus there is no clinical data yet to support the use of one formulation over another, which is an unsatisfactory state of affairs.
Conclusions
As pointed out in the introduction, PEA has been the subject of a number of reviews in recent years (e.g. 20, 22, 23), usually with a focus on the biochemistry of the endogenous compound, its variation in physiological and pathological conditions, and the preclinical pharmacology of exogenously administered PEA. Pharmacokinetic data has largely been neglected, and the clinical data has been listed and described, rather than subjected to close scrutiny. We have attempted to rectify this in the present article.
Our analysis of the pharmacokinetic properties of PEA suggests that the compound has a high volume of distribution. Perhaps the most intriguing finding was the concentration of label in the hypothalamus after oral dosing of PEA tritiated in the acyl side chain 31. It would clearly be of interest to confirm this finding and to identify potential novel PEA targets that are preferentially expressed in the hypothalamus.
With respect to the safety of PEA, our analysis suggests that too few patients have been treated for more than 60 days to argue that the compound lacks ADRs when given long term. This may well turn out to be the case, but further data is needed to allow a reasonable risk assessment.
The clinical studies investigated in detail in the present review are of variable quality. In all cases, the authors have focused on the change in VAS scores, rather than the proportion of subjects experiencing a reduction in pain to under a clinically meaningful cut‐off point, although this issue was addressed in survival analyses undertaken in the meta‐analysis 21. Further, comparative studies with current treatments are rare, although in the case of endometriosis, PEA did not perform as well as celecoxib 44.
The clinical data are clearly promising, but more clinical trials are necessary, ideally with publicly available study protocols. Study size, treatment lengths and choice of scales for primary outcome measures are all important considerations 48, as well as head‐to‐head comparisons of unmicronized vs. micronized formulations of PEA (in order to determine whether or not one formulation is clinically superior to the other), and comparisons vs. standard treatments. Given the promising data so far accrued with this compound, it is to be hoped that these data will be forthcoming.
After this article was accepted, Andresen et al. 67 have reported a well‐conducted double‐blind multicentre study comparing ultramicronised PEA (2 x 600 mg) and placebo as add‐on treatments in 73 patients with neuropathic pain following spinal cord injury. Over the 12 week period, no superiority over placebo was seen.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
The corresponding author thanks the Swedish Science Research council (Grant no. 12158) and the Research Funds of the Medical Faculty, Umeå University for research support.
Supporting information
Appendix S1 Further pharmacokinetic analysis of published data on PEA
Appendix S2 Search methodology used in the present review
Supporting info item
Supporting info item
Gabrielsson, L. , Mattsson, S. , and Fowler, C. J. (2016) Palmitoylethanolamide for the treatment of pain: pharmacokinetics, safety and efficacy. Br J Clin Pharmacol, 82: 932–942. doi: 10.1111/bcp.13020.
References
- 1. Coburn A, Graham C, Haninger J. The effect of egg yolk in diets on anaphylactic arthritis (passive arthus phenomenon) in the guinea pig. J Exp Med 1954; 100: 425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuehl F, Jacob T, Ganley O, Ormond R, Meisinger M. The identification of N‐(2‐hydroxyethyl)‐palmitamide as a naturally occurring anti‐inflammatory agent. J Am Chem Soc 1957; 79: 5577–8. [Google Scholar]
- 3. Hansen HS. Effect of diet on tissue levels of palmitoylethanolamide. CNS Neurol Disord Drug Targets 2013; 12: 17–25. [DOI] [PubMed] [Google Scholar]
- 4. Kahlich R, Klíma J, Cihla F, Franková V, Masek K, Rosicky M, et al. Studies on prophylactic efficacy of N‐2‐hydroxyethyl palmitamide (Impulsin) in acute respiratory infections. Serologically controlled field trials. J Hyg Epidemiol Microbiol Immunol 1979; 23: 11–24. [PubMed] [Google Scholar]
- 5. Aloe L, Leon A, Levi‐Montalcini R. A proposed autacoid mechanism controlling mastocyte behaviour. Agents Actions 1993; 39: C145–7. [DOI] [PubMed] [Google Scholar]
- 6. Mazzari S, Canella R, Petrelli L, Marcolongo G, Leon A. N‐(2‐hydroxyethyl)hexadecamide is orally active in reducing edema formation and inflammatory hyperalgesia by down‐modulating mast cell activation. Eur J Pharmacol 1996; 300: 227–36. [DOI] [PubMed] [Google Scholar]
- 7. Jaggar S, Sellaturay S, Rice A. The endogenous cannabinoid anandamide, but not the CB2 ligand palmitoylethanolamide, prevents the viscero‐visceral hyperreflexia associated with inflammation of the rat urinary bladder. Neurosci Letts 1998; 253: 123–6. [DOI] [PubMed] [Google Scholar]
- 8. Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature 1998; 394: 277–81. [DOI] [PubMed] [Google Scholar]
- 9. Calignano A, La Rana G, Piomelli D. Antinociceptive activity of the endogenous fatty acid amide, palmitylethanolamide. Eur J Pharmacol 2001; 419: 191–8. [DOI] [PubMed] [Google Scholar]
- 10. Capasso R, Izzo A, Fezza F, Pinto A, Capasso F, Mascolo N, et al. Inhibitory effect of palmitoylethanolamide on gastrointestinal motility in mice. Br J Pharmacol 2001; 134: 945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scarampella F, Abramo F, Noli C. Clinical and histological evaluation of an analogue of palmitoylethanolamide, PLR 120 (comicronized Palmidrol INN) in cats with eosinophilic granuloma and eosinophilic plaque: a pilot study. Vet Dermatol 2001; 12: 29–39. [DOI] [PubMed] [Google Scholar]
- 12. Farquhar‐Smith W, Jaggar S, Rice A. Attenuation of nerve growth factor‐induced visceral hyperalgesia via cannabinoid CB1 and CB2‐like receptors. Pain 2002; 97: 11–21. [DOI] [PubMed] [Google Scholar]
- 13. Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, et al. The nuclear receptor peroxisome proliferator‐activated receptor‐α mediates the anti‐inflammatory actions of palmitoylethanolamide. Mol Pharmacol 2005; 67: 15–9. [DOI] [PubMed] [Google Scholar]
- 14. Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G. The endogenous fatty acid amide, palmitoylethanolamide, has anti‐allodynic and anti‐hyperalgesic effects in a murine model of neuropathic pain: involvement of CB1, TRPV1 and PPARγ receptors and neurotrophic factors. Pain 2008; 139: 541–50. [DOI] [PubMed] [Google Scholar]
- 15. D'Agostino G, La Rana G, Russo R, Sasso O, Iacono A, Esposito E, et al. Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF‐κB nuclear signalling in dorsal root ganglia. Eur J Pharmacol 2009; 613: 54–9. [DOI] [PubMed] [Google Scholar]
- 16. De Filippis D, Luongo L, Cipriano M, Palazzo E, Cinelli MP, de Novellis V, et al. Palmitoylethanolamide reduces granuloma‐induced hyperalgesia by modulation of mast cell activation in rats. Mol Pain 2011; 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahmad A, Crupi R, Impellizzeri D, Campolo M, Marino A, Esposito E, et al. Administration of palmitoylethanolamide (PEA) protects the neurovascular unit and reduces secondary injury after traumatic brain injury in mice. Brain Behav Immun 2012; 26: 1310–21. [DOI] [PubMed] [Google Scholar]
- 18. Romero TRL, Duarte IDG. N‐palmitoyl‐ethanolamine (PEA) induces peripheral antinociceptive effect by ATP‐sensitive K+‐channel activation. J Pharmacol Sci 2012; 118: 156–60. [DOI] [PubMed] [Google Scholar]
- 19. Di Paola R, Impellizzeri D, Mondello P, Velardi E, Aloisi C, Cappellani A, et al. Palmitoylethanolamide reduces early renal dysfunction and injury caused by experimental ischemia and reperfusion in mice. Shock 2012; 38: 356–66. [DOI] [PubMed] [Google Scholar]
- 20. Keppel Hesselink JM. New targets in pain, non‐neuronal cells, and the role of palmitoylethanolamide. Open Pain J 2012; 5: 12–23. [Google Scholar]
- 21. Paladini A, Fusco M, Cenacci T, Schievano C, Piroli A, Varrassi G. Palmitoylethanolamide, a special food for medical purposes, in the treatment of chronic pain: a pooled data meta‐analysis. Pain Physician 2016; 19: 11–24. [PubMed] [Google Scholar]
- 22. Alhouayek M, Muccioli GG. Harnessing the anti‐inflammatory potential of palmitoylethanolamide. Drug Discov Today 2014; 19: 1632–9. [DOI] [PubMed] [Google Scholar]
- 23. Mattace Raso G, Russo R, Calignano A, Meli R. Palmitoylethanolamide in CNS health and disease. Pharmacol Res 2014; 86: 32–41. [DOI] [PubMed] [Google Scholar]
- 24. Lowin T, Apitz M, Anders S, Straub RH. Anti‐inflammatory effects of N‐acylethanolamines in rheumatoid arthritis synovial cells are mediated by TRPV1 and TRPA1 in a COX‐2 dependent manner. Arthritis Res Ther 2015; 17: 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Overton H, Babbs A, Doel S, Fyfe M, Gardner L, Griffin G, et al. Deorphanization of a G protein‐coupled receptor for oleoylethanolamide and its use in the discovery of small‐molecule hypophagic agents. Cell Metab 2006; 3: 167–75. [DOI] [PubMed] [Google Scholar]
- 26. Lan H, Vassileva G, Corona A, Liu L, Baker H, Golovko A, et al. GPR119 is required for physiological regulation of glucagon‐like peptide‐1 secretion but not for metabolic homeostasis. J Endocrinol 2009; 201: 219–30. [DOI] [PubMed] [Google Scholar]
- 27. Dalle Carbonare M, Del Giudice E, Stecca A, Colavito D, Fabris M, D'Arrigo A, et al. A saturated N‐acylethanolamine other than N‐palmitoyl ethanolamine with anti‐inflammatory properties: a neglected story. J Neuroendocrinol 2008; 20 (Suppl 1): 26–34. [DOI] [PubMed] [Google Scholar]
- 28. Vacondio F, Bassi M, Silva C, Castelli R, Carmi C, Scalvini L, et al. Amino acid derivatives as palmitoylethanolamide prodrugs: synthesis, in vitro metabolism and in vivo plasma profile in rats. PLoS One 2015; 10: e0128699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petrosino S, Schiano Moriello A, Cerrato S, Fusco M, Puigdemont A, De Petrocellis L, et al. The anti‐inflammatory mediator palmitoylethanolamide enhances the levels of 2‐arachidonoyl‐glycerol and potentiates its actions at TRPV1 cation channels. Br J Pharmacol 2016; 173: 1154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grillo SL, Keereetaweep J, Grillo MA, Chapman KD, Koulen P. N‐Palmitoylethanolamine depot injection increased its tissue levels and those of other acylethanolamide lipids. Drug Des Devel Ther 2013; 7: 747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Artamonov M, Zhukov O, Shuba I, Storozhuk L, Khmel T, Klimashevsky V, et al. Incorporation of labelled N‐acylethanolamine (NAE) into rat brain regions in vivo and adaptive properties of saturated NAE under x‐ray irradiation. Ukr Biokhim Zh 2005; 77: 51–62. [PubMed] [Google Scholar]
- 32. Moreno S, Farioli‐Vecchioli S, Cerù MP. Immunolocalization of peroxisome proliferator‐activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 2004; 123: 131–45. [DOI] [PubMed] [Google Scholar]
- 33. Stevens SS. On the theory of scales of measurement. Science 1946; 2684: 677–80. [DOI] [PubMed] [Google Scholar]
- 34. Joyce CR, Zutshi DW, Hrubes V, Mason RM. Comparison of fixed interval and visual analogue scales for rating chronic pain. Eur J Clin Pharmacol 1975; 6: 415–20. [DOI] [PubMed] [Google Scholar]
- 35. Murina F, Graziottin A, Felice R, Radici G, Tognocchi C. Vestibulodynia: synergy between palmitoylethanolamide + transpolydatin and transcutaneous electrical nerve stimulation. J Low Genit Tract Dis 2013; 17: 111–6. [DOI] [PubMed] [Google Scholar]
- 36. Song F, Hooper L, Loke YK. Publication bias: what is it? How do we measure it? How do we avoid it? Open Access J Clin Trials 2013; 5: 71–81. [Google Scholar]
- 37. Bacci C, Cassetta G, Emanuele B, Berengo M. Randomized split‐mouth study on postoperative effects of palmitoylethanolamide for impacted lower third molar surgery. ISRN Surg 2011; 2011: 917350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wen W, Sitar S, Lynch SY, He E, Ripa SR. A multicenter, randomized, double‐blind, placebo‐controlled trial to assess the efficacy and safety of single‐entity, once‐daily hydrocodone tablets in patients with uncontrolled moderate to severe chronic low back pain. Expert Opin Pharmacother 2015; 16: 1593–606. [DOI] [PubMed] [Google Scholar]
- 39. Lewis JA. Post‐marketing surveillance: how many patients? Trends Pharmacol Sci 1981; 2: 93–4. [Google Scholar]
- 40. Finniss DG, Kaptchuk TJ, Miller F, Benedetti F. Biological, clinical, and ethical advances of placebo effects. Lancet 2010; 375: 686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guida G, De Martino M, De Fabiani A, Cantieri LA, Alexandre A, Vassallo GM, et al. La palmitoilethanolamida (Normast®) en el dolor neuropático crónico por lumbociatalgia de tipo compresivo: estudio clínico multicéntrico. Dolor 2010; 25: 35–42. [Google Scholar]
- 42. Marini I, Bartolucci ML, Bortolotti F, Gatto MR, Bonetti GA. Palmitoylethanolamide versus a nonsteroidal anti‐inflammatory drug in the treatment of temporomandibular joint inflammatory pain. J Orofac Pain 2012; 26: 99–104. [PubMed] [Google Scholar]
- 43. Canteri L, Petrosino S, Guida G. Riducción del consumo di antiinfiammatorios y analgésicos en el tratamiento del dolor neuropático crónico en pacientes afectados por lombosciatalgia de tipo compresivo y en tratamiento con Normast® 300 mg. Dolor 2010; 25: 227–34. [Google Scholar]
- 44. Cobellis L, Castaldi MA, Giordano V, Trabucco E, De Franciscis P, Torella M, et al. Effectiveness of the association micronized N‐Palmitoylethanolamine (PEA)‐transpolydatin in the treatment of chronic pelvic pain related to endometriosis after laparoscopic assessment: a pilot study. Eur J Obstet Gynecol Reprod Biol 2011; 158: 82–6. [DOI] [PubMed] [Google Scholar]
- 45. Kriek R. Marketing messages in pharmacological papers and scientific chapters: The case of palmitoylethanolamide and its formulations. Pharmacol Res 2014; 85: 1–3. [DOI] [PubMed] [Google Scholar]
- 46. Aulton ME. Aulton's Pharmaceutics. The Design and Manufacture of Medicines, 32nd edn. London: Churchill Livingstone, 2006. [Google Scholar]
- 47. Impellizzeri D, Bruschetta G, Cordaro M, Crupi R, Siracusa R, Esposito E, et al. Micronized/ultramicronized palmitoylethanolamide displays superior oral efficacy compared to nonmicronized palmitoylethanolamide in a rat model of inflammatory pain. J Neuroinflammation 2014; 11: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain 2005; 113: 9–19. [DOI] [PubMed] [Google Scholar]
- 49. Gatti A, Lazzari M, Gianfelice V, Di Paolo A, Sabato E, Sabato AF. Palmitoylethanolamide in the treatment of chronic pain caused by different etiopathogenesis. Pain Med 2012; 9: 1121–30. [DOI] [PubMed] [Google Scholar]
- 50. Dominguez CM, Martin AD, Ferrer FG, Puertas MI, Muro AL, Gonzalez JM, et al. N‐Palmitoylethanolamide in the treatment of neuropathic pain associated with lumbosciatica. Pain Manag 2012; 2: 119–24. [DOI] [PubMed] [Google Scholar]
- 51. Del Giorno R, Skaper S, Paladini A, Varrassi G, Coaccioli S. Palmitoylethanolamide in fibromyalgia: results from prospective and retrospective observational studies. Pain Ther 2015; 4: 169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Giugliano E, Cagnazzo E, Soave I, Lo Monte G, Wenger JM, Marci R. The adjuvant use of N‐palmitoylethanolamine and transpolydatin in the treatment of endometriotic pain. Eur J Obstet Gynecol Reprod Biol 2013; 168: 209–13. [DOI] [PubMed] [Google Scholar]
- 53. Crestani F, Burato A, Michielan F. La palmitoilethanolamide micronizzata aumenta l'analgesia da agopuntura nel dolore da radicolopatia: studio pilota. G Ital Med Riabil MR 2013; 1: 49–54. [Google Scholar]
- 54. Cocito D, Peci E, Ciaramitaro P, Merola A, Lopiano L. Short‐term efficacy of ultramicronized palmitoylethanolamide in peripheral neuropathic pain. Pain Res Treat 2014; 2014: 854560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Desio P. Associazione tra pregabalin e palmitoiletanolamide per il trattamento del dolore neuropatico. Pathos 2010; 4: 9–14. [Google Scholar]
- 56. Schifilliti C, Cucinotta L, Fedele V, Ingegnosi C, Luca S, Leotta C. Micronized palmitoylethanolamide reduces the symptoms of neuropathic pain in diabetic patients. Pain Res Treat 2014; 2014: 849623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Conigliaro R, Drago V, Foster PS, Schievano C, Di Marzo V. Use of palmitoylethanolamide in the entrapment neuropathy of the median in the wrist. Minerva Med 2011; 102: 141–7. [PubMed] [Google Scholar]
- 58. Truini A, Biasiotta A, Di Stefano G, La Cesa S, Leone C, Cartoni C, et al. Palmitoylethanolamide restores myelinated‐fibre function in patients with chemotherapy‐induced painful neuropathy. CNS Neurol Disord Drug Targets 2011; 10: 916–20. [DOI] [PubMed] [Google Scholar]
- 59. Indraccolo U, Barbieri F. Effect of palmitoylethanolamide‐polydatin combination on chronic pelvic pain associated with endometriosis: preliminary observations. Eur J Obstet Gynecol Reprod Biol 2010; 1: 76–9. [DOI] [PubMed] [Google Scholar]
- 60. Keppel Hesselink JM. Chronic idiopathic axonal neuropathy and pain, treated with the endogenous lipid mediator palmitoylethanolamide: a case collection. Int Med Case Rep J 2013; 6: 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Keppel Hesselink JM, Kopsky DJ. Treatment of chronic regional pain syndrome type 1 with palmitoylethanolamide and topical ketamine cream: modulation of nonneuronal cells. J Pain Res 2013; 6: 239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kopsky DJ, Keppel Hesselink JM. Multimodal stepped care approach with acupuncture and PPAR‐alpha agonist palmitoylethanolamide in the treatment of a patient with multiple sclerosis and central neuropathic pain. Acupunct Med 2012; 1: 53–5. [DOI] [PubMed] [Google Scholar]
- 63. Calabrò RS, Gervasi G, Marino S, Mondo PN, Bramanti P. Misdiagnosed chronic pelvic pain: pudendal neuralgia responding to a novel use of palmitoylethanolamide. Pain Med 2010; 11: 781–4. [DOI] [PubMed] [Google Scholar]
- 64. Moore PA, Hersh EV. Combining ibuprofen and acetaminophen for acute pain management after third‐molar extractions. Translating clinical research to dental practice. J Am Dent Assoc 2013; 144: 898–908. [DOI] [PubMed] [Google Scholar]
- 65. Ramagopalan S, Skingsley AP, Handunnetthi L, Klingel M, Magnus D, Pakpoor J, et al. Prevalence of primary outcome changes in clinical trials registered on ClinicalTrials.gov: a cross‐sectional study. F1000Res 2014; 3: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rothwell PM. Treating individuals 2. Subgroup analysis in randomised controlled trials: importance, indications, and interpretation. Lancet 2005; 365: 176–86. [DOI] [PubMed] [Google Scholar]
- 67. Andresen SR, Bing J, Hansen RM, Biering‐Sørensen F, Johannesen IL, Hagen EM, et al. Ultramicronized palmitoylethanolamide in spinal cord injury neuropathic pain: A randomized, double‐blind, placebo‐controlled trial. Pain 2016; doi: 10.1097/j.pain.0000000000000623 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Further pharmacokinetic analysis of published data on PEA
Appendix S2 Search methodology used in the present review
Supporting info item
Supporting info item