

Abstract
Alzheimer′s disease has recently emerged as a possible field of application for PDE4D inhibitors (PDE4DIs). The great structure similarity among the various PDE4 isoforms and, furthermore, the lack of the full length crystal structure of the enzyme, impaired the rational design of new selective PDE4DIs. In this paper, with the aim of exploring new insights into the PDE4D binding, we tackled the problem by performing a computational study based on docking simulations combined with molecular dynamics (D‐MD). Our work uniquely identified the binding mode and the key residues involved in the interaction with a number of in‐house catechol iminoether derivatives, acting as PDE4DIs. Moreover, the new binding mode was tested using a series of analogues previously reported by us and it was used to confirm their key structural features to allow PDE4D inhibition. The binding model disclosed within the current computational study may prove to be useful to further advance the design and synthesis of novel, more potent and selective, PDE4D inhibitors.
Keywords: Phosphodiesterases, PDE4, PDE4D selective inhibitors, docking, molecular dynamics
1. Introduction
Cyclic nucleotide phosphodiesterases (PDEs) are a large family of enzymes able to catalyze the hydrolysis of second messengers cAMP and cGMP into the corresponding 5′‐nucleotide monophosphates. To date, eleven PDE sub‐families (further divided in more than 60 isoforms and splice variants) have been classified on the basis of their variable tissue localization, kinetic profile, structure similarity, regulation mechanism and different pharmacological effects explained by their inhibitors.1,2
In particular, the PDE4 family acts selectively on cAMP and displays a complex localization. Indeed, they are mainly found in inflammatory cells, brain, smooth muscle and cardiovascular tissue both at cytosolic and plasmatic membrane level.3,4
Within the PDE4 subfamily, four members have been identified (namely A, B, C and D), further classified in 25 subtypes. All of them can also be distinguished in “long”, “short” and “super‐short” forms, on the basis of the so‐called up‐stream conserved regions (UCRs).3, 4, 5, 6, 7 In particular, the long isoforms are characterized by two regulatory domains (UCR1 and UCR2), inserted between the N‐terminal portion and the catalytic domain; in the short forms UCR1 domain is missing, whereas the super‐short forms lack UCR1 and a UCR2 portion.
Two pivotal actions have been evidenced for UCR1 and UCR2 domains. Firstly, they promote the long forms dimerization in vivo, involving particularly the UCR1/catalytic domain interfaces.8, 9, 10, 11, 12 On the other hand, UCR2, closing across the active site, blocks the access to cAMP, thus exerting a partial enzyme inhibition.12,13
Concerning the active site of PDE4, it shows a 78 % of similarity among the four isoforms and it is formed by three main pockets: a metal pocket (M pocket) in which the two catalytic ions (Mg2+ and Zn2+) were placed, surrounded by a shell of coordinating‐waters, a solvent filled side pocket (S pocket) and an hydrophobic pocket. In that region (Q pocket) the natural substrate engages a bi‐dentate H‐bond with an invariant glutamine residue (Q369 in PDE4D) whose carbonyl oxygen makes an additional H‐bond with a conserved tyrosine residue (Y329 in PDE4D).14,15
PDE4 inhibition has been extensively studied, on account of the associated potential therapeutic application. Indeed, PDE4 inhibitors (PDE4Is) may be useful in the treatment of inflammatory diseases, in case of both respiratory disorders (COPD, asthma) and autoimmune pathologies (rheumatoid arthritis, multiple sclerosis, atopic dermatitis).[15–18] In addition, PDE4Is displayed positive effects on CNS disorders.19,20 In particular, several studies indicated the PDE4D isoforms as an interesting target for neurodegenerative pathologies such as Alzheimer′s Disease (AD).21
Up to now, the most exploited strategy to target PDE4 was to design competitive inhibitors of cAMP. Those compounds were active‐site‐directed and the key‐interactions with the enzyme always included an H‐bond with Q369 and, usually, the coordination to the catalytic metal ions in the active site.22 Two of them, Roflumilast and Apremilast have been recently approved as drug for COPD and psoriatic arthritis treatment, respectively.23,24
However, many studies have been recently focused on the role of UCR1‐UCR2 oligomers and also of the C terminal helix conserved region 3 (CR3) as negative regulatory domains. Gurney and co‐workers reported that proper modulators may promote the closure of both UCR2 and CR3 across the active site, explaining in this way the mechanism of inhibition for the PDE4.25 A new pattern of interactions has been described for those allosteric modulators: while still engaging the H‐bond with Q369, they push a moiety out of the active site to interact with either UCR2 or CR3 helix rather than showing interactions with the two metal ions of the catalytic pocket. Notably, both UCR2 and CR3 display differences in sequence between PDE4B and PDE4D isoforms. This fact can be exploited for the design of isoform‐selective inhibitors.25,26
In the last ten years, our research on PDE4Is led to numerous Rolipram‐related small molecules endowed with a selective inhibition of PDE4D isoform.27, 28, 29 Compound GEBR‐7b (1, Table 1), selected among them for behavioral studies, improved spatial memory both in wild type mice or rats and in the APPswe/PS1dE9 mouse model of AD.30,31 In addition, in the xylazine/ketamine test and taste reactivity test (two well‐known assays used as surrogate measures of emesis in non‐vomiting species) 1 did not induce emesis at doses up to 33–100 times higher than the pro‐cognitive dose.30
Moreover there is clear evidence that selective PDE4D inhibitors could improve memory without undesirable side effects.[32] Thus, due to these promising results, we were interested in the development of our PDE4DIs compound library. Preliminary computational studies performed by us on this PDE4DIs series did not seem to give an accurate picture of the ligand‐enzyme interaction, not affording a reliable model to guide the further synthesis.28,29
On this basis, with the aim to more precisely analyze this interaction and clarify the activity of our compound library, a computational protocol consisting of docking and molecular dynamics (MD) simulations has been applied. In details, a re‐docking and further dockings with a collected database of PDE4DIs, including some 3‐cyclopentyloxy‐4‐methoxyphenyl derivatives (compounds 1–5, Table 1),28 a number of pyridofuropyrimidines (compounds 6–18, Table 2),33 and quinolines (compounds 19–35, Table 3),34 have been performed to set and validate the docking protocol. Subsequently, two binding poses selected within this first phase were further investigated by means of MD. Finally, additional docking studies, performed on an external dataset of our structurally related inhibitors[29] highlighted the potentiality of the selected binding mode in supporting SAR considerations and disclosed the key elements for the further design of new inhibitors.
Table 1.
Molecular structure of compounds 1–5
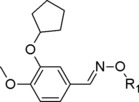
| Compound | R1 | PDE4D IC50 (μM) |
|---|---|---|
| 1 |
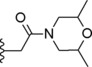
|
0.67 |
| 2 |
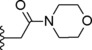
|
3.48 |
| 3 |
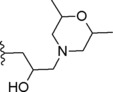
|
3.46 |
| 4 |
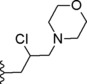
|
5.91 |
| 5 |

|
3.42 |
Table 2.
Molecular structure of compounds 6–18
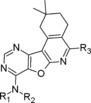
| Compound | R1 | R2 | R3 | PDE4D IC50 (nM) |
|---|---|---|---|---|
| 6 | −H |
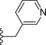
|
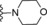
|
13 |
| 7 | −H |
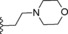
|
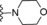
|
9.4 |
| 8 |
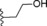
|
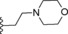
|
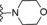
|
2.3 |
| 9 | −H |
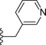
|
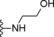
|
13.5 |
| 10 | −H |
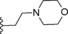
|
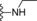
|
2.7 |
| 11 | −H |
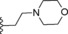
|
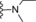
|
14 |
| 12 | −H |
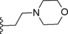
|
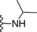
|
0.4 |
| 13 | −H |
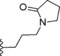
|
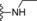
|
1.5 |
| 14 | −H |
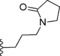
|
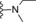
|
17 |
| 15 | −H |
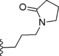
|
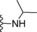
|
0.1 |
| 16 | −H |
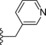
|
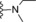
|
240 |
| 17 | −H |
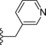
|
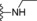
|
1.3 |
| 18 | −H |
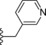
|
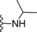
|
0.5 |
Table 3.
Molecular structure of compounds 19–35
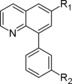
| Compound | R1 | R2 | PDE4D IC50 (nM) |
|---|---|---|---|
| 19 |
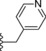
|
−OMe | 2.5 |
| 20 |
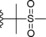
|
−OMe | 1.2 |
| 21 |
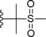
|
−OMe | 9.3 |
| 22 |
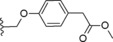
|
−OMe | 0.4 |
| 23 |
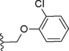
|
−OMe | 5.0 |
| 24 |
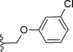
|
−OMe | 12.1 |
| 25 |
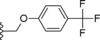
|
−OMe | 48.7 |
| 26 |
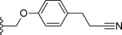
|
−OMe | 2.0 |
| 27 |
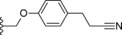
|
−H | 22.2 |
| 28 |
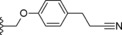
|
−OCF3 | 17.0 |
| 29 |
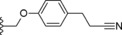
|
−F | 10.5 |
| 30 |
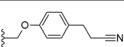
|
−OEt | 4.5 |
| 31 |
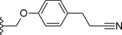
|
−Me | 18.3 |
| 32 |
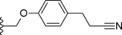
|
−OCHF2 | 0.9 |
| 33 |

|
−OCHF2 | 0.8 |
| 34 |
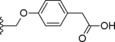
|
−OCHF2 | 0.2 |
| 35 |
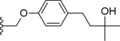
|
−OCHF2 | 0.6 |
2. Experimental
2.1. Data Set
The first dataset of thirty‐five PDE4D inhibitors was collected from literature and consisted of five 3‐cyclopentyloxy‐4‐methoxyphenyliminoether derivatives (1–5) [28] (compounds on which the studies were focused) , thirteen pyridofuropyrimidines (6–18) [33] and seventeen quinoline compounds (19–35) 34. It was used for the validation of the docking protocol. The second one, instead, was composed by already reported PDE4D inhibitors,28 analogues of 1–5, exploited to test the optimized binding mode retrieved by MD.
All the compounds were built, parameterized (Gasteiger‐Hückel method) and energy minimized within OpenEye Scientific Software41 using MMFF94s force field.
As regarding the protein, five human PDE4D X‐rays out of the thirty‐two deposited were collected from Protein Data Bank.43 Mutated structures and low resolution structures were discarded. Since molecular dynamic simulations were going to be exploited during our workflow, fragmented crystal structures (even when the portion was a pivotal regulatory domain) were not selected. Only crystal structures not displaying missing residues in the binding site of PDE4Is were considered.
PDE4D X‐ray complexes selected for the computational studies were: 1XOQ,14 1Y2E,35 3IAK,36 1OYN,37 and 1MKD.38 These X‐rays displayed a range of resolution from 1.83 Å to 2.90 Å and a various number of inner shield metal‐coordinating waters (from 1 to 6). For all calculations only one molecular chain (chain A) was kept and hydrogen atoms were added at a neutral pH.
2.2. Docking Studies
The validation of the docking protocol included re‐docking calculation and docking experiments of a suitable ligand dataset, following the standard procedure. For all the five PDE4D‐inhibitor complexes, re‐docking studies of the corresponding co‐crystallized ligands inside their own X‐ray active site have been performed using both AutoDock4.042 and MOE softwares.43 In particular, the docking protocol was considered successful if the RMSD between the optimum retrieved solution and the crystal pose proved to be smaller than 2.0 Å. Successively, the database including all the thirty‐five compounds, was submitted to docking procedure, that consisted of docking all compounds into the previous five X‐rays using AutoDock4.0.
Docking was performed to allow flexibility of the ligand but a rigid body protein approximation was used to speed up the calculation. Water molecules were considered fixed in the active site. The simulation used a smaller grid, focused on the binding region to predict the binding mode of the ligand dataset. In particular, affinity maps for all the atom types present, as well as an electrostatic map, were computed with a grid spacing of 0.375 Å. The search was carried out with the Lamarckian Genetic Algorithm, the number of generations, energy evaluations, and docking runs were set to 50000, 25000000 and 100, respectively. The docking complexes with the lowest binding energy and showing specific key interactions were used to analyze ligand pose.
According to the results of these simulations, two parameters were set for the further calculations: the crystal to be used and the number of coordinated waters to the metal ions. Calculations were performed using computing clusters, and the Grid computing infrastructure. Docking simulation was carried out on the European grid computing infrastructure (European Grid Infrastructure (EGI) http://www.egi.eu/), using an in‐house developed software for computation management in EGI. The software uses a pilot job strategy to allocate workers on the Grid infrastructure, and then allows an easy job submission, detection and automatic management of Grid failures, job scheduling, and near to‐constant throughput throughout the docking computation. Such approach allows to tackle large computations reliably on the Grid infrastructure and with highly predictable completion times.
As regarding the in silico model validation step, the twelve ligands were docked in the optimized PDE4D‐1 complex structure retrieved from the MD run. More in details the average structure of the complex was calculated from the MD trajectories by Amber Tools, considering only the frames where the RMSD of the complex was stable. Then, taking that structure as reference, the MD trajectory was analyzed to retrieve the frame with the lower RMSD and this latter was extracted as PDB file. The exploited docking protocol was the same described above.
2.3. PCA Analysis
Given the high amount of data generated by cross‐docking experiments, the results were analyzed by means of a Principal Component Analysis (PCA) performed with the R software.44
PCA allow reducing the dimensionality of the original data matrix into a small number of orthogonal principal components (PC). The result of this process is to map samples through scores and variables by the loadings in a new vector space defined by the relevant principal components (PC). In our particular case a single PCA analysis was performed for each inhibitor, considering the crystal structures as the samples and the number and type of interactions between the ligands and the amino acid residues as the independent variable. PC1 and PC2 resulted to be enough to explain from 60 to 70 % of the total variance of the data and were used to draw the further score plots. The preliminary analysis of the score plots well explained the effect of the different crystal structures on the retrieved docking results.
2.4. Molecular Dynamic Simulations
Molecular dynamic simulations of the complex PDE4D‐inhibitor were performed using AMBER12 package[45,46] starting the experiments from the docking solutions in order to assess their stability along the trajectories. The ligands were parameterized using the semiempirical quantum chemistry method AM1‐BCC to derive partial charges, while the other constants were derived by the Antechamber module for the GAFF force field.47,48 The protein was parametrized with the AMBER ff03 force field. Mg2+ and Zn2+ ions were treated according to the “non‐bonded” model method.[49] In order to remove all the possible bad contacts between atoms, preliminary minimization in vacuum of the system was run using a steepest descendent algorithm until energy convergence of 0.0001 kcal/mol. The inhibitor‐protein complex was solvated in a truncated octahedral periodic box with 8 Å of perimetral solvent thickness using TIP3P water model. Na+ ions were added to neutralize the whole system.
The simulations were performed at neutral pH, with histidines 164 and 200 protonated at δ position to coordinate the Zn2+ ion. The water molecule between the two ions was treated as hydroxide ion as suggested by studies of Li et al.,50 and by MD simulations studies on PDE11 performed by some of us.51 Since HIS 160, close to the hydroxide ion, can readily capture a proton, it was protonated at both δ and ϵ positions. All the bonds involving hydrogen atoms were constrained by the SHAKE algorithm,[52] and the time step was set to 2 fs. The non‐bonded cutoff distance was 8 Å and long range electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method. Four steps of minimization were performed keeping the position of protein and ligand restrained for four minimization steps, by a force constant of 500 kcal/mol/Å2, 100 kcal/mol/Å2 in the second, 10 kcal/mol/Å2 in the third and without ligand and protein restrained in the fourth. All the minimization steps were run till the system energy convergence of 0.0001 kcal/mol. Next the system was heated from 0 to 100 K in 30 ps using Langevin dynamics at constant volume, and from 100 to 300 K in 20 ps at constant pressure. After that, the system was equilibrated for 7 ns at constant pressure of 1 atm. Position restrains of 10 kcal/mol/Å were used on the ligand and protein during heating and equilibration steps. After equilibration, production molecular dynamics phase was performed at 300 K using constant pressure of 1 atm. The exploited method was validated performing a 20 ns MD simulation of the complex 1XOQ‐Roflumilast. Then 80 ns MD run were performed for the docking complexes. The resulting trajectories were analyzed using Amber Tools and VMD programs. The root mean square deviation (RMSD) was calculated for the protein backbone and ligand atoms using least‐squares fitting. The pmemd CUDA program of the AMBER12 package45 was used for MD simulations running on a cluster Tesla K20 Graphical Processing Unit (GPU).
3. Results and Discussion
3.1. Docking Protocol Setting
The docking protocol was initially validated by re‐docking five co‐crystallized ligands (Figure 1) in their crystal structure (1XOQ,14 1Y2E,35 3IAK,36 1OYN,37 and 1 MKD,38).
Figure 1.
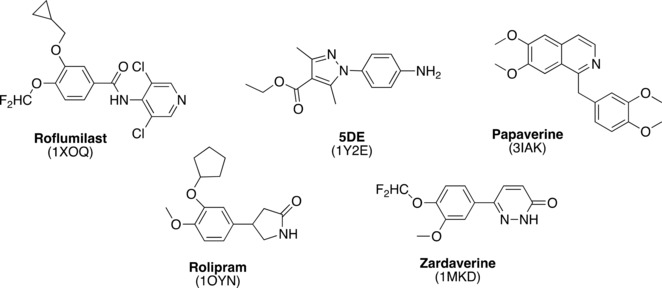
Molecular structure of co‐crystallized ligands.
For the selection of these five PDE4D X‐rays, a first filter on the thirty‐two X‐Ray structures of PDE4D deposited into PDB was applied, discarding those structures which gaps in their sequence, mutated residues in the binding pocket of the ligands, broken fragments (to allow the following MD studies). Then, according to the structure of our compounds, all characterized by a catechol moiety, only the PDE4s co‐crystallized with a catechol ligand were kept. The final four crystal structures (1MKD, 1OYN, 1XOQ, 3IAK) were selected considering in particular three factors: i) the inclusion of the metal‐coordinating waters in the enzyme 3D model during the calculations; ii) the number of these coordinated waters; iii) the resolution of the crystal structures (Table 4). Finally, one extra X ray complex, solved with a not‐catechol ligand was added (1Y2E), to verify if the PDE4 enzymatic pocket dimensions and side‐chain disposition was conditioned by the co‐crystallized ligand.
Table 4.
Re‐docking results according to the different PDE4 X rays.
| Autodock4 | ||||
|---|---|---|---|---|
| PDB code | R (Å) | n coord. waters | RMSD (no coord. waters) (Å) | RMSD (coord. waters) (Å) |
| 1XOQ | 1.8 | 6 | 4.60 | 0.73 |
| 1Y2E | 2.1 | 6 | 5.60 | 1.60 |
| 3IAK | 2.8 | 6 | 4.88 | 1.59 |
| 1OYN | 2.0 | 5 | 3.00 | 2.33 |
| MKD | 2.9 | 1 | 4.15 | 5.03 |
The goodness of this process was evaluated by the root mean square deviation value (RMSD) between the best docking solution (using the most populated cluster within 0.5 kcal/mol from best energy pose) and the crystal pose: the threshold value was set to 2.0 Å, according to the common docking validation protocols.39 As reported in Table 4, the inclusion of the six coordinated waters during the calculation reduced the RMSD substantially, suggesting their pivotal role in the interaction between the ligands and the enzyme model.
In particular 1XOQ displayed the lower RMSD (RMSD=0.73 Å) as well as the lower resolution (R=1.8 Å). Consistent results were obtained by docking a dataset of 35 PDE4DIs (compound 1–35, Tables 1–1, 2, 3 and Table 1S in the Supplementary Information Section) into the aforementioned five X‐rays structures and analyzing the results by Principal Component Analysis (PCA). In more details, a PCA for each compound was performed, taking the crystal structures as the samples. For each amino acid residue, the number and type of interactions engaged with the best pose (in terms of energy) of each docking solution cluster was calculated. Then, since the number of clusters was different among the compounds, the data were expressed as percentage, in order to be used as independent variable in the following analysis.. A score plot was then produced, displaying a common trend for all the PDE4DIs of the dataset: 1Y2E was not related to the other X‐rays, being significantly far from the other objects in the score plot space. Notably it was the only crystal structure with a non‐catechol ligand (Figure 1), thus revealing the effect of the co‐crystallized ligand chemotype in the PDE4 docking results. The difference between 1XOQ, 3IAK and 1OYN and 1MKD interaction pattern (Figure 2) could be associated with the different number of coordinated waters in the active site, in accordance with re‐docking calculations (for ligands 6–18, Figure 2B, the effect is less important, probably because they less interacted with the M pocket where water molecules are placed, see docking results below). As regards 1Y2E, not grouped with the other X‐rays, it was the only crystal structure with a non‐catechol ligand (Figure 1), thus suggesting to consider even the co‐crystallized ligand chemotype during the set‐up of PDE4 docking calculations. Taking together, the results highlighted the X‐ray resolution, the number of coordinated waters and the co‐crystal ligand chemotype as the key factors to be considered for the selection of the best crystal. Consequently, docking studies and PCA analysis converged in suggesting 1XOQ as the most suitable model for our further simulations with the catechol derivative dataset of interest (RMSD: 0.73 Å, R: 1.8 Å, number of coordinated waters: 6, ligand: Roflumilast).
Figure 2.

The score plot of compound 1, 15 and 34 (A, B, C, respectively) from the PCA analysis. In the axis the first two components are represented, explaining 68 %, 69 % and 62 % of the total variance of the system respectively. The red circle included the clustered objects in the space, while the other ungrouped objects are considered not correlated to the first group.
3.2. Docking Results
Regarding pyridofuropyrimidine and quinoline scaffolds (compounds 6–18 and 19–35 respectively), only a preliminary discussion about the docking results was reported, since docking protocol was properly set up for the catechol derivatives. Each series displayed a common single binding pose into PDE4 active site. For the first group of compounds (6–18, Table 2), the nitrogen atom of the pyridine ring and the oxygen atom of the furan portion are engaged in a bi‐dentate H‐bond with Q369 (Figure 3A). The different heterocycles at the R2 position extended their scaffold through the active site, towards the solvent, while the amine substituents at R1 interacted with Q pocket residues (Y159, N321, Y329, and T333). An additional H‐bond between the nitrogen atom in the R3 chain and the backbone oxygen of S368 was observed for the most potent compounds 15 and 18.
Figure 3.
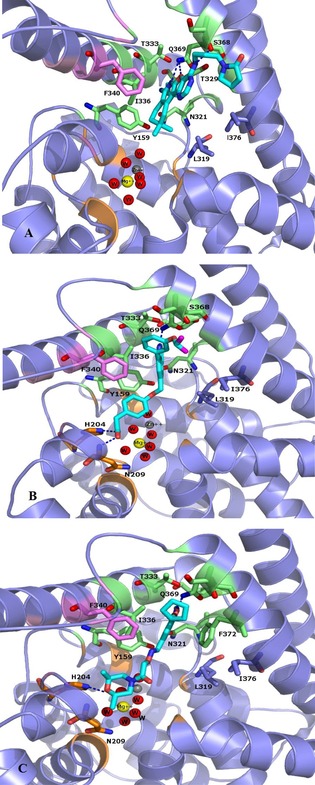
The putative binding mode of compounds 15 (Panel A), 34 (Panel B), 1 (Panel C) within PDE4D active site. Carbons atoms in 15, 34, 1, are coloured in cyan. The main amino acids residues are shown in sticks, the enzyme is shown in cartoon diagram. The residues forming the M, Q and S pockets displayed in cartoon diagram are coloured orange, pink and green respectively, Zinc and Magnesium ions and the oxygen of the water are represented as spheres and the H‐bonds between each ligand and the active site amino acids are shown in dashed line.
The quinoline derivatives 19–35 (Table 3) engaged an H‐bond with Q369 involving the heterocyclic nitrogen atom, while the aryloxy substituents in position 6 were oriented towards the M and S pockets (Figure 3B). The most active compounds in this series formed additional H‐bonds with the backbone of N209 and the side chain nitrogen of H204, two residues that in crystal structures are often involved in water mediated H‐bonds with the solvation shell of the two metal ions. More detailed information may be extracted by fine tuning the docking protocol for these two PDE4‐Is series, but the description lied beyond the aim of the present work.
Concerning compounds 1–5 (Table 1) the docking simulations afforded two binding poses within PDE4D active site, namely conformation A and B. They were characterized by similar energy and for some of them by similar cluster population distribution (Table 5), but displaying different pattern of interaction with the enzyme model: regarding conformation A (Figure 3C), the two catechol oxygen atoms engaged a bi‐dentate H‐bond with Q369, placing the aromatic ring into an hydrophobic clamp made of I336 and F37214 in order to engage a π−π interaction with the latter phenylalanine. The rest of the molecule extended inside the active site to make additional interactions with the metal binding pocket. Conformation B form again an H‐bond with Q369 this time involving the iminoether oxygen or nitrogen of the structure: as a result the aromatic ring was placed outside the active site, while the terminal cyclic amine of the structure did not reach the M pocket of the active site. Relying on the reported experimental data about catechol derivatives binding mode within PDE4 active site (e.g. Roflumilast in 1XOQ, Rolipram in 1OYN, Zardaverine in 1MKD, Papaverine in 3IAK), conformation A was initially selected by us as the common binding mode for all these inhibitors, deserving further and deeper evaluations on both binding modes by means of MD. Accordingly with our previous calculations,28 the docking poses of compounds 1 and 3 highlighted the presence of further H‐bonds with H160 and H204. Concerning compound 2 an additional H‐bond was displayed between the morpholine oxygen and the backbone nitrogen of N209. For compounds 4 and 5, isomers R and S showed a similar behavior within PDE4D active site: polar interactions were detected between the morpholine or dimethylmorpholine oxygen and the side chains of Q343 (see Table 1S).
Table 5.
Binding energy and cluster population from docking calculations
| CONFORMATION A | CONFORMATION B | |||
|---|---|---|---|---|
| Cmpd | cluster energy (kcal/mol) | cluster population | cluster energy (kcal/mol) | cluster population |
| 1 | −8.36 | 19 | −8.15 | 8 |
| 2 | −7.95 | 32 | −7.46 | 11 |
| 3 | −6.55 | 18 | −4.88 | 4 |
| 4 | −8.19 | 12 | −7.98 | 14 |
| 5 | −8.61 | 6 | −8.75 | 8 |
In summary, assuming conformation A as the proper one for catechol series, docking results suggested the key role of additional H‐bond with the M pocket (in particular with H160 and H204) to afford a proper potency of inhibition of the target.
Since key prerequisite for successful differentiation between active and non‐active ligands is the accurate prediction of their binding affinities, MD were subsequently applied. In fact, many studies have already shown rather poor correlations between docking scores and experimental binding affinities, suggesting the need of further refinements of the binding pose by means of more extensive calculations.40
3.3. Molecular Dynamic Simulations
Before running the MD simulations of the docking solutions, the MD method was evaluated performing a 100 ns MD on the crystal PDE4D‐Roflumilast complex 1XOQ. The stable RMSD values of the backbone of the protein and ligand, evaluated considering the RMSD value change from the starting conformation (Figure 4), demonstrated the suitability of the selected MD parameters for the further simulations.
Figure 4.
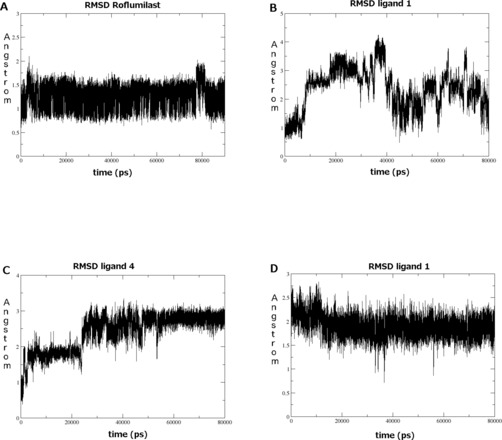
Root mean square deviation (RMSD) of Roflumilast, 1 and 4 during MD simulations. (A) Roflumilast starting from conformation A (crystallographic binding pose). (B) ligand 1 starting from conformation A. (C) ligand 4 starting from conformation A. (D) ligand 1 starting from conformation B.
Starting from the previously selected A conformation, we thus performed a first run of 80 ns MD simulations on all the five ligands‐receptor complexes. Unexpectedly, while Roflumilast resulted stable in A conformation (Figure 4A), for 1–5, the docking pose was not confirmed by MD results. In fact, compound 1 was unstable during all the simulation (such shown by RMSD value for the ligand, Figure 4B), immediately losing the H‐bond with H204 and after 40 ns the one with Q369. As regards compound 3, consistently with the behavior of compound 1, both isomers were unstable in their trajectories. Same conclusions could be detected for compounds 2 and 5. Finally even in the case of compound 4, the catechol oxygen atoms did not manage to keep the H‐bond with Q369 for the whole trajectory; in fact, after 50 ns the ligand assumed a second binding mode (Figure 4C), engaging a new H‐bond between the iminoether oxygen and Y329, which stabilize the compound till the end of the simulation. Notably this latter binding mode was comparable to the conformation B from docking calculations. In the context of the latter result and of the discussed docking solutions, a second MD simulation was performed for each inhibitor 1–5 starting from conformation B. In addition, since for all compounds isomers R and S similarly interacted with PDE4D both in docking and in the first MD simulations, the following studies were performed involving only S isomer.
Notably, these complexes resulted stable (see as example ligand 1, Figure 4D), unless the loss of the H bond with Q369, which was conserved during all the simulation only by compound 1, involving its carbonyl oxygen (Figure 5). A detailed analysis of the key interactions for all the complexes submitted to MD was performed, considering the most representative structure for each complex, calculated from the MD trajectories applying cluster analysis where the RMSD of the complex was stable. The complexes highlighted some common features which helped us to better define the binding mode corresponding to conformation B. In detail, some interactions were shared by all the compounds, in particular the van der Waals contacts with Y159, L319, N321, P322, Y329, W332, T333, I336, M357, S368. Moreover each ligand formed an H bond between its iminoether portion and an active site residue and definitively did not occupy the metal pocket and the solvent filled side pocket of the active site.
Figure 5.
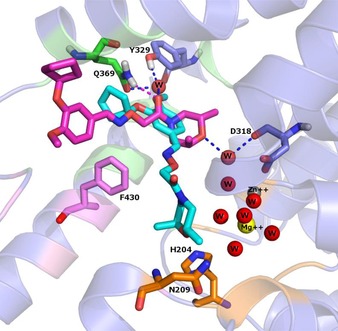
The conformations of compound 1 before (cyan) and after (magenta) the MD simulation. The main amino acids residues are shown in sticks, the enzyme is shown in cartoon diagram. The residues forming the M, Q and S pockets displayed in cartoon diagram are coloured orange, pink and green respectively. Zinc and Magnesium ions are represented as spheres and the H‐bonds between each ligand and the active site amino acids are shown in magenta dashed line. The water is represented as sphere and water bridged H‐bonds are shown as blue dashed lines.
As written above, compound 1 (Figure 5), the most potent and selective inhibitor among the data‐set, was the only able to form a wide net of stable H‐bond interactions, mainly in the nearby of Q369 and Y329 residues. More in detail, the carbonyl oxygen of 1, involved in an H‐bond with the amidic nitrogen of Q369, was engaged also in two additional water mediated H‐bonds, one with Y329, and one again with Q369, supporting a water molecule to bridge between these two residues. This pattern of interaction should be probably able to reinforce the stabilising H‐bond between Q369 and Y329, always observable in the PDE4D crystal structures. A further water mediated H‐bond, performed between the morpholine oxygen and the carbonyl oxygen of D318 backbone, increased the complex stability. Compound 2 displayed an H‐bond between its carbonyl oxygen and Y329, plus a π−π stacking interaction with F372. Concerning the remaining compounds, none of them showed the same H‐bonds stability of 1 and 2, thus the following interactions were observed only for modest percentages of frames. Concerning compound 4, its conformation B performed a water bridge mediated H‐bond, between the iminoether oxygen and Y329 and the only direct H‐bond present in the latest 40 ns was the one with Q369 (Figure 6). Compound 5 displayed a similar pattern of interactions, with a reduced number of frames involved in water bridge H‐bonds with Q369 and D318, plus a π−π stacking interaction with F340. No strong and direct H‐bonds interactions were detected also for compound 3, even if the ligand RMSD was stable and Q369 held its H‐bond with Y329.
Figure 6.
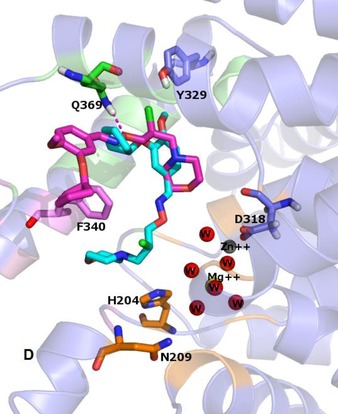
The conformations of compound 4 before (in cyan) and after (in magenta) the MD simulation. The main amino acids residues are shown in sticks, the enzyme is shown in cartoon diagram. The residues forming the M, Q and S pockets displayed in cartoon diagram are coloured orange, pink and green respectively. Zinc and Magnesium ions and oxygen of the water are represented as spheres and the H‐bonds between each ligand and the active site amino acids are shown in dashed line.
Consistently with biological data related to the analyzed compounds, the sole sub‐micromolar compound 1 was the one able to form the highest number of H‐bond interactions with the binding site. This pattern of interaction suggested the H‐bond with Q369 as the pivotal element to gain potency in PDE4 inhibition, in accordance with literature14, and underlined the importance of the interactions with the water molecules inside the active site.
In conclusion, these computational results highlight a new binding mode for catechol inhibitors, conformation B, within the human PDE4D enzyme and the interaction with residue Q369, Y329, and F340, often in combination, resulted to be essential for the complex stability. In particular, the ligands have been predicted to differently occupy the space of the active site if compared with previous PDE4DIs, since no direct contacts with the metal pocket have been revealed. The great number of the interactions involved amino acid residues close to residue Q369 and the aromatic ring of their catechol moiety, according to our simulations, seems oriented toward the region of the active site where UCR2 and CR3 may potentially close over. Since the closure of these two domains is a dynamic process, the simulation of how the interaction with the studied compounds may occur is not possible without the availability of a complete PDE4 crystal structure. The preliminary observation is that the interaction with these regulatory domains may allow the selectivity of action on PDE4D among the other isoforms.
3.4. In Silico Model Validation
To preliminary validate conformation B as binding mode for the iminoether series of PDE4D inhibitors, new docking calculations were made: the new dataset consisted of our 12 reported analogues of 1 28 (compounds 36–47, Table 6), which have been already docked onto 1XOQ (published data)28. In the new calculations the optimized PDE4D conformation with 1 after MD runs (the one discussed above), was used as enzyme model. Here the interaction with Q369 became essential to distinguish active from inactive compounds (in accordance with literature which highlighted the invariant glutamine as the pivotal residue for PDE4D inhibition). Indeed, docking solutions with the optimized PDE4D model displayed 36–47 in conformation B and showed that the only active compounds of the dataset (compound 38, 40 and 41) managed to form a bi‐dentate H‐bond between Q369 and their iminoether portion. Moreover 38 formed an additional H‐bond with D318. Notably 42–47 didn't have the iminoether group, thus preventing the formation of the H‐bond with Q369, and probably affecting in this way the inhibition activity. The pivotal role of this bi‐dentate interaction may be suggested even by the inactivity of 39 which displayed an acetylate hydroxyl group on the iminoether chain. Finally inactive compound 36 and 37, the longer analogues of 1, showed only one H‐bond with Q369, since the longer chain didn't manage to involve the carbonyl oxygen in the interaction, thus not forming an enough stable interaction with PDE4D active site. Notably, the conformation A solutions retrieved by the reported previous docking calculations28 allowed the formation of the double Q369 H‐bond for all the compounds, thus explaining the difference in potency only with little different patterns of interactions at the M pocket level.
Table 6.
Molecular structure of compounds 36–47
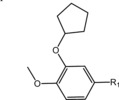
| Compound | R1 | PDE4D % inhib. at 10 µM |
|---|---|---|
| 36 |
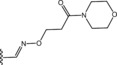
|
49 |
| 37 |

|
24 |
| 38 |
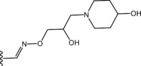
|
65 |
| 39 |
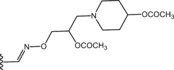
|
12 |
| 40 |
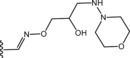
|
56 |
| 41 |
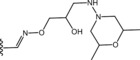
|
80 |
| 42 |
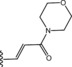
|
25 |
| 43 |
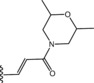
|
16 |
| 44 |
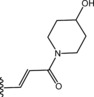
|
22 |
| 45 |
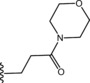
|
7 |
| 46 |
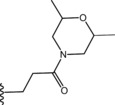
|
10 |
| 47 |
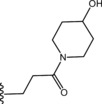
|
12 |
Thus, we suggest that conformation B more accurately promote SAR considerations on this series of compounds, pointing out the key elements to allow PDE4 inhibition: i) the iminoether moiety as H‐bond acceptor anchor point ii) the presence of a second polar group on the chain in the proper position to form an additional H‐bond with Q369 iii) a terminal ring with polar atoms to make additional interactions with the active site. However, since the lack of a crystal PDE4D‐iminoether analogue complex with UCR1, UCR2 and CR3, the role of the aromatic ring is still to be defined.
4. Conclusions
Up to now, several studies indicated the PDE4D isoform as an interesting target for neurodegenerative pathologies such as Alzheimer′s disease. Moreover, it has been clearly demonstrated that selective PDE4D inhibitors could improve memory and synaptic functions without undesirable side effects such as nausea and sedation. The lack of total length PDE4D crystal structure has impaired until now the rational design of new more selective compounds. On these bases, in order to deeply understand the interaction of our previously developed PDE4DIs and their enzymatic target, we have applied a computational study that involved docking and molecular dynamic simulations.
Any information coming from the just the docking studies yielded two recurring binding modes for our inhibitors (conformation A, conformation B). Interestingly, the results of molecular dynamic simulations performed as a subsequent step on our compounds 1–5 highlighted conformation B as the most stable, therefore identifying the putative PDE4DIs active pose for these data‐set. Moreover the new binding mode was more consistent with the biological profile of already published compounds of the same series (36–47), revealing the essential features for PDE4D inhibition. This result could be useful to rationally guide the design and synthesis of new potent and selective PDE4DIs in the future.
Abbreviations
- AD
Alzheimer′s disease
- cAMP
cyclic adenosine monophosphate
- CNS
central nervous system
- COPD
chronic obstructive pulmonary disease
- CR3
C terminal helix conserved region 3
- CREB
cAMP response element‐binding protein
- cGMP
cyclic guanosine monophosphate
- GPU
graphical processing unit
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- MD
molecular dynamic
- PC
principal components
- PCA
principal components analysis
- PDE
phosphodiesterases
- PDE4Is
PDE4 inhibitors
- RMSD
root mean square deviation
- SAR
structure activity‐ relationship
- UCR
upstream conserved regions
Conflict of Interest
None declared.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Alzheimer Association is gratefully acknowledged for financial support to this study (IIRG 11‐208306) together with the Italian Ministry of Education and Research through the Flagship InterOmics (PB05) and HIRMA (RBAP11YS7K) projects, and the European MIMOmics (305280) project. SG, EC, PF and OB thank Mr. V. Ruocco for his support to calculations.
P. D'Ursi, S. Guariento, G. Trombetti, A. Orro, E. Cichero, L. Milanesi, P. Fossa, O. Bruno, Mol. Inf. 2016, 35, 369.
References
- 1. Bender A. T., Beavo J. A., Pharmacol. Rev. 2006, 58, 488. [DOI] [PubMed] [Google Scholar]
- 2. Beavo J. A., Heaslip R. J., Mol. Pharmacol. 1994, 46, 399. [PubMed] [Google Scholar]
- 3. Houslay M. D., Adams R. D., Biochem. J. 2003, 370, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ghosh R., Sawant O., Ganpathy P., Pitre S., Kadam V., Int. J. Pharm. Tech. Res. 2009, 1, 1148. [Google Scholar]
- 5. Houslay M. D., Baillie G. S., Maurice D. H., Circ. Res. 2007, 100, 950. [DOI] [PubMed] [Google Scholar]
- 6. Conti M., Iona S., Cuomo M., Swinnen J. V., Odeh J., Svoboda M. E., Biochem. 1995, 34, 7979. [DOI] [PubMed] [Google Scholar]
- 7. Houslay M. D., Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 249. [DOI] [PubMed] [Google Scholar]
- 8. Richter W., Conti M., J. Biol. Chem. 2002, 277, 40212. [DOI] [PubMed] [Google Scholar]
- 9. Richter W., Conti M., J. Biol. Chem. 2004, 279, 30338. [DOI] [PubMed] [Google Scholar]
- 10. Xie M., Blackman B., Scheitrum C., Mika D., Blanchard E., Lei T., Conti M., Richter W., Biochem. J. 2014, 459, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bolger G. B., Dunlop A. J., Meng D., Day J. P., Klussmann E., Baillie G. S., Adams D. R., Houslay M. D., PNAS 2015, 112, E1414. [Google Scholar]
- 12.P. Cedervall, A. Aulabaugh, K. F. Geoghegan, T. M. McLellan, J. Pandit, Engineered stabilization and structural analysis of the autoinhibited conformation of PDE4, PNAS, 112 (2015) E1414–E1422. [DOI] [PMC free article] [PubMed]
- 13. Burgin A. B., Magnusson O. T., Singh J., Witte P., Staker B. L., Bjornsson J. M., Thorsteinsdottir M., Hrafnsdottir S., Hagen T., Kiselyov A. S., Stewart L. J., Gurney M. E., Nat. Biotechnol. 2010, 28, 63. [DOI] [PubMed] [Google Scholar]
- 14. Card G. L., England B. P., Suzuki Y., Fong D., Powell B., Lee B., Luu C., Tabrizizad M., Gillette S., Ibrahim P. N., Artis D. R., Bollag G., Milburn M. V., Kim S. H., Sclessinger J., Zhang K. Y., Structure 2004, 12, 2233. [DOI] [PubMed] [Google Scholar]
- 15. Houslay M. D., Schafer P., Zhang K. Y. J., Drug Discovery Today 2005, 10, 1503. [DOI] [PubMed] [Google Scholar]
- 16. Lipworth B. J., Lancet 2005, 365, 167. [DOI] [PubMed] [Google Scholar]
- 17. Lerner A., Epstein P. M., Biochem. J. 2006, 393, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang Z., Mancini J. A., Curr. Med. Chem. 2006, 13, 3253. [DOI] [PubMed] [Google Scholar]
- 19. O'Donnell J. M., Zhang H. T., Trends Pharmacol. Sci. 2004, 25, 158. [DOI] [PubMed] [Google Scholar]
- 20. Kanes S. J., Tokarczyk S. J., Bilker W., Abel T., Kelly M. P., Neuroscience 2007, 144, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gurney M. E., D′Amato E. C., Burgin A. B., Neurotherapeutics 2015, 12, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang H., Robinson H., Ke H., J. Mol. Biol. 2007, 371, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DALIRESP (roflumilast) tablet [Forest Laboratories, Inc.]. DailyMed. Forest Laboratories, Inc. August 2013. Retrieved 13 November 2013.
- 24.Brooks, M. (21 March 2014). FDA Clears Apremilast (Otezla) for Psoriatic Arthritis. Medscape Medical News (WebMD). Retrieved 28 March 2014.
- 25. Gurney M. E., Burgin A. B., Magnusson O. T., Stewart L. J., Handb. Exp. Pharmacol. 2011, 204, 167. [DOI] [PubMed] [Google Scholar]
- 26. Fox 3rd D., Burgin A. B., Gurney M. E., Cell Signal. 2014, 26, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bruno O., Brullo C., Arduino N., Schenone S., Ranise A., Bondavalli F., Ottonello L., Dapino P., Dallegri F., Farmaco 2004, 59, 223. [DOI] [PubMed] [Google Scholar]
- 28. Bruno O., Romussi A., Spallarossa A., Brullo C., Schenone S., Bondavalli F., Vanthuyne N., Roussel C., J. Med. Chem. 2009, 52, 6546. [DOI] [PubMed] [Google Scholar]
- 29. Brullo C., Massa M., Rocca M., Rotolo C., Guariento S., Rivera D., Ricciarelli R., Fedele E., Fossa P., Bruno O., J. Med. Chem. 2014, 57, 7061. [DOI] [PubMed] [Google Scholar]
- 30. Bruno O., Fedele E., Prickaerts J., Parker L. A., Canepa E., Brullo C., Cavallero A., Gardella E., Balbi A., Domenicotti C., Bollen E., Gijselaers H. J., Vanmierlo T., Erb K., Limebeer C. L., Argellati F., Marinari U. M., Pronzato M. A., Ricciarelli R., Br. J. Pharmacol. 2011, 164, 2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sierksma A. S., Van den Hove D. L., Pfau F., Philippens M., Bruno O., Fedele E., Ricciarelli R., Steinbusch H. W., Vanmierlo T., Prickaerts J., Neuropharmacology 2014, 77, 120. [DOI] [PubMed] [Google Scholar]
- 32. Brullo C., Massa M., Villa C., Ricciarelli R., Rivera D., Pronzato M. A., Fedele E., Barocelli E., Bertoni S., Flammini L., Bruno O., Bioorg. Med. Chem. 2015, 23, 3426. [DOI] [PubMed] [Google Scholar]
- 33. Taltavull J., Serrat J., Gràcia J., Gavaldà A., Còrdoba M., Calama E., Montero J. L., Andrés M., Mirelpeix M., Vilella D., Hernandez B., Beleta J., Ryder H., Pagès L., Eur. J. Med. Chem. 2011, 46, 4946. [DOI] [PubMed] [Google Scholar]
- 34. Aspiotis R., Deschenes D., Dubè D., Girard Y., Huang Z., Lalibertè F., Liu S., Pappa R., Nicholson D. W., Young R. N., Bioorg. Med. Chem. Lett. 2010, 20, 5502. [DOI] [PubMed] [Google Scholar]
- 35. Card G. L., Blasdel L., England B. P., Zhang C., Suzuki Y., Gillette S., Fong D., Ibrahim P. N., Artis D. R., Bollag G., Milburn M. V., Kim S. H., Schlessinger J., Zhang K. Y. J., Nat. Biotechnol. 2005, 23, 201. [DOI] [PubMed] [Google Scholar]
- 36.R. K. Y. Cheng, L. Crawley, J. Barker, M. Wood, B. Felicetti, M. Whittaker, 3IAK PDE4D X-rays, deposited and accepted in PDB on 08-09-2009.
- 37. Huai Q., Wang, Sun H. Y., Kim H. Y., Liu Y., Ke H., Structure 2003, 11, 865. [DOI] [PubMed] [Google Scholar]
- 38. Lee M. E., Markowitz J., Lee J. O., Lee H., Febs Lett. 2002, 530, 53. [DOI] [PubMed] [Google Scholar]
- 39. Jain A. N., J. Comput. Aided Mol. Des. 2008, 22, 201. [DOI] [PubMed] [Google Scholar]
- 40. Atkovska K., Samsonov S. A., Paszkowski-Rogacz M., Pisabarro M. T., Int. J. Mol. Sci. 2014, 15, 2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.OpenEye Scientific Software, Santa Fe, New Mexico, USA, HYPERLINK “http://www.eyesopen.com/”www.eyesopen.com.
- 42. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J., J. Comput. Chem. 2009, 16, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MOE, Chemical Computing Group Inc., Montreal; H3A 2R7 Canada, http://www.chemcomp.com.
- 44.R-based chemometric software developed by the Group of Chemometrics of the Division of Analytical Chemistry of the Italian Chemical Society, www.gruppochemiometria.it.
- 45.D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossvary, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M. J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko, P. A. Kollman, 2012 AMBER 12, University of California, San Francisco.
- 46. Goetz A. W., Williamson M. J., Xu D., Poole D., Le Grand S., Walker R. C., J. Chem. Theory Comput. 2012, 8, 1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jakalian A., Jack D. B., Bayly C. I., J. Comput. Chem. 2002, 23, 1623. [DOI] [PubMed] [Google Scholar]
- 48. Wang J., Wang W., Kollman P. A., Case D. A., J. Comput. Chem. 2005, 25, 1157. [DOI] [PubMed] [Google Scholar]
- 49. Stote R. H., Karplus M., Proteins 1995, 23, 12. [DOI] [PubMed] [Google Scholar]
- 50. Li Z., Cai Y. H., Cheng Y. K., Lu X., Shao Y. X., Li X., Liu M., Liu P., Luo H. B., J. Chem. Inf. Model. 2013, 53, 972. [DOI] [PubMed] [Google Scholar]
- 51. Cichero E., D′Ursi P., Moscatelli M., Bruno O., Orro A., Rotolo C., Milanesi L., Fossa P., Chem. Biol. Drug Des. 2013, 82, 718. [DOI] [PubMed] [Google Scholar]
- 52. Shuichi M., Kollman P. A., J. Comput. Chem. 1992, 8, 952. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary