

ABSTRACT
Azido‐functionalized poly(ethylene glycol) (PEG) derivatives are finding ever‐increasing applications in the areas of conjugation chemistry and targeted drug delivery by their judicious incorporation into nanoparticle‐forming polymeric systems. Quantification of azide incorporation into such PEG polymers is essential to their effective use. 1H Nuclear Magnetic Resonance (NMR) analysis offers the simplest approach; however, the relevant adjacent azide‐bearing methylene protons are often obscured by the PEG manifold signals. This study describes the synthesis of 1,2,3‐triazole adducts from their corresponding PEG azides via a convenient, mild click reaction, which facilitates straightforward NMR‐based quantitative end‐group analysis.This method was found to be compatible with many examples of bifunctional azido PEGs with molecular weights ranging from 2 to 18 kDa bearing a variety of functional groups. © 2016 The Authors. Journal of Polymer Science Part A: Polymer Chemistry Published by Wiley Periodicals, Inc. J. Polym. Sci., Part B: Polym. Phys. 2016, 54, 2888–2895
Keywords: azides, click chemistry, NMR spectroscopy, poly(ethylene glycol), triazoles, calculations, hydrophilic polymers, water‐soluble polymers
INTRODUCTION
Poly(ethylene glycol) (PEG) derivatives have been applied in bioconjugation chemistry through the modification of peptides and proteins;1, 2 and have found broad use in immunology,3 virology,4 and drug delivery5 systems including liposomal6 and micelle systems.7, 8 The incorporation of PEG‐containing moieties (PEGylation) into such systems generally improves pharmacological properties9 including increased water solubility, enhanced resistance to protein hydrolysis/degradation, improved bioavailability (circulation half‐life),10 and reduced antigenicity.11 Bioconjugation of PEG derivatives is most often achieved via reactive end groups; typically NHS ester, aldehyde, benzotriazole carbonate, p‐nitrophenyl carbonate, maleimide, vinyl sulfone, thiol, amine, hydrazide, aminoxy, alkyne, and azide groups are employed.12, 13, 14, 15 Ideally the bioconjugation reaction will be high yielding, chemoselective, and proceed under mild, often aqueous, reaction conditions. These are all characteristics of the Huisgen 1,3‐dipolar cycloaddition between an azide and an alkyne to form a 1,2,3‐triazole linkage.16 This quintessential “click” reaction,17, 18 particularly employing an azido PEG derivative, has been utilized in polymer chemistry,19, 20 peptide conjugation,21 and to attach other biomolecules.22 Proper stoichiometry between the dipolarophile and the 1,3‐dipole is often essential for the successful implementation of these 1,2,3‐triazole forming reactions. Thus, it is imperative that the extent of PEG functionalization be verified. Many functional groups incorporated into PEG can be easily quantified by 1H NMR spectroscopy. Azides incorporated into PEGs prove to be more challenging as their adjacent methylene (N3—CH2—) proton signals can be obscured by the often pervasive PEG manifold signals. Although syntheses of azido PEGs have been described before,23, 24, 25 no robust method to quantitate the degree of azide incorporation has been disseminated. This publication describes the synthesis of numerous examples of PEG azides and outlines the development of a straightforward click derivatization that facilitates quantitative end group 1H NMR analysis of azido PEG derivatives (Scheme 1).
Scheme 1.
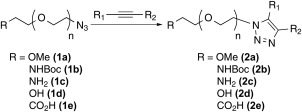
Triazole forming click reaction.
EXPERIMENTAL
Materials
Anhydrous dichloromethane (DCM) and tetrahydrofuran (THF) were purchased from Sigma Aldrich. Absolute ethanol was obtained from Decon Laboratories. Diethyl ether (Et2O), methyl tert‐butyl ether (MTBE), and sodium azide were purchased from Fisher Scientific. Triethylamine (NEt3) and methanesulfonyl chloride (MsCl) were obtained from Acros and distilled from CaH2 and P2O5, respectively. Trifluoroacetic acid (TFA) was purchased from Oakwood Products. Pearlman's catalyst (Pd(OH)2/C), 2,2,6,6‐tetramethylpiperidine‐1‐oxyl (TEMPO), phenylacetylene (PA), diphenylacetylene (DPA), dimethyl acetylenedicarboxylate (DMAD), and diethyl acetylenedicarboxylate (DEBD) were purchased from Sigma Aldrich and used as received. Potassium bromide and di‐tert‐butyl dicarbonate ((Boc)2O) were purchased from Acros and used as received.
Instrumentation
Gel permeation chromatography (GPC) measurements were carried out using a Waters 515 isocratic pump connected in series to a Waters guard column (200 Å, 6 × 40 mm, 6 μm), two Ultrahydrogel 250 columns (7.8 × 300 mm, 6 μm), an Ultrahydrogel 500 column (7.8 × 300 mm, 10 μm), a Waters 2487 UV detector, a Wyatt Dawn Heleos light scattering detector, and a Wyatt Optilab rEX refractive index detector. A H2O:CH3CN (60:40, v/v, with 0.1% TFA) mixture used as the eluent at a flow rate of 0.7 mL/min at 35.5 °C. 1H NMR spectra were measured with a Varian VNMRS 400 MHz spectrometer. Chemical shifts are reported in parts per million using DMSO‐d 6 or CD3CN as an internal standard. 1H NMR spectra can be found in the Supporting Information.
Synthesis of mPEG‐OMs (4)
mPEG‐OH (3) (100 g, 1 eq.) was dried via azeotropic distillation with toluene (500 mL, 0.2 g PEG/mL) at 60 °C under reduced pressure. Once the majority of toluene had been removed and the starting material solidified, the temperature was reduced to 25 °C and reduced pressure was maintained for 14 h. The starting material dissolved in anhydrous DCM (166 mL, 0.6 g PEG/mL) with the help of a heat gun. The resulting mixture was cooled in an ice water bath prior to the addition of NEt3 (2.3 eq.) followed by MsCl (3.4 eq.) and the reaction was allowed to warm to room temperature slowly over 14 h. The reaction was considered complete via TLC analysis (100:10:1, CHCl3:MeOH:25% aq. NH4OH; Dragendorff stain). The reaction mixture was diluted with DCM (833 mL, 0.12 g PEG/mL); washed with 2 M HCl:brine (1:1 v/v, 4 × 200 mL, 0.5 g PEG/mL); brine (1 × 200 mL, 0.5 g PEG/mL); dried with MgSO4 and 4 Å molecular sieves; and then concentrated under reduced pressure. The resulting crude solid was dissolved in DCM (400 mL, 0.25 g PEG/mL, with the gentle assistance of a heat gun) and precipitated by the addition of excess Et2O (10× v/v, ∼4 L). The precipitation was cooled to 4 °C for ∼12 h before the solids were collected by vacuum filtration and washed with additional Et2O. The product was dried under reduced pressure to yield mPEG‐OMs (4) as a white solid (yield typically >90%).
1H NMR (400 MHz, DMSO‐d 6, δ, ppm): 4.32–4.29 (m, 2H), 3.58–3.40 (m, ∼n × 4H), 3.24 (s, 3H), 3.18 (s, 3H).
Synthesis of mPEG‐N3 (1a)
A round‐bottom flask fitted with a condenser was charged with mPEG‐OMs (4) (100 g, 1 eq.), absolute ethanol (800 mL, 0.125 g PEG/mL), and then sodium azide (10 eq.). The resulting suspension was heated to a gentle reflux (85 °C) overnight. The reaction was temporarily cooled to room temperature and an aliquot (1 mL) was taken and filtered through a small plug of MgSO4/SiO2 into a 7 mL scintillation vial using CHCl3 to rinse the plug. The sample was concentrated to half the initial volume and then precipitated with Et2O (∼5 mL). The solids were collected by centrifugation and dried under reduced pressure prior to NMR analysis. The disappearance of the CH2OMs methylene (1H NMR, δ 4.31 ppm, CD3CN) was used to monitor the progress of the reaction. After the reaction was complete (typically 16 h), the reaction mixture was concentrated on a rotary evaporator. DCM (500 mL, 0.2 g PEG/mL) was added to the crude solid material, and the resulting suspension was concentrated on a rotary evaporator. The process was repeated one additional time with the same amount of DCM. To the subsequent crude material was added DCM:MeOH (10:1, v/v, 800 mL, 0.125 g/mL). This suspension was filtered through a plug of SiO2/MgSO4 using additional DCM:MeOH to rinse the plug. The filtrate was concentrated on a rotary evaporator to a volume of ∼400 mL (0.25 g PEG/mL) before the addition of Et2O (10× v/v, ∼4 L). The precipitation was cooled to 4 °C for ∼12 h before the solids were collected by vacuum filtration and washed with additional Et2O. The product was dried under reduced pressure to yield mPEG‐N3 (1a) as a white solid (yield typically >90%).
1H NMR (400 MHz, CD3CN, δ, ppm): 3.64–3.44 (m, ∼n × 4H), 3.38–3.36 (m, 2H overlapped with 13C satellites), 3.29 (s, 3H).
Synthesis of H2N‐PEG‐OH (6)
Bn2N‐PEG‐OH (5) (50 g, 1 eq.) was dissolved in H2O (250 mL, 0.2 g PEG/mL) before the addition of Pd(OH)2 (2.5 g, 5 % wt). The flask was placed under reduced pressure until gas could no longer be seen evolving from the reaction mixture. The flask was backfilled with H2 and then re‐evacuated under reduced pressure. The process was repeated two additional times. The reaction was left to stir at room temperature under H2 (1 atmosphere, balloon pressure). After 24 h, an aliquot (∼0.7 mL) was taken and filtered through a 0.45 μm syringe‐driven filter into a small vial to remove as much catalyst as possible. The majority of the water was removed on a rotary evaporator and the vial was left to dry in vacuo. Reaction progress was monitored via 1H NMR (DMSO‐d 6) for the disappearance of the starting material R—CH2NBn2 methylene (δ 2.55) and aromatic (δ ∼7.2–7.3) signals and appearance of the fully deprotected R—CH2NH2 methylene (δ 2.63) signals. When the reaction was complete, NaCl (90 g, to give ∼0.36 g/mL) was added and mixture was stirred until dissolution. The reaction mixture was then filtered through a Millipore Durapore™ 0.45 μm membrane to remove the catalyst. The flask and filter were rinsed with brine. The pH of the clear filtrate was adjusted to ∼12–13 with 1 M NaOH and then extracted with DCM (10 × 250 mL, 0.2 g PEG/mL). The combined organic layers were stirred with a generous amount of Na3PO4 and MgSO4 for 30 min before filtering. The clear filtrate was concentrated on a rotary evaporator to a volume of ∼200 mL (0.25 g PEG/mL) and precipitated with the addition of Et2O (10× v/v, ∼2 L). The precipitation was cooled to 4 °C for ∼12 h before the solids were collected by vacuum filtration and washed with additional Et2O. The product was dried under reduced pressure to yield H2N‐PEG‐OH (6) as a white solid (yield typically >90%).
1H NMR (400 MHz, DMSO‐d 6, δ, ppm): 4.52 (br s, 1H), 3.72–3.26 (m, ∼n × 4H), 2.63 (t, J = 5.6 Hz).
Synthesis of BocNH‐PEG‐OH (7)
H2N‐PEG‐OH (6) (50 g, 1 eq.) was dissolved in aqueous 0.1 g/mL K2CO3 (400 mL, 0.125 PEG g/mL, pH ∼11–12) before the addition of di‐tert‐butyl dicarbonate (1.5 eq.). After stirring overnight at room temperature, TLC analysis (100:10:1, CHCl3:MeOH:25% aq. NH4OH; Dragendorff stain) indicated complete consumption of starting material. NaCl (144 g, to give ∼0.36 g/mL) was added and mixture was stirred until dissolution. The reaction mixture was extracted with DCM (5 × 250 mL, 0.2 g PEG/mL) and the combined organic layers were dried over MgSO4 and filtered. The clear filtrate was concentrated on a rotary evaporator to a volume of ∼250 mL (0.2 g PEG/mL) and precipitated with the addition of Et2O (10× v/v, ∼2.5 L). The precipitation was cooled to 4 °C for ∼12 h before the solids were collected by vacuum filtration and washed with additional Et2O. The product was dried under reduced pressure to yield BocNH‐PEG‐OH (7) as a white solid (yield typically >90%).
1H NMR (400 MHz, DMSO‐d 6, δ, ppm): 6.74 (br s, 1H), 4.57 (t, J = 5.5 Hz, 1H), 3.69–3.31 (m, ∼n × 4H), 3.06 (q, J = 6.0 Hz, 2H), 1.37 (s, 9H).
Synthesis of BocNH‐PEG‐OMs (8)
Prepared from Boc‐PEG‐OH (7) following the same method described above for the synthesis of mPEG‐OMs (4).
1H NMR (400 MHz, DMSO‐d 6, δ, ppm): 6.75 (br s, 1H), 4.32–4.29 (m, 2H) 3.69–3.31 (m, ∼n × 4H), 3.18 (s, 3H), 3.05 (q, J = 6.0 Hz, 2H), 1.37 (s, 9H).
Synthesis of BocNH‐PEG‐N3 (1b)
Prepared from Boc‐PEG‐OMs (8) following the same method described above for the synthesis of mPEG‐N3 (1a).
1H NMR (400 MHz, DMSO‐d 6, δ, ppm): 6.72 (br s, 1H), 3.68–3.27 (m, ∼n × 4H), 3.04 (q, J = 6.0 Hz, 2H), 3.37–3.34 (m, 2H overlapped with 13C satellites), 1.35 (s, 9H).
Synthesis of H2N‐PEG‐N3 (1c)
NHBoc‐PEG‐N3 (1b) (50 g, 1 eq.) was dissolved in DCM (75 mL, 0.67 g PEG/mL) with the assistance of a heat gun. Once cooled to ambient temperature, TFA (75 mL) was added. After 2 h, an aliquot (∼0.7 mL) was transferred to a 7 mL scintillation vial and precipitated by the addition of cold Et2O (5 mL). The solids were collected by centrifugation, washed with additional Et2O, and dried under reduced pressure. Reaction progress was monitored via 1H NMR (DMSO‐d 6) for the disappearance of the starting material tert‐butyl singlet (δ 1.35). Once complete, the reaction mixture was concentrated on a rotary evaporator to remove the majority of the DCM. The resulting crude syrup was precipitated by the addition of Et2O (10× v/v, ∼1.5 L). The precipitation was cooled to 4 °C before the solids were collected by vacuum filtration and washed with additional Et2O to yield a sticky white solid (TFA‐amine salt). The crude material was transferred to a separatory funnel using saturated K2CO3:brine (1:1, v/v, 500 mL, 0.1 g PEG/mL) to assist in the transfer. The aqueous mixture was extracted with DCM (6 × 250 mL, 0.2 g PEG/mL). The combined organic layers were stirred with a generous amount of 4 Å activated powdered molecular sieves and MgSO4 for 30 min before filtering. The clear filtrate was concentrated on a rotary evaporator to a volume of ∼200 mL (0.25 g PEG/mL) and precipitated with the addition of Et2O (10× v/v, ∼2 L). The precipitation was cooled to 4 °C for ∼12 h before the solids were collected by vacuum filtration and washed with additional Et2O. The product was dried under reduced pressure to yield H2N‐PEG‐N3 (1c) as a white solid (yield typically >80%).
1H NMR (400 MHz, DMSO‐d 6, δ, ppm): 3.70–3.30 (m, ∼n × 4H), 3.41–3.36 (m, 2H overlapped with 13C satellites), 2.69 (t, J = 5.7 Hz, 2H).
Synthesis of trityl‐PEG‐OMs (10)
Prepared from trityl‐PEG‐OH (9) following the same method described above for the synthesis of mPEG‐OMs (4).
1H NMR (400 MHz, CD3CN, δ, ppm): 7.47–7.45 (m, 6H), 7.35–7.24 (m, 9H), 4.32–4.30 (m, 2H), 3.62–3.48 (m, ∼n × 4H), 3.15–3.13 (m, 2H), 3.06 (s, 3H).
Synthesis of trityl‐PEG‐N3 (11)
Prepared from trityl‐PEG‐OMs (10) following the same method described above for the synthesis of mPEG‐N3 (1a).
1H NMR (400 MHz, CD3CN, δ, ppm): 7.47–7.44 (m, 6H), 7.35–7.24 (m, 9H), 3.64–3.50 (m, ∼n × 4H), 3.38–3.35 (m, 2H overlapped with 13C satellites), 3.15–3.13 (m, 2H).
Synthesis of HO‐PEG‐N3 (1d)
Trityl‐PEG‐N3 (11) (100 g, 1 eq.) was dissolved in 0.1 M H2SO4 (1000 mL, 0.1 g PEG/mL) and stirred at room temperature. Within a few minutes the reaction became hazy with a white precipitant (trityl‐OH). After a total of 2.5 h, the reaction mixture was neutralized with 2 M NaOH. NaCl (360 g, to give ∼0.36 g/mL) was added, the mixture was stirred for 30 min, and then filtered through a Millipore Durapore™ 0.45 μm membrane. The clear aqueous filtrate was extracted with DCM (15 × 200 mL, 0.5 PEG g/mL). The combined organic layers were dried with MgSO4 and 4 Å molecular sieves, filtered, concentrated to ∼400 mL (0.25 g PEG/mL), and then precipitated with MTBE (10× v/v, ∼4 L). The precipitation was cooled to 4 °C for ∼12 h before the solids were collected by vacuum filtration and washed with additional MTBE. The product was dried under reduced pressure to yield HO‐PEG‐N3 (1d) as a white solid (yield typically >95%).
1H NMR (400 MHz, CD3CN, δ, ppm): 3.64–3.47 (m, ∼n × 4H), 3.38–3.35 (m, 2H overlapped with 13C satellites), 2.78 (t, J = 5.8 Hz, 1H).
Synthesis of CO2H‐PEG‐N3 (1e)
HO‐PEG‐N3 (1d) (20 g, 1 eq.) was dissolved in H2O (100 mL, 0.2 PEG g/mL) with the assistance of a heat gun. Once cooled to room temperature, KBr (0.5 eq.) was added. The reaction vessel was fitted with a thermometer and a pH meter. The internal temperature of the reaction was lowered to 2–5 °C with an ice‐water bath. The reaction was charged with 2,2,6,6‐tetramethylpiperidine‐1‐oxyl (TEMPO, 0.007 eq.) as a CH3CN solution (200 mg/mL) followed by the addition of cold 5% bleach (125 mL, adjusted to pH ∼10 using NaH2PO4). The temperature of the exothermic reaction was kept below 15 °C with the ice‐water bath. The pH of the reaction was maintained between 9.5 and 10.5 using 2 M K3PO4. After 3 h (once the pH stabilized), the reaction was quenched by the addition of 12 mL of ethanol (∼10% of total bleach volume). The pH was maintained between 9.5 and 10.5 using 2 M NaOH during the quench. Afterwards, the reaction was allowed to warm to room temperature. The pH of the reaction was adjusted to ∼2 using 4 M HCl prior to the addition of NaCl (90 g, to give ∼0.36 g/mL). After the majority of the NaCl had dissolved (∼20 min) the reaction was extracted into DCM (15 × 100 mL, 0.2 g PEG/mL). The combined organic layers were dried with MgSO4 and 4 Å molecular sieves, filtered, concentrated to ∼100 mL (0.2 g PEG/mL) and precipitated with MTBE (12× v/v, ∼1.2 L). The precipitation was cooled to 4 °C for ∼12 h before the solids were collected by vacuum filtration and washed with additional cold MTBE. The product was dried under reduced pressure to yield CO2H‐PEG‐N3 (1e) as a white solid (yield typically >90%).
1H NMR (400 MHz, CD3CN, δ, ppm): 4.05 (s, 2H), 3.66–3.49 (m, ∼n × 4H), 3.38–3.35 (m, 2H overlapped with 13C satellites).
Synthesis of mPEG‐Triazole(CO2Et)2 (2a)
A round‐bottom flask fitted with a condenser was charged with mPEG‐N3 (1a) (500 mg, 1 eq.) and CH3CN (5.0 mL, 100 mg PEG/mL). After dissolution, DEBD (2 eq.) was added and the reaction mixture was heated to reflux. To monitor the reaction: an aliquot (0.3 mL) was taken, concentrated on a rotary evaporator, and further dried under reduced pressure. Reaction progress was monitored by changes in the ratio of the starting material R—OCH3 (1H NMR, δ 3.29 ppm, CD3CN) to product CH2‐triazole (1H NMR, δ 4.74 ppm, CD3CN). After completion, the reaction mixture was concentrated on a rotary evaporator. The resulting crude material was dissolved in DCM (1.5 mL, 333 mg PEG/mL) and precipitated with Et2O (20×, v/v, ∼30 mL). The solids were collected by vacuum filtration and washed with additional Et2O. The product was dried under reduced pressure to yield mPEG‐triazole(CO2Et)2 (2a) as a white solid (yield typically >85%).
1H NMR (400 MHz, CD3CN, δ, ppm): 4.74 (t, J = 5.1 Hz, 2H), 4.38 (dq, J 1 = 7.1, J 2 = 17.8 Hz, 4H), 3.82 (t, J = 5.1 Hz, 2H), 3.60–3.44 (m, ∼n × 4H), 3.29 (s, 3H), 1.34 (q, J = 7.1 Hz, 6H).
Synthesis of BocNH‐PEG‐Triazole(CO2Et)2 (2b)
A round‐bottom flask fitted with a condenser was charged with BocNH‐PEG‐N3 (1b) (500 mg, 1 eq.) and CH3CN (5.0 mL, 100 mg PEG/mL). After dissolution, DEBD (2 eq.) was added and the reaction mixture was heated to reflux. To monitor the reaction: an aliquot (0.3 mL) was taken, concentrated on a rotary evaporator, and further dried under reduced pressure. Reaction progress was monitored by changes in the ratio of the starting material R—CH2NHBoc (1H NMR, δ 3.28 ppm, CD3CN) to product CH2‐triazole (1H NMR, δ 4.74 ppm, CD3CN). After completion, the reaction mixture was concentrated on a rotary evaporator. The resulting crude material was dissolved in DCM (1.5 mL, 333 mg PEG/mL) and precipitated with Et2O (20×, v/v, ∼30 mL). The solids were collected by vacuum filtration and washed with additional Et2O. The product was dried under reduced pressure to yield BocNH‐PEG‐triazole(CO2Et)2 (2b) as a white solid (yield typically >85%).
1H NMR (400 MHz, CD3CN, δ, ppm): 5.37 (br s, 1H), 4.74 (t, J = 5.1 Hz, 2H), 4.38 (dq, J 1 = 7.1, J 2 = 17.8 Hz, 4H), 3.82 (t, J = 5.1 Hz, 2H), 3.64–3.35 (m, ∼n × 4H), 3.17 (q, J = 5.7 Hz, 2H), 1.40 (s, 9H), 1.34 (q, J = 7.1 Hz, 6H).
Synthesis of HO‐PEG‐Triazole(CO2Et)2 (2d)
A round‐bottom flask fitted with a condenser was charged with HO‐PEG‐N3 (1d) (500 mg, 1 eq.) and CHCl3 (5.0 mL, 100 mg PEG/mL). After dissolution, DEBD (2–5 eq.) was added and the reaction mixture was heated to reflux. To monitor the reaction: an aliquot (0.3 mL) was taken, concentrated on a rotary evaporator, and further dried under reduced pressure. Reaction progress was monitored by changes in the ratio of the starting material R—OH (1H NMR, δ 2.78 ppm, CD3CN) to product CH2‐triazole (1H NMR, δ 4.74 ppm, CD3CN). After completion, the reaction mixture was concentrated on a rotary evaporator. The resulting crude material was dissolved in DCM (1.5 mL, 333 mg PEG/mL) and precipitated with Et2O (20×, v/v, ∼30 mL). The solids were collected by vacuum filtration and washed with additional Et2O. The product was dried under reduced pressure to yield HO‐PEG‐triazole(CO2Et)2 (2d) as a white solid (yield typically >80%).
1H NMR (400 MHz, CD3CN, δ, ppm): 4.74 (t, J = 5.2 Hz, 2H), 4.38 (dq, J 1 = 7.1, J 2 = 17.8 Hz, 4H), 3.82 (t, J = 5.2 Hz, 2H), 3.64–3.40 (m, ∼n × 4H), 2.79 (t, J = 5.5 Hz, 1H), 1.34 (q, J = 7.1 Hz, 6H).
Synthesis of CO2H‐PEG‐Triazole(CO2Et)2 (2e)
A round‐bottom flask fitted with a condenser was charged with CO2H‐PEG‐N3 (1e) (500 mg, 1 eq.) and CHCl3 (5.0 mL, 100 mg PEG/mL). After dissolution, DEBD (5 eq.) was added. To monitor the reaction: an aliquot (0.3 mL) was taken, concentrated on a rotary evaporator, and then further dried under reduced pressure. Reaction progress was monitored by changes in the ratio of the starting material CH2CO2H (1H NMR, δ 4.05 ppm, CD3CN) to product CH2‐triazole (1H NMR, δ 4.74 ppm, CD3CN). After completion, the reaction mixture was concentrated on a rotary evaporator and further dried under reduced pressure. The resulting crude material was dissolved in DCM (1.5 mL, 333 mg PEG/mL) and precipitated with Et2O (20×, v/v, ∼30 mL). The solids were collected by vacuum filtration and washed with additional Et2O. The product was dried under reduced pressure to yield CO2H‐PEG‐triazole(CO2Et)2 (2e) as a white solid (yield typically >85%).
1H NMR (400 MHz, CD3CN, δ, ppm): 4.74 (t, J = 5.1 Hz, 2H), 4.38 (dq, J 1 = 7.1, J 2 = 17.8 Hz, 4H), 4.05 (s, 2H), 3.82 (t, J = 5.1 Hz, 2H), 3.68–3.40 (m, ∼n × 4H), 1.34 (q, J = 7.1 Hz, 6H).
RESULTS AND DISCUSSION
Synthesis of PEG Azides
The present study utilized PEG starting materials encompassing three structural types: mPEG‐OH (3), Bn2N‐PEG‐OH (5), and trityl‐PEG‐OH (9).26 Within each class, PEGs of varying molecular weights (2–20 kDa, n ≈ 47–454) with tightly controlled PDIs (1.01–1.05 by GPC) were prepared. These PEG starting materials gave access to five classes of azides 1a‐e (Scheme 2). Synthesis of azide class 1a (mPEG‐N3) required transformation of the terminal hydroxyl in 3 to the corresponding azide through a mesylate intermediate 4. Initial mesylate syntheses attempts suffered from by‐products that were not easily removed by PEG precipitation and thus required flash column chromatography (FCC). Three changes to the synthesis eliminated the need for FCC. First, the starting PEG was dried via azeotropic distillation with toluene to remove trace amounts of moisture. Second, the reactants were dried and purified via vacuum distillation; mesyl chloride from P2O5 and triethylamine from CaH2. Finally, the crude reaction mixture was washed with brine‐saturated 1 M HCl to remove any mesylate and triethylamine salts. Precipitation of the crude product from CH2Cl2/Et2O or MTBE (1:10 v/v) yielded the pure mesylate intermediate 4 in high yield. Displacement of the mesylate with excess sodium azide in refluxing ethanol proceeded smoothly to yield a series of azides 1a. Synthesis of azide class 1b (BocNH‐PEG‐N3) began with the heterogeneous catalytic hydrogenation of dibenzyl protected PEG amine 5 using Pearlman's catalyst under aqueous conditions. The subsequent free amine 6 was reacted with di‐tert‐butyl dicarbonate in an aqueous solution of K2CO3 to form Boc‐amine 7. An identical procedure previously described provided azide 1b through mesylate 8. Deprotection with TFA in methylene chloride (1:1, v/v) provided azide class 1c (H2N‐PEG‐N3). Azide class 1d and 1e were both prepared from triphenylmethyl (trityl) protected PEG 9 that was converted to its azide derivative 11 via mesylate 10 without issue. Dilute sulfuric acid facilitated the hydrolysis of the trityl group to provide azide class 1d (HO‐PEG‐N3). The final azide class (1e, CO2H‐PEG‐N3) was prepared via an oxidation protocol employing a catalytic amount of 2,2,6,6‐tetramethylpiperidine 1‐oxyl (TEMPO) and in situ generated hypobromite as the regenerating oxidant.27
Scheme 2.
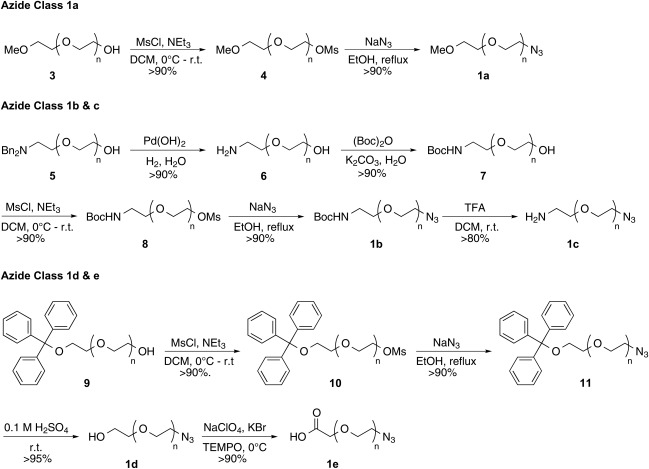
Syntheses of azido PEG derivatives.
For simplicity, we wanted to assess the level of azide incorporation in our PEG derivatives directly by 1H NMR spectroscopy. Unfortunately, regardless of the azide class or molecular weight, we found it difficult to measure the degree of azide incorporation in our polymers by routine NMR analysis. The chemical shifts of the newly formed PEG‐CH 2—N3 methylene protons tended to overlap with the 13C satellite peaks associated with the PEG manifold typically observed at δ ∼ 3.35–3.40 ppm (Fig. 1). This issue was especially pronounced with the higher molecular weight (∼10–20 kDa) PEG analogs that have larger satellite signals. Changing NMR solvents did not completely alleviate this issue. High Performance Liquid Chromatography (HPLC) was considered, however, these methods often failed to adequately resolve subtle functional group changes (e.g., R—OMs to R—N3) in these macromolecules. To overcome this limitation, we sought to identify a simple, rapid, and robust derivatization procedure that would afford products whose 1H NMR signals were distinctly different and shifted far away from the interfering PEG proton manifold.
Figure 1.
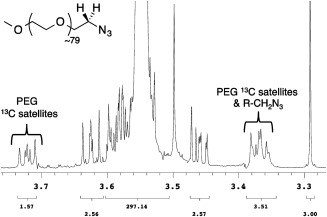
1H NMR (CD3CN) of mPEG3.5K‐N3.
We envisioned that a click reaction between our azido PEGs and a suitable dipolarophile would provide a triazole derivative amenable to standard NMR quantification methods. Ideally, the cycloaddition would proceed without the need for a metal catalyst, which could complicate work ups and interfere with the NMR spectrum. As a model, we screened mPEG3.5K‐N3 (class 1a) against four dipolarophiles: diphenyl‐ and phenylacetylene (DPA and PA respectively); and dimethyl‐ and diethyl acetylenedicarboxylate (DMAD and DEBD respectively). All uncatalyzed reactions with the two aryl alkynes (Table 1, Entries 1–3), returned starting material. DMAD reactions with D2O or MeOH (Entries 4–5) did yield the desired product, albeit alongside by‐products detected by NMR. Shorter reaction time at 70 °C in D2O alleviated this issue (Entry 6); although, PEG work ups from aqueous conditions are more complicated than those from organic solvents. Ultimately, DEBD proved to be the most efficient dipolarophile screened. Reactions in ethanol (Entries 7–8), with and without copper wire catalysis, provided the desired triazole, although the former suffered from lower conversion and contained by‐products. DEBD reactions in aprotic solvents (Entries 9–10) were also successful, with acetonitrile proceeding in a significantly shorter time than deuterated chloroform.
Table 1.
Survey of Dipolarophiles and Reaction Conditions 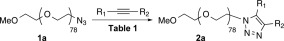
| Entry | Alkyne | Solvent | Temp. (time) | Conv. (%) |
|---|---|---|---|---|
| 1 | DPA | CH3CN | reflux (26h) | 0 |
| 2 | DPA | EtOH | reflux (32h) | 0 |
| 3 | PA | CH3CN | reflux (24h) | 0 |
| 4 | DMAD | MeOH | reflux (4 h) | 93a |
| 5 | DMAD | D2O | r.t. (12 h) | 50a |
| 6 | DMAD | D2O | 70 °C (3 h) | >99 |
| 7b | DEBD | EtOH | 30 °C (96 h) | 86a |
| 8 | DEBD | EtOH | reflux (10h) | >99 |
| 9 | DEBD | CDCl3 | reflux (32h) | >99 |
| 10 | DEBD | CH3CN | reflux (4 h) | >99 |
By‐products detected in NMR.
Cu wire catalyst added.
The 1H NMR (400 MHz, CD3CN) spectrum of the resultant click adduct showed several pronounced new signals that were clearly resolved from the PEG proton manifold. Particularly useful are the R—CH2‐triazole protons and the ester methylene protons (R—CO2CH2CH3) located at δ 4.74 and 4.38, respectively (Fig. 2, protons a and b/c). These distinct signals are far enough downfield to avoid interference from the PEG manifold and satellites protons. Azide substitution was determined by comparing the reference signals from the original azido PEG starting material (i.e., R‐OMe, protons He) to the new triazole related protons (H a‐d,f‐g) and averaging the corresponding values. This gives an approximate incorporation value of >99% for this azido PEG derivative.
Figure 2.
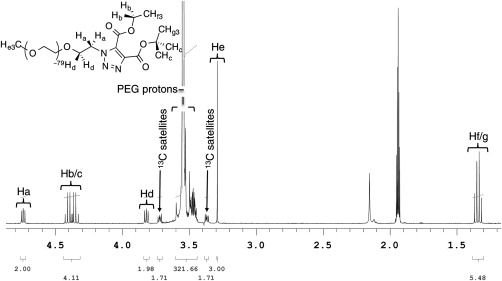
1H NMR (CD3CN, 400 MHz) of DEBD click adduct.
With suitable mild reaction conditions in hand, we surveyed PEGs of various molecular weights across the five azide classes using DEBD as the dipolarophile (Table 2). Each of the methoxy and NHBoc azido PEGs (Entries 1–4) screened were easily converted to their corresponding triazoles and were found to have >99% azide substitution. At ambient temperature, the primary amines tested did not yield cycloaddition products; at elevated temperatures, Michael addition products between the amine and the alkyne were detected (Entries 5–6). The hydroxyl azido PEGs produced minor inseparable by‐products in acetonitrile and THF above room temperature (Entries 8–10); substituting chloroform as the solvent alleviated this issue. The carboxylate PEGs also worked best in chloroform as long as the reaction was not heated (Entries 16–17). The degree of azide substitution was found to be >99% for each of the hydroxyl and carboxylate azido PEGs measured. In all cases, the substitution values derived will always suffer from some imprecision due to the inherent error in NMR integration which is dependent on the instrument used and its optimization. Click adducts from larger PEG azides (>15 kDa) are best analyzed by 600 MHz 1H NMR which helps reduce signal‐to‐noise problems.
Table 2.
Azido PEG Reactions with DEBD
| Entry | R | Mol. Wt. | DEBD Eq. | Solvent | Temp. (time) | Yield (%) | N3 Substitution (%) |
|---|---|---|---|---|---|---|---|
| 1 | OMe | 3.5 | 2.0 | CH3CN | reflux (4 h) | 91 | >99 |
| 2 | OMe | 18.6 | 2.0 | CH3CN | reflux (36 h) | 97 | >99 |
| 3 | NHBoc | 2.1 | 2.0 | CH3CN | reflux (16 h) | 93 | >99 |
| 4 | NHBoc | 12.3 | 2.0 | CH3CN | reflux (27 h) | 89 | >99 |
| 5 | NH2 | 2.1 | 2.0 | CH3CN | reflux (15 h) | –a | – |
| 6 | NH2 | 12.5 | 2.0 | CH3CN | reflux (38 h) | –a | – |
| 7 | OH | 5.0 | 2.0 | CH3CN | reflux (8 h) | 89b | >99 |
| 8 | OH | 11.1 | 1.2 | CH3CN | 40 °C (36 h) | –b | – |
| 9 | OH | 11.1 | 2.0 | CH3CN | 75 °C (36 h) | –b | – |
| 10 | OH | 11.1 | 2.0 | THF | 60 °C (16 h) | –b | – |
| 11 | OH | 2.1 | 2.0 | CHCl3 | 40 °C (72 h) | 83 | >99 |
| 12 | OH | 2.1 | 5.0 | CHCl3 | reflux (24 h) | 88 | >99 |
| 13 | OH | 11.1 | 1.2 | CHCl3 | 40 °C (72 h) | 88 | >99 |
| 14 | OH | 11.1 | 3.0 | CHCl3 | 65 °C (48 h) | 85 | >99 |
| 15 | CO2H | 2.1 | 5.0 | CHCl3 | reflux (24 h) | –b | – |
| 16 | CO2H | 2.1 | 5.0 | CHCl3 | r.t. (36 h) | 85 | >99 |
| 17 | CO2H | 11.1 | 5.0 | CHCl3 | r.t. (36 h) | 96 | >99 |
Mixture of click and Michael addition products.
By‐products detected in NMR.
CONCLUSIONS
In summary, a simple and robust method to determine the level of azide incorporation in PEG derivatives was demonstrated. Conversion of PEG azides to their corresponding 1,2,3‐triazoles followed by simple 1H NMR integration provides a convenient quantification method that may find utility in this field as well as other areas of synthetic chemistry. DEBD was found to be the ideal dipolarophile for this protocol; offering the optimal combination of catalysis‐free reactivity, reaction rate, and yield. The 1H NMR spectra of the resulting click adducts contain distinct signals isolated downfield of the PEG manifold protons and satellites. This allows for the quantification of azide incorporation in azido PEG derivatives. The method described is compatible with a variety of functional groups including methoxy, hydroxyl, Boc‐protected amines, and carboxylates. In addition to the examples presented herein, we envision extension of this protocol to azido PEGs of higher or lower molecular weights than those demonstrated, bearing alternative functional groups not included in this study.
Supporting information
Supporting Information
REFERENCES AND NOTES
- 1. Hamley I. W., Biomacromolecules 2014, 15, 1543–1559. [DOI] [PubMed] [Google Scholar]
- 2. Veronese F. M., Biomaterials 2001, 22, 405–417. [DOI] [PubMed] [Google Scholar]
- 3. Chapman A. P., Adv. Drug Deliv. Rev. 2002, 54, 531–545. [DOI] [PubMed] [Google Scholar]
- 4. Shepherd J., Brodin H., Cave C., Waugh N., Price A., Gabbay J., Health Technol. Assess. 2004, 8. [DOI] [PubMed] [Google Scholar]
- 5. Jain A., Jain S. K., Crit. Rev. Ther. Drug Carrier Syst. 2008, 25. [DOI] [PubMed] [Google Scholar]
- 6. Immordino M. L., Franco D., Cattel L., Int. J. Nanomed. 2006, 1, 297–315. [PMC free article] [PubMed] [Google Scholar]
- 7. Barkey N. M., Preihs C., Cornnell H. H., Martinez G., Carie A., Vagner J., Xu L., Lloyd M. C., Lynch V. M., Hruby V. J., Sessler J. L., Sill K. N., Gillies R. J., Morse D. L., J. Med. Chem. 2013, 56, 6330–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kataoka K., Harada A., Nagasaki Y., Adv. Drug Deliv. Rev. 2001, 47, 113–131. [DOI] [PubMed] [Google Scholar]
- 9. Veronese F. M., Pasut G., Drug Discov. Today 2005, 10, 1451–1458. [DOI] [PubMed] [Google Scholar]
- 10. Paolo C., Veronese F. M., Adv. Drug Deliv. Rev. 2003, 55, 1261–1277. [DOI] [PubMed] [Google Scholar]
- 11. Garay R. P., Labaune J. P., Open Conf. Proc. J. 2011, 2, 104–107. [Google Scholar]
- 12. Roberts M. J., Bentley M. D., Harris J. M., Adv. Drug Deliv. Rev. 2012, 64, 116–127. [Google Scholar]
- 13. Giorgi E. M., Agusti R., de Lederkremer R. M., Beilstein J. Org. Chem. 2014, 10, 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Monfardini C., Schiavon O., Caliceti P., Morpurgo M., Harris J. M., Veronese F. M., Bioconjugate Chem. 1995, 6, 62–69. [DOI] [PubMed] [Google Scholar]
- 15. Morpurgo M., Veronese F. M., Kachensky D., Harris J. M., Bioconjugate Chem. 1996, 7, 363–368. [DOI] [PubMed] [Google Scholar]
- 16. Huisgen R., Angew. Chem. 1963, 75, 604–637. [Google Scholar]
- 17. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- 18. Majid M. H., Hamidi H., Zadsirjan V., Curr. Org. Synth. 2014, 11, 647–675. [Google Scholar]
- 19. Luisa G., Molteni G., Tetrahedron Lett. 2003, 44, 1133–1135. [Google Scholar]
- 20. Tunca U., J. Polym. Sci. Part A: Polym. Chem. 2014, 52, 3147–3165. [Google Scholar]
- 21. Tang W., Becker M. L., Chem. Soc. Rev. 2014, 43, 7013–7039. [DOI] [PubMed] [Google Scholar]
- 22. Presolski S. I., Hong V. P., Finn M. G., Curr. Protoc. Chem. Biol. 2011, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hiki S., Kataoka K., Bioconjugate Chem. 2010, 21, 248–254. [DOI] [PubMed] [Google Scholar]
- 24. Parrish B., Breitenkamp R. B., Emrick T., J. Am. Chem. Soc. 2005, 127, 7404–7410. [DOI] [PubMed] [Google Scholar]
- 25. Breitenkamp K., Sill K. N., Skaff H.. U.S. Patent 8,212,083, July 3, 2012.
- 26. Cardoen G., Burke B., Sill K., Mirosevich J., J. Polym. Res. 2012, 19, 1–10. [Google Scholar]
- 27. Li X. Q., Meng F. T., Su Z. G., J. Chem. Res. 2005, 5, 280–281. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information