

ABSTRACT
A genetic recombinant Saccharomyces cerevisiae starter with high ethanol tolerance capacities was constructed. In this study, the gene of trehalose-6-phosphate synthase (encoded by tps1), which catalyzes the first step in trehalose synthesis, was cloned and overexpressed in S. cerevisiae. Moreover, the gene of neutral trehalase (encoded by nth1, trehalose degrading enzyme) was deleted by using a disruption cassette, which contained long flanking homology regions of nth1 gene (the upstream 0.26 kb and downstream 0.4 kb). The engineered strain increased its tolerance against ethanol and glucose stress. The growth of the wild strain was inhibited when the medium contained 6 % or more ethanol, whereas growth of the engineered strain was affected when the medium contained 10 % or more ethanol. There was no significant difference in the ethanol yield between the wild strain and the engineered strain when the fermentation broth contained 10 % glucose (p > 0.05). The engineered strain showed greater ethanol yield than the wild type strain when the medium contained more than 15 % glucose (p < 0.05). Higher intracellular trehalose accumulation by overexpression of tps1 and deletion of nth1 might provide the ability for yeast to protect against environmental stress.
KEYWORDS: Ethanol tolerance, neutral trehalase, PCR-based gene disruption, Saccharomyces cerevisiae, trehalose-6-phosphate synthase, Trehalose
Introduction
Saccharomyces cerevisiae converts glucose to ethanol under anaerobic conditions. S. cerevisiae is widely used as a starter for production of ethanol; generally, S. cerevisiae shows a threshold level of 4–6 % ethanol.1 Further, Rose 2 showed that the growth of S. cerevisiae was inhibited when the medium contained an ethanol concentration as low as 2 %. When yeasts cell are exposed to external ethanol, ethanol not only changes the lipid composition of the plasma membrane, resulting in the destruction of its integrity, but also impairs the biosynthesis of endogenic proteins, therefore affecting transportation of nutrients as a result of inefficient fermentation.3 Apart from the effects of external ethanol on yeast cells, yeast cells decrease in viability and ethanol production due to accumulation of intracellular ethanol during ethanol fermentation.4 Levels of ethanol tolerance in yeast cells are related to the structure of cell membranes, activity of mitochondria, induction of heat shock proteins, and the concentration of trehalose.5 To diminish the latter processes for ethanol production, such as distillation and concentration, by improving ethanol tolerance in fermentation starters, many researchers have studied the relationship of ethanol tolerance and trehalose concentration in yeast cells.6,7
Trehalose is a non-reducing disaccharide that plays several important bio-functions in yeast, bacteria, fungi, insects, and plants.8 Several researchers demonstrated that trehalose is not only a storage carbohydrate but a protectant in yeast cells. When yeast suffers from environmental stress during growth, yeast regulates synthesis of trehalose; in the meantime, trehalose functions as a potential protectant by preserving the integrity of the plasma membrane and stabilizing proteins in cells.9 In other words, concentration of trehalose in cells is an index of stress tolerance.10,11
In yeast cells, the concentration of trehalose is regulated by 2 synthesis enzymes, trehalose-6-phosphate synthase (TPS, encoded by tps1) and trehalose-6-phosphate phosphatase (TPP, encoded by tps2 in yeast), and 2 degradation enzymes, neutral trehalases encoded by nth1 and nth2 and acid trehalase encoded by ath1.12,8 To accumulate high levels of trehalose in yeast cells, genetic strains with overexpression of tps1 or tps2 or deletion of nth1, nth2 or ath1 were constructed.13,14 In the present study, a S. cerevisiae recombinant with tps1 overexpression and nth1 deletion exhibited higher trehalose accumulation during growth conditions with or without stress. This engineered strain was able to tolerant high ethanol concentration and achieved higher ethanol yields in presence of high initial glucose concentrations.
Materials and methods
Strains and media
All of the strains used in this study are listed in Table 1. Escherichia coli TOP10F’ was purchased from Novagen Inc. (Wisconsin, USA). S. cerevisiae (BCRC 21685) was purchased from Bioresource Collection and Research Center, Food Industry Research and Development Institution, Hsinchu, Taiwan. E. coli was maintained and cultivated in Luria-Bertani (LB) medium, which contains 10 g/L peptone, 10 g/L NaCl, and 5 g/L yeast extract at 37°C. Yeast strains were maintained by YPD (10 g/L yeast extract, 20 g/L peptone, 20 g/L dextrose) and cultivated in YPD liquid broth at 28°C. To select the E. coli transformants based on the zeocin resistance, low salt LB (10 g/L peptone, 5 g/L NaCl, and 5 g/L yeast extract) plates were supplemented with 25 mg/L Zeocin (Invitrogen Corp., Carlsbad, California, USA) and for kanamycin resistance, 50 mg/L kanamycin sulfate (USB, Cleveland, USA) was added to LB plate.15 For selection of zeocin and G418 resistance after yeast transformations, the YPD plates were supplemented with 100 mg/L Zeocin and 200 mg/L G418 (Sigma Chemical Co., St Louis, MO), respectively. DNA manipulations were performed according to standard protocols.16
Table 1.
Strains used in this study.
| Strain Name | Properties or product | Reference or source |
|---|---|---|
| E. coli TOP10F' | F’ [lacIq Tn10(Tetr)] mcrA (mrr-hsdRMS-mcrBC) 80lacZM15lacX74 recA1araD139 (ara-leu)7697 galUgalKrpsL (Strr) endA1 nupG | Invitrogen |
| S. cerevisiae (BCRC 21685) (SC) | Wild type | Bioresources Collection and Research Center, Taiwan |
| S. cerevisiae with overexpression of tps1 (SCT) | Carrying pGAPZC-tps1 vector | This study |
| S. cerevisiae with overexpression of tps1 and deletion of nth1 gene (SCTΔN) | Carrying pGAPZC-tps1 vector and gene disruption cassette (Upstream nth1 region-kanMX6-Downstream nth1 region replaces the nth1 gene). | This study |
Plasmids and genomic DNA
The expression vector pGAPZαC was purchased from Invitrogen (Carlsbad, CA, USA) and used for tps1 gene overexpression study. The pFA6a-kanMX6 vector, a gift from Dr. John Pringle's lab, was used to amplify the kanr marker gene.17 The pGEMT vector (Promega Corporation, Madison, WI, USA) with a single 3´-terminal thymidine at both ends was used for ligation of PCR products. Recombinant vector was multiplied in E. coli TOP10F’. Plasmids within the host cells were extracted using Gene-spin miniprep plasmid purification kit (Protech Technology, Taipei, Taiwan).
Yeast genomic DNA was prepared according to the manufacturer's protocol (Genomic DNA purification kit, BioKit, Miaoli, Taiwan).
Primers
Table 2 lists the primers used in this study. These primers were designed according the website of NCBI (National Center for Biotechnology Information) and Saccharomyces Genome Deletion Project (http://www-sequence.stanford.edu/group/yeast_deletion_project/project_desc.html) for nucleotide sequences of tps1 gene (GenBank ID:NM_001178474.1), nth1 gene (GenBank ID:NM_001180309.1) and genomic DNA of S. cerevisiae and pFA6a-kanMX6, respectively.
Table 2.
Oligonucleotides used in this study.
| Oligonucleotide | Sequence 5′–3′ | Primer design | Purpose | Source |
|---|---|---|---|---|
| Tps1-F | ACTTTCGAAACGATGGGTACTACGGATAACGCTAAGGCGCAACTGACCT | BstBI | Amplification of tps1 gene | This study |
| Tps1-R | ACTGGTACCTCAGTGATGGTGATGGTGATGGTTTTTGGTGGCAGAGGAG | KpnI | ||
| VTF | TTCGAAACGATGGGTACTAC | Positioned from the start codon of the tps1 gene | Verification of tps1 gene insertion | |
| VR | AGTGATGGTGATGGTGATGG | 6xHis-tag sequence | ||
| L1 | CACAAGGTTATCAATATGAAACTTGCGCGATCACCGATTTA | Positioned 265 bp from the start codon of the nth1 gene | To construct upstream region of nth1 gene | |
| L2 | GTCGACCTGCAGCGTACGTGGACCCTTGTGGTACTGTCAGAGACCTCGTGGACATCCATTCTACTGATTTTTTT | 5′ end homologous to 5′ kanMX region | ||
| L3 | CGAGCTCGAATTCATCGATGTTGAGCGTGTGTTAATACCCTACGAGACCGACACCGTAGTTGAGATAATATATA | 5′ end homologous to 3′ kanMX region | To construct downstream region of nth1 gene | |
| L4 | AAGAAAAAGAAACTGTTTTGTCCTTGGAAAAAAAGCACTACCTA | Positioned 391 bp from the stop codon of the nth1 gene | ||
| kanMX-F | AAAAAAATCAGTAGAATGGATGTCCACGAGGTCTCTGACAGTACCACAAGGGTCCACGTACGCTGCAGGTCGAC | 5′ end had 18 bases extensions homologous to the 3′ side of the UP nth1 region. | To construct kanMX region | Saccharomyces Genome Deletion Project. |
| kanMX-R | TATATATTATCTCAACTACGGTGTCGGTCTCGTAGGGTATTAACACACGCTCAACATCGATGAATTCGAGCTCG | 5′ end had 18 bases extensions homologous to the 5′ side of the DOWN nth1 region | ||
| DC-F | CCTATCGTTTTCCGTAGAGT | Located within the UP nth1 region | For amplification of disruption cassette | This study |
| DC-R | CCTGAAGTCGAGTAAACAGA | Located within the DOWN nth1 region | ||
| A1 | CAGGAGCATCGTGTAGGAC | Positioned 373 bp from the start codon of the nth1 gene | Gene deletion verification primer | This study |
| D1 | AAAAGGTCACATGCTTATAATCAAC | Positioned 514 bp from the stop codon of the nth1 gene | ||
| B | AAACGACGATTACCTTGACTACTTG | Located within the coding region of the nth1 gene | Gene deletion verification primer | Saccharomyces Genome Deletion Project. |
| kanB | CTGCAGCGAGGAGCCGTAAT | Positioned 250 bp downstream of the 5 ′-end of the kanMX region | ||
| C | TTGTGGTTGAAAAATATGATGTCAC | Located within the coding region of the nth1 gene | ||
| kanC | TGATTTTGATGACGAGCGTAAT | Positioned 587 bp upstream of the 3 ′ end of the kanMX region | ||
| Nth1F | ATGAGTCAAGTTAATACAAGCCAAG | Positioned from the start codon of the nth1 gene | For amplification of nth1 gene | This study |
| Nth1R | TAGTCCATAGAGGTTTCTTTCTTG | Positioned from the stop codon of the nth1 gene |
The underlined bases encode restriction site and italicized bases encode 6xHis-tag.
Saccharomyces Genome Deletion Project. (http://www-sequence.stanford.edu/group/yeast_deletion_project/project_desc.html)
Construction of tps1 overexpression strain
Construction of recombinant pGAPZC-tps1 vector
Total RNA was isolated from the S. cerevisiae wild strain, using a TRIzol reagent (Life Technologies, Inc., Grand Island, NY) and reverse transcription of RNA was performed by using M-MLV Reverse Transcriptase enzyme (Promega Corporation, Madison, WI, USA) according to the manufacturer's instructions. The resulting cDNA (cDNA) was used to amplify tps1 gene by PCR using primer Tps1-F and Tps1-R.
The PCR reaction mixture contained 1x Pfu Turbo Cx buffer, 0.2 µM each primer, 0.2 mM each dNTP (Promega Corporation, Madison, WI, USA) and 2.5 units Pfu Turbo Cx polymerase (Agilent Technologies CA, USA). PCR amplifications were carried out as; pre-incubation at 95°C for 2 min, then 30 cycles of 30 sec at 95°C, 30 sec at 58°C, and 2 min at 72°C, and 10 min of extension at 72°C. DNA thermal cycler (Labcycler Gradient, SENSQUEST, Gottingen, Germany) was used to obtain the PCR product and the PCR product was analyzed by agarose gel electrophoresis and purified with a Clean/Gel Extraction Kit (BioKit, Miaoli, Taiwan).
The BstBI-KpnI fragment carrying the tps1 gene and the expression vector pGAPZαC were digested with BstBI and KpnI and ligated together. Ligation products were transformed into CaCl2-treated E. coli TOP10F’ according to Hanahan and Meselson.18 The resulting vector was designated as pGAPZC-tps1 and used for yeast transformation
Integration of recombinant vector pGAPZC-tps1 into S. cerevisiae
The recombinant vector pGAPZC-tps1 was linearized at the AvrII site prior to transformation. Yeast transformation was performed by electroporation method according to the manufacture's protocol (MicroPulser electroporation apparatus, Bio-Rad Laboratories, Hercules, CA, USA). The positives colonies were selected by plating transformants on YPDS plates (10 g/L yeast extract, 20 g/L peptone, 20 g/L dextrose, 1 M sorbitol) containing 100 mg/L Zeocin. Transformants were confirmed by PCR using VTF as forward primers and VR as reverse primer. The engineered strain with tps1 overexpression was preserved, named as SCT and further used for nth1 gene deletion.
Construction of nth1 deletion strain
Construction of recombinant pGEMT-kanMX vector
The Recombinant pGEMT-kanMX vector was constructed by method of overlapping PCR. In each reaction, 2 types of DNA polymerase, Taq polymerase lacking 3′–5′ exonuclease activity and Pfu polymerase exhibiting 3′–5′ exonuclease activity, were used simultaneously to increase the efficiency of overlapping.19 The reaction mixture contained 0.2 mM of each dNTP (Promega Corporation, Madison, WI, USA), 0.2 µM of each primer, 1 U of GoTaq DNA Polymerase (Promega Corporation, Madison, WI, USA), and 0.2 U of Pfu Turbo Cx polymerase (Agilent Technologies, CA, USA).
In the first round of PCR, the upstream sequence of the nth1 gene (5′ UP nth1), the kanr marker gene and the downstream sequence of the nth1 gene (3′ DOWN nth1) were amplified independently (Fig. 1). Genomic DNA sequences of S. cerevisiae were used as templates for amplifications of the 5′ UP nth1 region (0.26 kb) with primer combination of L1 and L2, and the 3′ DOWN nth1 region (0.4 kb) with primer combination of L3 and L4. The pFA6a-kanMX6 vector was used for amplification of the kanMX region (1.6 kb) using primers kanMX-F and kanMX-R.
Figure 1.
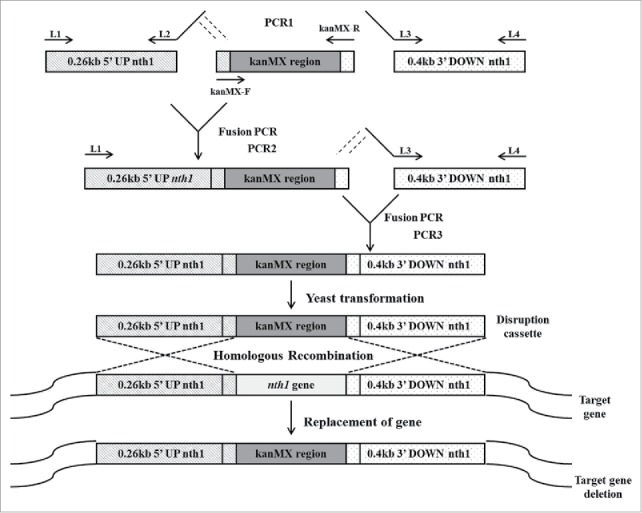
Construction of deletion cassette with long flanking homology regions by overlapping PCR. In the first round of PCR, the upstream sequence of the nth1 gene, the kanr marker gene and the downstream sequence of the nth1 gene were amplified in 3 different PCR reaction tubes. S. cerevisiae genomic DNA was used as template for amplification of 0.26 kb 5′ UP nth1 region and 0.4 kb 3′ DOWN nth1 region by using primers L1, L2, and L3, L4, respectively. The pFA6a-kanMX6 vector was used as a template for amplification of kanMX region by using primers kanMX-F and kanMX-R. In the second round of PCR, the 5′ UP nth1 region and kanMX region were fused together by using primers L1 and kanMX-R. In the third round of PCR, the 3′ DOWN nth1 region and second round PCR product (5′ UP nth1 region-kanMX region) were fused together by using primers L1 and L4. The third round PCR product (5′ UP nth1 region- kanMX region - 3′ DOWN nth1 region) was cloned into pGEMT vector for construction of the disruption cassette. After transformation of the disruption cassette into S. cerevisiae, homologous recombination takes place with the deletion of target gene.
In the second round of PCR, the 5′ UP nth1 region and the kanMX region were fused. L1 and kanMX-R were used as primers, and the PCR products of 5′ UP nth1 and kanMX from the first PCR round served as DNA templates.
In the third round of PCR, 3′ DOWN nth1 region (the PCR product resulting from the first round of PCR) and 5′ UP nth1 region-kanMX region (the PCR product resulting from the second round of PCR) were fused using primers L1 and L4.
All PCR mixtures were pre-incubated at 95°C for 2 min, and reactions were performed for 30 cycles of 30 sec at 95°C, 30 sec at 55°C, and 2 min at 72°C, and 10 min of extension at 72°C in a DNA thermal cycler (Labcycler Gradient, SENSQUEST, Gottingen, Germany).
The 5′ UP nth1-kanMX-3′ DOWN nth1, obtained from the third round of PCR, was ligated with pGEMT vector (Promega Corporation, Madison, WI, USA) and transformed into CaCl2-treated E. coli TOP10F’ according to Hanahan and Meselson.18 The transformed host was screened by blue/white selection. The resulting plasmid was designated as pGEMT-kanMX vector and sequenced using T7 and SP6 as sequencing primers.
Construction of disruption cassette for nth1 gene deletion
To construct the disruption cassette for nth1 deletion, the recombinant pGEMT-kanMX vector was used as a template and the PCR reaction was conducted using Pfu Turbo Cx polymerase (Agilent Technologies, CA, USA) with primers, DC-F and DC-R (Table 2). After a pre-incubation of 95°C for 2 min, the disruption cassette was cloned with 30 cycles of PCR, which was initiated by 30 sec of denaturation at 95°C, 30 sec of annealing at 46°C, and 2 min of extension at 72°C, and then by 10 min extension at 70°C in a DNA thermal cycler (SENSQUEST, Gottingen, Germany). All PCR products were analyzed by agarose gel electrophoresis and purified with a Clean/Gel Extraction Kit (BioKit, Miaoli, Taiwan).
Integration of disruption cassette into S. cerevisiae
Aliquots of the disruption cassette were used for transformation of the S. cerevisiae by electroporation (MicroPulser electroporation apparatus, Bio-Rad Laboratories, Hercules, CA, USA). The colonies were selected by plating on YPDS plates (10 g/L yeast extract, 20 g/L peptone, 20 g/L dextrose, 1 M sorbitol) containing 200 mg/L G418 (Sigma Chemical Co., St Louis, MO, USA).
Yeast sporulation and separation of haploid spores
The G418-resistant transformants were grown on freshly prepared GNA presporulation plates (50 g/L dextrose, 30 g/L Nutrient broth, 10 g/L yeast extract, 20 g/L agar) at 30°C overnight prior to transfer to the sporulation medium (10 g/L potassium acetate, 0.05 g/L zinc acetate).20 Sporulation was induced by incubating cells at 25°C with shaking (50 rpm) for 5 d. To obtained haploid spores, tetrad-cells were washed with sterile water and lysed with lyticase (0.05 U/µL) at 37°C for 20 min. To grow diploid yeast cells, spores were spread on YPD plates containing 200 mg/L G418.
Confirmation of nth1 gene deletion
To validate the nth1 deletion in diploid yeasts, PCR was performed with GoTaq DNA Polymerase (Promega Corporation, Madison, WI, USA), and 4 sets of primers were used (Table 2 and Fig. 2). The appropriate replacement of the nth1 gene with kanMX was verified in the mutants by the appearance of PCR products of the expected size using different sets of primers (Fig. 2). Four ORF-specific confirmation primers (A1, B, C, and D1 primers) were used. The A1 and D1 primers were positioned 373 bp from the start and 514 bp from the stop codons of the nth1 gene, respectively. The B and C primers were located within the coding region of the ORF of nth1 gene. The KanB and KanC primers were the internal sequences of the KanMX module. The other 2 sets of primers (Nth1F and R, and L1 and L4) were used to distinguish between heterozygous diploid and homozygous diploid yeast cells. The Nth1F and Nth1R primers were in the interior position of the nth1 gene and were used to confirm the complete deletion of the nth1 gene. A primer set of L1 and L4 was positioned at the upstream and downstream region of the target gene, respectively.
Figure 2.
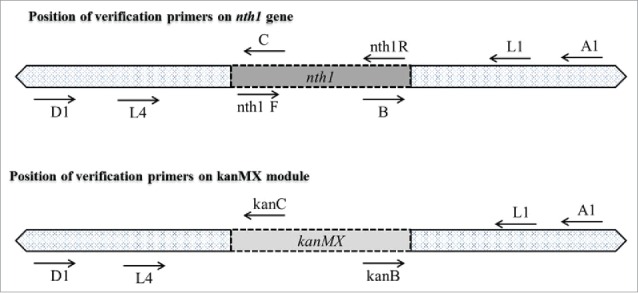
Schematic diagram of verification primer position.
The genomic DNA of transformed yeasts was used as a template. All PCR mixtures were pre-incubated at 95°C for 2 min, and then reactions were performed for 30 cycles of 30 sec at 95°C, 30 sec at 50°C, and 2 min at 72°C, and 10 min of extension at 72°C.
Measurement of protein expression
Frozen culture (1 mL) was incubated in 9 mL YPD broth at 30°C with shaking (150 rpm) overnight. The overnight culture was transferred into 100 mL of YPD broth to achieve an OD620 value of 0.3 and incubated in a 500 mL baffled Erlenmeyer Flask at 30°C with shaking (150 rpm) for 24 h. To evaluate the protein expression level, yeast pellet (100 mg) was used for extraction of proteins.21 SDS PAGE was performed according to Laemmli.22 The protein bands on the gel were transferred to polyvinylidene fluoride (PVDF) membrane (Perkin Elmer, Foster City, CA, USA) using Mini-Trans-Blot system (BioRad Laboratory, Inc., USA) at 4°C with a constant current of 30 V for overnight followed by 60 V for 90 min. The membrane was incubated with 5% dried milk powder in Tris-buffered saline Tween (TBST) (20 mM Tris, 140 mM NaCl, and 0.1% (v/v) Tween 20) to block non-specific binding. The membrane was incubated with primary antibody Anti-His Tag, clone HIS. H8 (Millipore, Schwalbach, Germany) and secondary antibody Peroxidase-conjugated AffiniPure Goat Anti-Mouse IgG (Jackson immunoresearch, West Grove, PA, USA) in a ratio of 1:5000. Chemiluminescence detection was performed with Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA) and the image was taken by a CCD-camera (Fusion-SL 3500.WL; Peqlab Biotechnologie, Erlangen, Germany).
Measurement of trehalose concentration
Frozen culture (1 mL) was incubated in 9 mL YPD broth at 30°C with shaking (150 rpm) overnight. To measure the intracellular trehalose concentration during growth, the overnight culture was transferred into 100 mL of YPD broth to achieve an OD620 value of 0.3 and incubated in 500 mL baffled Erlenmeyer Flasks at 30°C with shaking (150 rpm) for 1, 24, 48, and 72 h.
To measure the intracellular trehalose concentration under ethanol stress, the overnight culture was transferred into 100 mL of YPD broth to achieve an OD620 value of 0.3 and incubated in a 500 mL baffled Erlenmeyer Flask at 30°C with shaking (150 rpm) for 24 h. After incubation, cells were collected by centrifugation and transferred into fresh YPD broth (100 mL) containing ethanol (0 %, 10 %, and 15 %) and incubated in 500 mL baffled Erlenmeyer Flasks at 30°C with shaking (150 rpm) for another 1 h.
Cells were collected by centrifugation at 7000 xg for 5 min and dried at 100°C for 12 h. To extract trehalose, a pellet of 40 mg was mixed with 2 mL ethanol (99.5 %) and incubated in a boiling water bath for 1 h. For HPLC analysis of trehalose, extracts were suspended in 0.5 mL of acetonitrile:water (1:1). Acetonitrile:water (7:3) was used as the mobile phase. The HPLC system was equipped with a refractive index detector and a Lichrocart® 250–4 Purospher® Star NH2 column (5 µm) (Merck, Darmstadt, Germany).23
Ethanol tolerance
Frozen culture (0.5 mL) was incubated in 4.5 mL YPD broth at 30°C with shaking (150 rpm) overnight. To obtain cells in mid-exponential phase, the overnight culture was transferred into a 125 mL baffled Erlenmeyer flask containing 5 mL YPD and left for 3–4 h until an OD620 of 1.0 was reached. Cells were adjusted to an OD620 value of 0.05 with YPD containing 0–18 % ethanol and incubated in the wells of a 96-well flat bottom polystyrene microtiter plate (Costar, Corning Inc., Corning, NY, USA) sealed with a gas-permeable sealing membrane (Breathe Easy membrane, Sigma Chemical Co., St Louis, MO, USA) at 30°C.24 The growth of yeasts was monitored by measurement of OD620 with a microplate reader (Fluostar optima, BMG Labtech, Germany).
Ethanol fermentation
Frozen culture (1 mL) was incubated in 9 mL YPD broth at 30°C with shaking (150 rpm) overnight. Cells were collected and centrifuged at 4000 xg for 5 min. The collected cells (3 × 107 cells/mL) were transferred into 250-mL Erlenmeyer flasks containing 100 mL of YP broth (10 g/L yeast extract, 20 g/L peptone) and glucose (10 %, 15 %, 20 %, 25 % and 30 %). The flasks were incubated statically at 30°C and 1 mL samples were withdrawn at the indicated time intervals.
Ethanol concentration (g/L) was determined by gas chromatography using an Agilent 7890A (Agilent Inc., Palo Alto, CA, USA) equipped with an Agilent FID detector and Rtx-1301 (30 m × 3.0 μm, ID 0.53 mm) column (RESTK Corporation, Bellefonte, PA, USA) with helium as the carrier gas. The temperatures used for compound separation were 35°C (hold 5 min) to 155°C at 20°C/min for column oven, 200°C for injection and 240°C for detection. The ethanol yield was calculated according to the following equation.25
Viable cell count
Sample was serially diluted as required and spread in YPD agar plates and plates were incubated at 30°C for 72 h. Viable cells were counted as colony forming units (CFU)/mL sample.
Glucose concentration
The DNS method26 was employed to estimate the concentration of glucose in the sample. The absorbance was measured at 540 nm. The glucose content of the sample was estimated from the glucose standard curve.
Statistical analysis
Trehalose concentration, ethanol concentration, glucose concentration and biomass values were evaluated by one-way ANOVA, followed by Duncan's new multiple-range test to determine the differences among means or Student's t-test when only 2 groups were compared using the Statistical Analysis System (SAS institute, Cary, NC, USA). A significance level of 5 % was adopted for all comparisons.
Results
nth1 gene deletion of S. cerevisiae
A gene deletion cassette was constructed by overlapping PCR and a target amplicon containing, in sequence: 0.26 kb sequence upstream of the nth1 gene, 1.6 kb kanr marker gene, and 0.4 kb sequence downstream of the nth1 gene. After transformation, a total of 136 G418-resistant colonies were obtained. Positive PCR results appeared in all primer combinations used for identification of either the nth1 gene (Fig. 3B, lines 1, 4, and 6) or the kanr gene (Fig. 3B, lines 2, 3, and 5) in all selected diploid yeast cells. The above results indicated that one of the 2 copies of the nth1 gene had been replaced by the deletion cassette in these transformants. In other words, these cells were heterozygous diploid yeast and its haploid carried the disrupted allele.27
Figure 3.
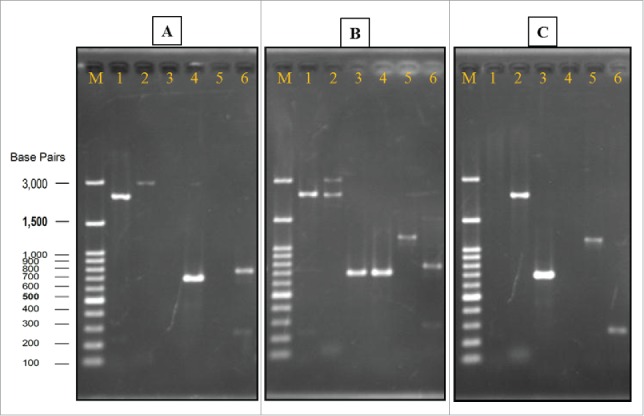
Verification of gene deletion strain. Genomic DNA of wild strain, heterozygous strain, and homozygous strain (Agarose gel image A, B, and C, respectively) were used as templates for PCR with different sets of primers. In all of the gels, Lane M: Gene-Direx 100 bp DNA Ladder, lane 1: primers Nth1-F and R, lane 2: primers L1 and L4, lane 3: primers A1 and kan B, lane 4: primers A1 and B, lane 5: primers D1 and kan C, lane 6: primers D1 and C.
To obtain the homozygous diploid yeast cells, transformants were induced to sporulate. A sporulation medium, which lacked nitrogen sources and contained non-fermentable carbon sources, was used to direct cells toward pathways of meiosis and sporulation. The formation of spores involves a form of cell division that is radically different from the budding process in mitotic cells.28 In sporulation, the chromosomes were segregated by meiosis, resulting in production of 4 haploid nuclei in the ascus called the tetrad. After germination of the G418 selected spore, the diploid yeast cells were obtained. A total of 24 colonies were selected randomly, and 16 colonies were homozygous diploid cells (data not shown). One of the homozygous cell colonies was preserved and named as tps1 overexpression plus nth1 deleted strain (SCTΔN) for further study. As results showed in Fig. 3C (lines 1, 4, and 6), primer combinations for detection of the nth1 gene did not generate a PCR product. On the other hand, DNA bands with expected sizes were observed (Fig. 3C, lines 2, 3, and 5) when primer combinations revealing kanr gene were applied.
Expression of trehalose-6-phosphate synthase
After gene deletion PCR and western blot were used to verify the insertion of tps1 in wild strain (SC) and recombinant strain (SCTΔN). SCTΔN obtained a PCR product of about 1.5 kb (tps1 gene), whereas wild strain SC did not generate PCR product (Fig. 4A). Western blot showed one protein band about 56 kDa (trehalose-6-phasphate synthase) from SCTΔN, whereas no protein band was observed from wild strain SC (Fig. 4B).
Figure 4.
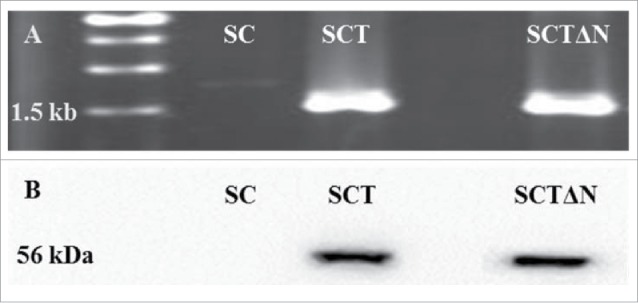
PCR confirmation of the recombinant expression vector pGAPZC-tps1 (A) and Over-expression of TPS (B) in SC (wild strain), SCT (tps1 overexpression strain) and SCTΔN (tps1 overexpression and nth1 deletion strain).
Trehalose concentration of nth1 gene deleted S. cerevisiae
Table 3 shows the intracellular trehalose contents of SC, SCT and SCTΔN when they grew in a YPD broth without external ethanol. In all strains, the trehalose concentration increased during 72 h of growth. Compared with the wild strain, the genetic modified strain, SCTΔN, synthesized significantly higher amounts of trehalose in its cells (p < 0.05).
Table 3.
Intracellular trehalose content of yeast cells during growth and under stress.
| Intracellular trehalose content of cell (mg/g dry weight) during growth | ||||
|---|---|---|---|---|
| Incubation time (h)1 | 1 | 24 | 48 | 72 |
| SC | 9.2 ± 0.2ca-d | 20.9 ± 0.1ba-d | 39.3 ± 0.7aa-d | 38.7 ± 0.7aa-d |
| SCT | 12.2 ± 0.6da-d | 22.7 ± 0.2ca-d | 40.1 ± 1.3ba-d | 48.1 ± 2.9aa-d |
| SCTΔN | 24.9 ± 1.4c* | 27.0 ± 0.1c* | 58.8 ± 1.4b* | 75.4 ± 3.5a* |
| Intracellular trehalose content of cell (mg/g dry weight) under stress | ||||
| Ethanol concentration2 | 0% | 10% | 15% | |
| SC | 9.2 ± 0.2ca-d | 25.3 ± 2.5ba-d | 63.7 ± 0.9aa-d | |
| SCT | 12.2 ± 0.6ca-d | 33.0 ± 1.8ba-d | 67.8 ± 0.7aa-d | |
| SCTΔN | 24.9 ± 1.4c* | 59.9 ± 1.6b* | 85.1 ± 0.2a* | |
Overnight grown cells were incubated for different times in YPD medium without adding ethanol.
Overnight grown cells were incubated for another 1 h in YPD medium with addition of different amounts of ethanol.
SC: wild type Saccharomyces cerevisiae
SCT: S. cerevisiae with overexpression of tps1
SCTΔN: S. cerevisiae with overexpression of tps1 and deletion of nth1 gene
Means in the same column are significantly different. One-way ANOVA, Duncan's multiple range test, p < 0.05.
Means in the same row followed by different letters are significantly different. One-way ANOVA, Duncan's multiple range test, p < 0.05.
When incubated under ethanol-induced environmental stress for 1 h, all strains raised its intracellular trehalose, and this increased amount was greater in YPD containing 15 % ethanol than in YPD containing 10 % ethanol (Table 3). Under ethanol stress, trehalose was accumulated much faster than without stress. SCTΔN produced more trehalose than SC and SCT before and after stress induction. The above results concluded that deletion of the nth1 gene enhanced the level of intracellular trehalose in yeast cells. When yeast cells encountered stress in their medium, a significant amount of trehalose was generated in a very short period, 1 h in this study, indicating that synthesis of trehalose functions as a very efficient defense mechanism. By considering the results above, the engineered strain SCTΔN with tps1 overexpression and nth1 gene deletion was used for further study.
Ethanol tolerance of nth1 gene-deleted S. cerevisiae
The effect of tps1 overexpression and nth1 deletion on cell growth under environmental stress was evaluated by comparing the growth of SCTΔN with that of SC in YPD medium containing 0 to 18 % (v/v) ethanol after incubation for 24 h (Fig. 5). When SC and SCTΔN grew under non ethanol stress (0 %, v/v, ethanol), there was no significant difference in the biomass level between them (p > 0.05). However, when the medium contained 6 % or more ethanol, SCTΔN showed superior growth over SC (p < 0.05). The growth of SCTΔN was inhibited by ethanol concentrations greater than 10 %, while 6 % could slow the growth of SC. When the media contained ethanol concentrations in the range of 14 to 18 %, SC showed no growth in 24 h. Compared with initial growth, only SCTδN showed significantly higher growth at 14 % and 16 % concentrations (p < 0.05).
Figure 5.
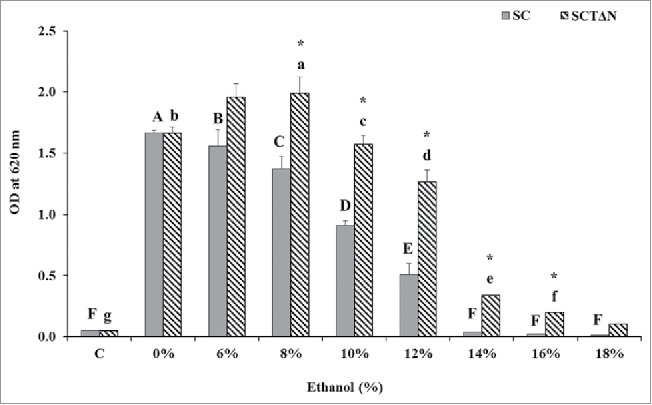
Effect of ethanol concentrations on 24 h growth of wild strain (SC) and engineered strain (SCTΔN). Control (C) is the initial OD of strains. Data are presented as the means ± SD (n = 3). *Significantly higher than the other group. One-way ANOVA, Student's t test, P < 0.05. Bar values with the different letters (A, B, C, D, E, F for SC, while a, b, c, d, e, f, g for SCTΔN) are significantly different. One-way ANOVA, Duncan's multiple range test, P < 0.05.
Ethanol production and cell growth
When the fermentation broth contained 10 % glucose, there was no significant difference in the ethanol concentrations between SC and SCTΔN during 96 h incubation (40.24 ± 0.10 g/L and 40.55 ± 0.10 g/L, respectively), except that SCTΔN produced more ethanol than SC at 12 h and 24 h. Ethanol concentrations of SC at 12 h and 24 h were 17.68 ± 0.03 g/L and 34.48 ± 0.02 g/L, respectively. Moreover, SCTΔN showed the concentrations of ethanol 18.77 ± 0.04 g/L and 36.38 ± 0.04 g/L after 12 h and 24 h, respectively. (Fig. 6A). When media contained 15 %, 20 %, 25 % and 30 % glucose, there was a significant difference in the ethanol concentrations between SC and SCTΔN during 96 h incubation (p < 0.05). SCTδN produced more ethanol than SC after 24 h incubation (Fig. 6B∼E). When media contained glucose in the range of 10 to 30 %, the highest amount of ethanol was produced at 25 % glucose for SC (61.35 ± 0.09 g/L) and at 30 % for SCTΔN (107.83 ± 0.77 g/L).
Figure 6.

Ethanol production capacities of wild strain (SC) and engineered strain (SCTΔN). Ethanol concentration in g/L (triangle), biomass Log CFU/mL (square) and residual glucose concentration in % (circle) of SC (wild strain) (closed) and SCTΔN (tps1 overexpression and nth1 deleted strain) (open) grown in YP broth (10 g/L yeast extract, 20 g/L peptone) containing different concentrations of glucose. A: 10% glucose; B: 15% glucose; C: 20% glucose; D: 25% glucose; E: 30% glucose; F: Ethanol yield (%) after 96 h of incubation. Data are presented as the means ± SD (n = 3). *Significantly higher than the other group. One-way ANOVA, Student's t test, P < 0.05. Bar values of the ethanol yield (%) with the different letters (a, b, c, d for SC and A, B, C, D for SCTΔN) are significantly different. One-way ANOVA, Duncan's multiple range tests, P < 0.05.
Theoretical ethanol yields of SC and SCTδN were compared after incubation for 96 h (Fig. 6F). There was no significant difference in the ethanol yields when the growth medium contained 10 % glucose; however, when the glucose was increased from 15 to 30 %, ethanol yields of SCTΔN were significantly higher than that of SC (p < 0.05). In the case of SC, as glucose concentration of media increased from 10 to 30 %, a dramatic decrease in ethanol yield was observed, from 78.75 ± 1.51 to 36.11 ± 0.01 %, respectively. Meanwhile, a disastrous drop in ethanol yield occurred when SCTΔN were cultured in a medium containing 30 % glucose.
Residual glucose in the media was determined during 96 h growth. When the medium contained 10–30 % glucose, the decrease of glucose concentration in the medium culturing SCTΔN was significantly faster than that of SC. When the medium contained 10–25 % glucose, SCTΔN depleted all glucose after growth for 96 h; however, when the medium contained 30 % glucose, 3.67 ± 0.32 % glucose was left over in the medium (Fig. 6E). On the other hand, SC could use up all of the glucose only at glucose concentrations of 10 and 15 % after incubation for 96 h. When the medium contained 25 and 30 % glucose, high residual glucose was detected, 5.12 ± 0.05 % and 8.19 ± 0.03 %, respectively, indicating that the presence of 25 and 30 % initial glucose in the medium induced stress to SC.
For both SC and SCTΔN, biomasses reached their peaks at 12 h incubation (Fig. 6A–E). In all tested glucose levels, SCTΔN grew significantly faster than SC (p < 0.05).
Discussion
Trehalose is one of the most chemically unreactive sugars. Its strong stability is a result of the very low energy (1 kcal mol−1) of the glycoside oxygen bond joining the 2 hexose rings. It does not dissociate into 2 reducing monosaccharidic constituents unless exposed to extreme hydrolytic conditions or to the action of trehalase. Therefore, trehalose is a unique and ubiquitous sugar capable of protecting biomolecules against environmental stress.29 Many studies took interest in the correlation between stress tolerance and trehalose accumulation in yeast under various stress conditions, such as ethanol,6 freezing,30 saline,9 and multiple environmental stresses.31 Researchers stated that the higher the trehalose content in the yeast cell, the higher its tolerance against environmental pressure, particularly to ethanol.10,11 In Sharma's study,6 a trehalose proficient strain (1.43 mg/mg protein) showed greater ethanol tolerance than a trehalose deficient strain, and Sharma6 suggested that trehalose maintained the bilayer structure of the plasma membrane in a fluid condition. Mansure et al.7 concluded that intracellular trehalose prevents the ethanol-induced electrolyte leakage in yeast cells and in a carboxyfluorescein-trapped liposome model system. However, Ratnakumar and Tunnacliffe32 suggested that there is no consistent relationship between intracellular trehalose concentration and desiccation tolerance in S. cerevisiae.
In this study, the wild strain (SC) and engineered strain (SCTΔN) increased intracellular trehalose concentrations during 72 h growth, and this might be due to stimulation of endogenic ethanol produced during growth.5 On the other hand, when external ethanol was high, 10 % and 15 % in this study, yeast accumulated trehalose extremely fast, 1 h in this study. SCTΔN exhibited significantly higher ethanol tolerance than SC and this might be due to SCTΔN synthesising more trehalose regardless of ethanol stress.
Many studies have reported that the genes encoding trehalose and biosynthetic enzymes are upregulated under various stresses.33,34 On the other side, Van Dijck et al.10 suggested that relative levels of trehalose seemed to be induced mainly by ethanol formation and decreases in sugar levels during fermentation, rather than by external ethanol. Our results showed that external ethanol stimulated trehalose biosynthesis at a very fast rate. Dasari et al.35 suggested that endogenic and external ethanol caused different toxicity in S. cerevisiae. In the present study, SCTΔN showed greater tolerance against both endogenic and external ethanol compared with SC and concurrently SCTΔN accumulated higher levels of trehalose. Our results confirmed that trehalose serves as a major substance in defense of ethanol toxicity, whether caused by external or endogenic ethanol.
Trehalose is synthesized by a 2-step process. In the first step of its biosynthesis pathway, trehalose-6-phosphate is formed from UDP-glucose and glucose-6-phosphate. This reaction is catalyzed by trehalose-6-phosphate synthase (TPS, encoded by tps1 in yeast). Then, this compound is converted to trehalose by trehalose-6-phosphate phosphatase (TPP, encoded by tps2 in yeast).8 Trehalose is hydrolyzed into glucose by trehalose degradation enzymes, neutral trehalases encoded by nth1 and nth2 or acid trehalase encoded by ath1.12 The yeast mutant with nth1 deletion13 and ath1 deletion14 exhibits a higher trehalose accumulation under stress conditions.
In this study, the nth1 gene (encoding neutral trehalase) of S. cerevisiae was deleted. As we expected, deletion of nth1 led to increased cell tolerance against environmental stress by preserving trehalose and this result agreed with many reports, such as Garre and Matallana 36 and Klaassen et al.37 To avoid illegitimate integrations, an overlapping PCR technique was used to generate disruption cassettes with long flanking sequences which were homologous to the 0.26 kb 5′ UP and 0.4 kb 3′ DOWN regions of the target nth1 yeast gene according to Wach.15 The disruption cassette is sourced from the upstream and downstream sequences of the target gene and heterologous kanMX marker gene. To obtain the homozygous diploid in which 2 copies of nth1 were replaced by kanMX, heterozygous diploid yeast cells, which were selected from YPD plates containing G418 (200 mg/L),27 were induced to sporulate. The resulting mutant, with an absence of neutral trehalase, exhibits higher trehalose accumulation, ethanol tolerance, and ethanol production compared with the wild strain.
Cell growth and ethanol production were not affected by tps1 overexpression and nth1 deletion when the medium contained 10 % glucose. At the time that glucose concentration was in the range of 15 to 30 %, SCTΔN showed significantly greater growth and higher ethanol production than SC (p < 0.05). Overexpression of tps1 (trehalose-6-phosphate synthase) and removal of nth1 (neutral trehalase) improved not only ethanol tolerance, but cell viability as well as ethanol production under glucose stress (osmotic stress). Maristela Freitas and Laluce5 stated that improvement in cell viability at high external concentrations of initial ethanol (above 6 %) seems to be an important aspect of the alcohol-tolerant strains.
Previous studies were reported the correlation between trehalose content and stress resistance.9,14,31 In this study, neutral trehalase, responsible for trehalose hydrolysis in intact cells was deleted and heterozygous strain was further used to obtain homozygous diploid strain. It was necessary step because after transformation of diploid yeast cells, one of the 2 copies of the target endogenous gene had been replaced by the disruption cassette. These heterozygous diploid yeast cells grew normally on selective medium, whereas nontransformants cells were not. After sporulation of heterozygous diploid cells, half the haploid spores only carried the disrupted allele. These spores were separated and germinated in rich medium containing glucose (YPD) and G418 to find out homozygous cells. Difference between heterozygous and homozygous cells was confirmed by PCR.
By the strategy of tps1 overexpression and nth1 deletion, S. cerevisiae exhibited increased viability and ethanol yield under ethanol stress, and this strategy might provide a preferable reuse of starters as immobilized cells as well as provide applications in continuous processes.
Conclusion
In this study, wild strain was improved by overexpression of tps1 gene and entire deletion of nth1 gene. A disruption cassette with long flanking homology regions of nth1 gene was applied to disrupt the gene of nth1 in S. cerevisiae. We also found that the procedures of sporulation and selection of haploid spores were necessary to obtain the strain with homozygous deletion of the target endogenous gene. Compared with the wild strain, the engineered strain obtained in this study exhibited higher trehalose accumulation with or without ethanol stress and showed greater ethanol and glucose tolerances. The strain constructed in this study will be promising to improve ethanol production with increased tolerance to different environmental stresses.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are grateful to Dr. John Pringle from University of North Carolina at Chapel Hill for the generous gift of pFA6a-kanMX6 vector.
Funding
This research was supported by the Ministry of Science and Technology, R.O.C. Taiwan (NSC 100-2313-B-126-001-MY3). Its financial support is greatly appreciated
References
- [1].Piper PW. The heat shock and ethanol stress responses of yeast exhibit extensive similarity and functional overlap. FEMS Microbiol Lett 1995; 134:121-7; PMID:8586257; http://dx.doi.org/ 10.1111/j.1574-6968.1995.tb07925.x [DOI] [PubMed] [Google Scholar]
- [2].Rose AH. Composition of the envelope layers of Saccharomyces cerevisiae in relation to flocculation and ethanol tolerance. J Appl Bacteriol [Internet] 1993; 74:110S-118S; PMID:8349530; http://dx.doi.org/ 10.1111/j.1365-2672.1993.tb04347.x [DOI] [PubMed] [Google Scholar]
- [3].Chi Z, Arneborg N. Relationship between lipid composition, frequency of ethanol-induced respiratory deficient mutants, and ethanol tolerance in Saccharomyces cerevisiae. J Appl Microbiol 1999; 86:1047-52; PMID:10389252; http://dx.doi.org/ 10.1046/j.1365-2672.1999.00793.x [DOI] [PubMed] [Google Scholar]
- [4].Ingram LO, Buttke TM. Effects of Alcohols on Micro-Organisms [Internet]. In: Rose AH, T DWTB, eds: Advances in Microbial Physiology. Academic Press; 1985, pp. 253–300. Available from: http://www.sciencedirect.com/science/article/pii/S0065291108602945 [DOI] [PubMed] [Google Scholar]
- [5].Maristela Freitas SP, Laluce C. Ethanol tolerance of thermotolerant yeasts cultivated on mixtures of sucrose and ethanol. J Ferment Bioeng [Internet] 1998; 85:388-97. Available from: http://www.sciencedirect.com/science/article/pii/S0922338X98800822; http://dx.doi.org/ 10.1016/S0922-338X(98)80082-2 [DOI] [Google Scholar]
- [6].Sharma SC. A possible role of trehalose in osmotolerance and ethanol tolerance in Saccharomyces cerevisiae. FEMS Microbiol Lett 1997; 152:11-5; PMID:9228764; http://dx.doi.org/ 10.1111/j.1574-6968.1997.tb10402.x [DOI] [PubMed] [Google Scholar]
- [7].Mansure JJC, Panek AD, Crowe LM, Crowe JH. Trehalose inhibits ethanol effects on intact yeast cells and liposomes. Biochim Biophys Acta - Biomembr 1994; 1191:309-16; PMID:8172916; http://dx.doi.org/ 10.1016/0005-2736(94)90181-3 [DOI] [PubMed] [Google Scholar]
- [8].Elbein AD, Pan YT, Pastuszak I, Carroll D. New insights on trehalose: A multifunctional molecule. Glycobiology 2003; 13:17-27; PMID:12626396; http://dx.doi.org/ 10.1093/glycob/cwg047 [DOI] [PubMed] [Google Scholar]
- [9].Mahmud SA, Nagahisa K, Hirasawa T, Yoshikawa K, Ashitani K, Shimizu H. Effect of trehalose accumulation on response to saline stress in Saccharomyces cerevisiae. Yeast [Internet] 2009; 26:17-30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19180643; PMID:19180643; http://dx.doi.org/ 10.1002/yea.1646 [DOI] [PubMed] [Google Scholar]
- [10].Van Dijck P, Colavizza D, Smet P, Thevelein JM, Van Dijck P, Colavizza D, Smet P. Differential importance of trehalose in stress resistance in fermenting and nonfermenting Saccharomyces cerevisiae cells. Appl Environ Microbiol [Internet] 1995; 61:109-15. Available from: papers3://publication/uuid/8BF3B7BF-B326-4D9D-8658-003721D73EF1\nfile:///Users/SAHARA/Library/Mobile; PMID:7887593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Odumeru J, D'Amore T, Russell I, Stewart G. Alterations in fatty acid composition and trehalose concentration of Saccharomyces brewing strains in response to heat and ethanol shock. J Ind Microbiol [Internet] 1993; 11:113-9; PMID:NOT_FOUND; http://dx.doi.org/ 10.1007/BF01583683 [DOI] [Google Scholar]
- [12].Nwaka S, Holzer H. Molecular biology of trehalose and the trehalases in the yeast Saccharomyces cerevisiae. Prog Nucleic Acid Res Mol Biol [Internet] 1998; 58:197-237. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9308367; PMID:9308367; http://dx.doi.org/ 10.1016/S0079-6603(08)60037-9 [DOI] [PubMed] [Google Scholar]
- [13].Dong J, Wang G, Zhang C, Tan H, Sun X, Wu M, Xiao D. A two-step integration method for seamless gene deletion in baker's yeast. Anal Biochem [Internet] 2013; 439:30-6; PMID:23597844; http://dx.doi.org/ 10.1016/j.ab.2013.04.005 [DOI] [PubMed] [Google Scholar]
- [14].Wang P-M, Zheng D-Q, Chi X-Q, Li O, Qian C-D, Liu T-Z, Zhang X-Y, Du F-G, Sun P-Y, Qu A-M, et al.. Relationship of trehalose accumulation with ethanol fermentation in industrial Saccharomyces cerevisiae yeast strains. Bioresour Technol [Internet] 2014; 152:371-6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24316480; PMID:24316480; http://dx.doi.org/ 10.1016/j.biortech.2013.11.033 [DOI] [PubMed] [Google Scholar]
- [15].Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast [Internet] 1996; 12:259-65. Available from: http://doi.wiley.com/10.1002/(SICI)1097-0061(19960315)12:33.0.CO;2-C; PMID:8904338; http://dx.doi.org/ 10.1002/(SICI)1097-0061(19960315)12:3%3c259::AID-YEA901%3e3.0.CO;2-C [DOI] [PubMed] [Google Scholar]
- [16].Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual [Internet]. Second. New York: Cold Spring Harbor Laboratory Press; 1989. Available from: http://www.molecularcloning.org/ [Google Scholar]
- [17].Bähler J, Wu J-Q, Longtine MS, Shah NG, McKenzie A, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast [Internet] 1998; 14:943-51. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9717240; PMID:9717240; http://dx.doi.org/ 10.1002/(SICI)1097-0061(199807)14:10%3c943::AID-YEA292%3e3.0.CO;2-Y [DOI] [PubMed] [Google Scholar]
- [18].Hanahan D, Meselson M. Plasmid screening at high colony density. Gene 1980; 10:63-7; PMID:6997135; http://dx.doi.org/ 10.1016/0378-1119(80)90144-4 [DOI] [PubMed] [Google Scholar]
- [19].Barnes WM. PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc Natl Acad Sci U S A 1994; 91:2216-20; PMID:8134376; http://dx.doi.org/ 10.1073/pnas.91.6.2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lin CA, Ellis SR, True HL. The Sua5 protein is essential for normal translational regulation in yeast. Mol Cell Biol 2010; 30:354-63; PMID:19884342; http://dx.doi.org/ 10.1128/MCB.00754-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Horvath A, Riezman H. Rapid protein extraction from Saccharomyces cerevisiae. Yeast 1994; 10:1305-10; PMID:7900419; http://dx.doi.org/ 10.1002/yea.320101007 [DOI] [PubMed] [Google Scholar]
- [22].Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227:680-5; PMID:5432063; http://dx.doi.org/ 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- [23].Ferreira JC, Paschoalin VMF, Panek AD, Trugo LC. Comparison of three different methods for trehalose determination in yeast extracts. Food Chem 1997; 60:251-4; http://dx.doi.org/ 10.1016/S0308-8146(96)00330-5 [DOI] [Google Scholar]
- [24].Tran TMT, Stanley GA, Chambers PJ, Schmidt SA. A rapid, high-throughput method for quantitative determination of ethanol tolerance in Saccharomyces cerevisiae. Ann Microbiol 2013; 63:677-82; http://dx.doi.org/ 10.1007/s13213-012-0518-4 [DOI] [Google Scholar]
- [25].Hatzis C, Riley C, Philippidis GP. Detailed material balance and ethanol yield calculations for the biomass-to-ethanol conversion process. Appl Biochem Biotechnol 1996; 57-58:443-59; http://dx.doi.org/ 10.1007/BF02941725 [DOI] [Google Scholar]
- [26].Miller GL. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem [Internet] 1959; 31:426-8. Available from: http://nsrdec.natick.army.mil/LIBRARY/59-69/R59-36.pdf\nhttp://pubs.acs.org/doi/abs/10.1021/ac60147a030; http://dx.doi.org/ 10.1021/ac60147a030 [DOI] [Google Scholar]
- [27].Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular Cell Biology [Internet]. Fourth. 2000. Available from: http://www.ncbi.nlm.nih.gov/books/NBK21475/ [Google Scholar]
- [28].Kupiec M, Byers B, Esposito R, Mitchell A. Meiosis and Sporulation in Saccharomyces cerevisiae [Internet]. In: The molecular and cellular biology of the yeast Saccharomyces: Cell Cycle & Cell Biology. Cold Spring Harbor Laboratory Press; 1997. Available from: https://cshmonographs.org/index.php/monographs/article/view/4686 [Google Scholar]
- [29].Schiraldi C, Di Lernia I, De Rosa M. Trehalose production: Exploiting novel approaches. Trends Biotechnol 2002; 20:420-5; PMID:12220904; http://dx.doi.org/ 10.1016/S0167-7799(02)02041-3 [DOI] [PubMed] [Google Scholar]
- [30].Nakamura T, Takagi H, Shima J. Effects of ice-seeding temperature and intracellular trehalose contents on survival of frozen Saccharomyces cerevisiae cells. Cryobiology [Internet] 2009; 58:170-4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19126409; PMID:19126409; http://dx.doi.org/ 10.1016/j.cryobiol.2008.11.012 [DOI] [PubMed] [Google Scholar]
- [31].Mahmud SA, Hirasawa T, Shimizu H. Differential importance of trehalose accumulation in Saccharomyces cerevisiae in response to various environmental stresses. J Biosci Bioeng 2010; 109:262-6; PMID:20159575; http://dx.doi.org/ 10.1016/j.jbiosc.2009.08.500 [DOI] [PubMed] [Google Scholar]
- [32].Ratnakumar S, Tunnacliffe A. Intracellular trehalose is neither necessary nor sufficient for desiccation tolerance in yeast. FEMS Yeast Res 2006; 6:902-13; PMID:16911512; http://dx.doi.org/ 10.1111/j.1567-1364.2006.00066.x [DOI] [PubMed] [Google Scholar]
- [33].Li L, Ye Y, Pan L, Zhu Y, Zheng S, Lin Y. The induction of trehalose and glycerol in Saccharomyces cerevisiae in response to various stresses. Biochem Biophys Res Commun [Internet] 2009; 387:778-83; PMID:19635452; http://dx.doi.org/ 10.1016/j.bbrc.2009.07.113 [DOI] [PubMed] [Google Scholar]
- [34].Parrou JL, Teste MA, François J. Effects of various types of stress on the metabolism of reserve carbohydrates in Saccharomyces cerevisiae: Genetic evidence for a stress-induced recycling of glycogen and trehalose. Microbiology 1997; 143:1891-900; PMID:9202465; http://dx.doi.org/ 10.1099/00221287-143-6-1891 [DOI] [PubMed] [Google Scholar]
- [35].Dasari G, Worth MA, Connor MA, Pamment NB. Reasons for the apparent difference in the effects of produced and added ethanol on culture viability during rapid fermentation by Saccharomyces cerevisiae. Biotechnol Bioeng [Internet] 1990; 35:109-22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18592500; PMID:18592500; http://dx.doi.org/ 10.1002/bit.260350202 [DOI] [PubMed] [Google Scholar]
- [36].Garre E, Matallana E. The three trehalases Nth1p, Nth2p and Ath1p participate in the mobilization of intracellular trehalose required for recovery from saline stress in Saccharomyces cerevisiae. Microbiology [Internet] 2009; 155:3092-9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19520725; PMID:19520725; http://dx.doi.org/ 10.1099/mic.0.024992-0 [DOI] [PubMed] [Google Scholar]
- [37].Klaassen P, Blankwater YJ, van Marrewijk LP, Osinga KA, Baankreis R. Effects of the NTH1 gene disruption in bakers' yeast. Folia Microbiol (Praha) 1994; 39:524-6; PMID:8550012; http://dx.doi.org/ 10.1007/BF02814086 [DOI] [PubMed] [Google Scholar]