

Abstract
Objective
Research in acute illness often requires an exception from informed consent (EFIC). Few studies have assessed the views of patients enrolled in EFIC trials. This study was designed to assess the views of patients and their surrogates of EFIC enrollment in a randomized, placebo-controlled trial of an investigational agent for traumatic brain injury.
Design
Interactive interview study.
Setting
Nested within the Progesterone for the Treatment of Traumatic Brain Injury (ProTECT III) trial, a Phase III randomized controlled trial in acute traumatic brain injury (TBI).
Participants
Patients and surrogates (for patients incapable of being interviewed) enrolled in ProTECT III under EFIC at 12 sites.
Measurements
Interviews focused on respondents’ acceptance of EFIC enrollment in ProTECT, use of placebo and randomization, understanding of major study elements, and views regarding regulatory protections. Descriptive statistical analysis was performed; textual data were analyzed thematically.
Main Results
85 individuals were interviewed. 84% had positive attitudes toward ProTECT III inclusion. 78% found their inclusion under EFIC acceptable, and 72% found use of EFIC in ProTECT III acceptable in general. Only 2 respondents clearly disagreed with both personal and general EFIC enrollment. The most common concerns (26%) related to absence of consent. 80% and 92% were accepting of placebo use and randomization, respectively. Though there were few black respondents (n=11), they were less accepting of personal EFIC enrollment than white respondents (55% vs 83%, p= 0.0494).
Conclusions
Acceptance of EFIC in this placebo-controlled trial of an investigational agent was high and exceeded acceptance among community consultation participants. EFIC enrollment appears generally consistent with patients’ preferences.
Keywords: Research ethics, Bioethics, Informed consent, Acute care research, Emergency research, Traumatic Brain Injury
Introduction
Informed consent is never sufficient to make research ethical and is not always possible (1). When acutely ill patients are unconscious or severely impaired, as in conditions such as cardiac arrest, shock, and traumatic brain injury, treatment must take place before a surrogate decision-maker can be identified. Treatment for many of these conditions remains suboptimal, and improvement depends on clinical research. An exception from informed consent (EFIC) is permitted in these situations (2–4), and there has been notable expansion in EFIC research in recent years. This expansion represents important progress in addressing the needs of critically ill patients. However, EFIC research raises important ethical challenges.
The views of patients enrolled in EFIC studies are important to understand. Patients should not be enrolled in trials simply because they cannot speak for themselves, and the ethical justification for EFIC research would be threatened if significant objections to enrollment were prevalent among enrollees (5). Providing insight into potential subjects’ views is one reason that U.S. regulations require community consultation before approval of EFIC studies (3). To date, however, only two studies have examined the views of EFIC enrollees. The Patients’ Experiences in Emergency Research (PEER-RAMPART) study found over 70% of enrolled patients and surrogates were accepting of their own EFIC enrollment in a trial of prehospital management of status epilepticus (6). Over 95% of Australian participants in a trial of glucose control in critical illness reported they would have agreed if they could have been asked for consent (7). Other studies of patients (or surrogates) who have survived critical illnesses also suggest potentially high rates of EFIC acceptance (8–10). However, feedback from community consultation reports has varied, and questions remain regarding respondents’ understanding of EFIC research, whether community consultants’ views reflect patients’ views, and whether particular groups have distinct concerns (11–15).
No study has assessed the views of patients enrolled in a placebo-controlled EFIC trial of an investigational agent. However, this design represents the greatest departure from clinical care and could raise concerns on the part of subjects that are not present, for example, in comparative trials of existing treatments (9, 16). The Progesterone for the Treatment of Traumatic Brain Injury (ProTECT™ III, NCT00822900) trial is a Phase III, placebo-controlled study of a 4-day infusion of progesterone in addition to standard medical care in acute moderate to severe traumatic brain injury. Primary outcomes include mortality and neurologic status at 6 months. The Patients’ Experiences in Emergency Research-ProTECT (PEER-ProTECT) study interviewed enrolled patients and surrogates.
MATERIALS AND METHODS
Objective and Population
This study was designed to provide quantitatively meaningful in-depth assessment of patients’ and surrogates’ views regarding EFIC enrollment in ProTECT III. PEER-ProTECT was nested within the ProTECT III trial, conducted within the Neurological Emergencies Treatment Trial (NETT) network. NETT contains 17 “hub sites” (hereafter referred to as sites), 12 of which referred patients for PEER-ProTECT.
All patients enrolled in ProTECT III under EFIC at referring sites were eligible for inclusion. If a patient could not be interviewed due to impairment or death, a surrogate was eligible. Notably, surrogates of patients who had died after EFIC enrollment, as well as individuals who withdrew trial consent after EFIC enrollment, were eligible. Because of their interactive nature, interviews were conducted only in English.
Recruitment and Interview Conduct
Potential participants were identified locally (Figure 1) during initial hospitalization or during follow-up contact with the primary research team. Local staff either asked participants for permission to be contacted by PEER-ProTECT interviewers or sent a letter stating they would be contacted in the future unless they wished otherwise (not all sites utilized the opt-out letter). Initial contact by PEER-ProTECT staff was made approximately 3 months after initial enrollment, with the goal of conducting interviews 3 to 6 months post-enrollment. Potential participants were intentionally not contacted prior to this point in an effort to maximize the chance that a patient (rather than a surrogate) had recovered and could be interviewed.
Figure 1.
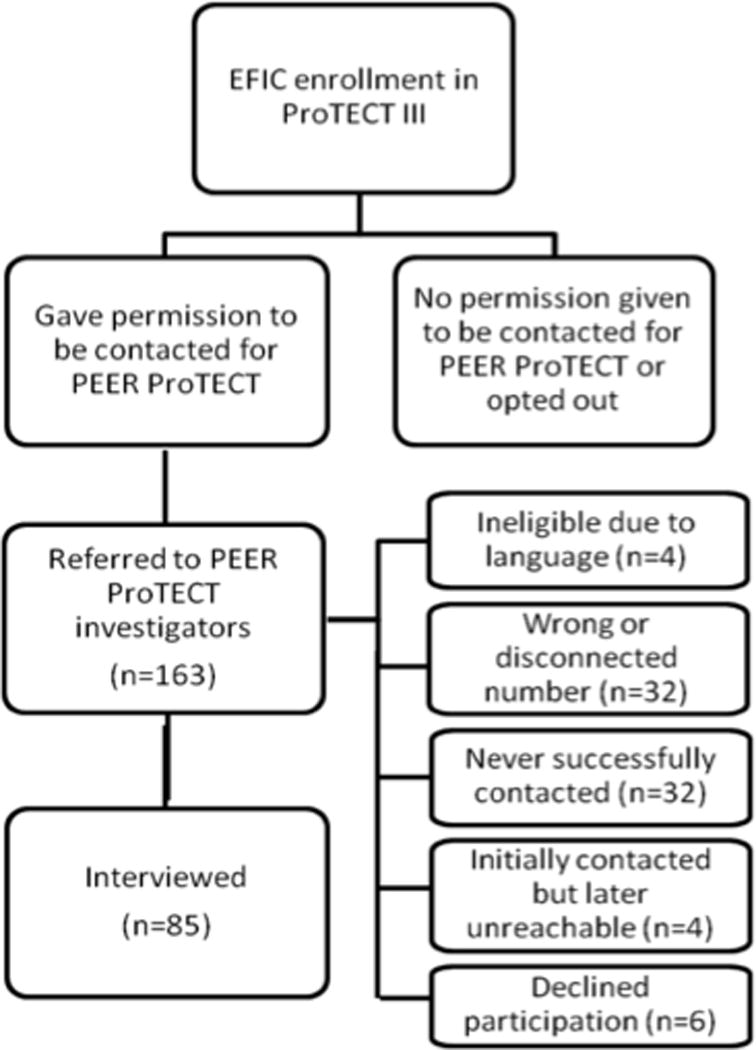
Study Enrollment
All interviews were conducted by telephone. A contracted partner, APCO Insight, maintained trained interviewers and recruited participants. Oral informed consent was obtained by PEER-ProTECT interviewers before or at the time of the interview. Participants were paid $20 for participation. PEER-ProTECT was reviewed and approved by the Emory University School of Medicine Institutional Review Board (IRB) and participating sites’ IRBs. Several IRBs determined the study did not require review at their site because their site only referred potential participants.
Interview Methods
The interview guide contained open and closed-ended questions designed to: 1) provide prevalence estimates of particular views; and 2) assess reasons for responses and subjects’ understanding. This interactive strategy- used in PEER-RAMPART- suits the nature of EFIC research. The guide was designed to maximize respondents’ understanding of key content and to allow respondents time to ask questions and develop views on complicated, unfamiliar content (6, 9, 17). The interview guide (Supplemental Digital Content- Appendix 1) was adapted from PEER-RAMPART and reviewed by content and methodological experts.
The interview guide contained 11 domains (Supplemental Digital Content- Appendix 1): 1) prior research experience, 2) knowledge of ProTECT III, 3) views on personal EFIC inclusion, 4) views on EFIC usage in general for ProTECT III, 5) placebo use, 6) randomization, 7) interactions with study staff, 8) community consultation and public disclosure, 9) trust in researchers, 10) demographic information, and 11) perceived side effects. Core domains (domains 3–6) featured an introduction and opportunity for clarification using open-ended questions, followed by a five-point Likert Scale question and probes regarding the response. Interviews generally lasted 20–30 minutes.
Data Management
Interviews were recorded, transcribed, and redacted. Questions with pre-defined response categories were coded real-time using Computer-Assisted Telephone Interviewing (CATI). Entries were checked for errors and corrected by one author (VS), with discrepancies resolved by two authors (VS and ND). For Likert-scale questions, a numerical value was assigned if respondents did not specify a numerical response but gave a clear verbal answer. For example, if a respondent indicated strong agreement by saying “I absolutely agree,” a “1” was assigned. If a respondent indicated agreement but did not indicate strength, a “2” was assigned. If agreement or disagreement was unclear, the response was considered “unknown.”
Data Analysis
Quantitative data were analyzed using SAS 9.3 (SAS Institute Inc., Cary, NC, USA). Descriptive statistics and bivariate analyses (Chi-Square and Fisher’s Exact test for proportions and Kruskal-Wallis test for ordinal categorical answers) were calculated, consistent with hypothesis-generating goals.
MAXQDA 10 (VERBI GmbH, Berlin, Germany) was used to facilitate descriptive analysis of text data. Data were primarily coded by one author (VS). Primary aims were to identify salient and prevalent themes within and across interview domains and to contextualize quantitative responses.
A multi-level coding strategy was used, consistent with the method of template analysis (18). Domain-based codes based on the interview guide allowed topical sorting, and a priori codes were developed based on expected responses. Inductive codes were then developed in multiple steps. First, all transcripts were read by two authors (ND and VS), and preliminary codes were developed. The codebook was refined during analysis, with review and re-coding of all transcripts using the final codebook. Finally, instances of major codes were reviewed to ensure that coded segments reflected a coherent theme. Instances of uncertainty or conflict were reviewed by three authors (VS, ND, and RP) and resolved by consensus.
Limited data are reported from two comparison populations. Demographic and total enrollment data from the ProTECT III trial are included to allow an assessment of representativeness. Previously published demographic data and attitudes toward EFIC among community consultation participants for ProTECT III are included where relevant (19).
RESULTS
Study population
Interviews were completed with 85 respondents (Figure 1). Median time between ProTECT enrollment and interview was 192 days (IQR 95-347). The majority of respondents (64%) were surrogates (Table 1). Most surrogates were either a spouse (24%) or a parent (43%) of the enrolled patient. Overall, 54% of respondents were female, though 70% of surrogate respondents were female. The majority (69%) were white; about half (52%) had completed education beyond high school.
Table 1.
PEER-ProTECT participants, compared to ProTECT III study and community consultation participants
| PEER ProTECT Population | ProTECT III Trial Population | Entire ProTECT III Community Consultation Population | |
|---|---|---|---|
| Total Participants | 85 | 882 | 8835 |
| Participant Type | |||
| Patient | 31 (36%) | 882 (100%) | N/A |
| Surrogate | 54 (64%) | 0 (0%) | N/A |
| Gender* | |||
| Female | 46 (54%) | 232 (26%) | 4470 (58%) |
| Male | 39 (46%) | 650 (74%) | 3280 (42%) |
| Race* | |||
| White | 59 (69%) | 659 (75%) | 5799 (78%) |
| Black | 11 (13%) | 134 (15%) | 1007 (14%) |
| Other | 15 (18%)† | 89 (10%) | 581 (8%) |
| Ethnicity* | |||
| Hispanic | 6 (7%) | 125 (14%) | 395 (7%) |
| Age* | |||
| Mean (SD) | 46 (16) | 39 (17) | 41 (16) |
| < 30 | 15 (18%) | N/A | N/A |
| 31–50 | 31 (36%) | N/A | N/A |
| 51–70 | 33 (39%) | N/A | N/A |
| > 70 | 5 (6%) | N/A | N/A |
| Not reported | 1 (1%) | N/A | N/A |
| Education* | |||
| ≤ High School | 41 (48%) | N/A | 1289 (18%) |
| > High School | 44 (52%) | N/A | 5837 (82%) |
PEER ProTECT demographic data is reported for patients and surrogates together
“Other” racial group in PEER ProTECT includes self-identified Latinos/Hispanics as they were subsequently not asked for race after ethnicity question
Our sample reflected the ProTECT III population (Table1) with the exception of being more often female and slightly older, likely due to inclusion of surrogates. Respondents were also similar to ProTECT III community consultation participants with the exception of being less well-educated (18% vs. 48% having high school or less education) and somewhat older. Five respondents were surrogates of patients who had died. No respondent had withdrawn consent for ProTECT III after EFIC enrollment. Only 5 of 296 EFIC-enrolled subjects across all 17 ProTECT III sites during the PEER-ProTECT study period withdrew consent.
All respondents (or the patient for whom the respondent was a surrogate) were officially enrolled under EFIC. However, 24 (28%) indicated a belief that a surrogate had given consent prior to study initiation. Thirteen of 24 described a time sequence of decision-making that supported this belief. For example, in some cases, patients were enrolled and randomized under EFIC because a surrogate was not identified within the defined window for prospective consent; however, a surrogate was identified and agreed to enrollment before drug infusion. In other cases, formal consent was obtained after infusion had begun, but surrogates felt they had agreed at the outset because the infusion was just starting. Still others (n=11), often patients, knew a surrogate gave consent but were unclear about the sequence of consent and study initiation. Respondents’ uncertainty about consent timing is understandable in this study, as ProTECT III involves a 4-day infusion and not a discrete event. The interviewer informed respondents that they were officially enrolled under EFIC but did not have patient-specific enrollment details. The interviewer thus did not actively dispute respondents’ claims about whether consent was obtained before drug administration.
Attitudes Toward EFIC and Enrollment in ProTECT III
Almost all respondents (95%) agreed TBI research is important (Table 2), and 84% were “glad that they/their family members were included.” 78% agreed it was “okay for researchers to include them/their family members in the ProTECT III research study without asking for permission first,” and 72% agreed it was “okay for researchers to include people (in general) in the ProTECT III research study without asking them for permission first.” Only 2 respondents clearly disagreed with both personal and general EFIC enrollment. Among 14 respondents who were neutral regarding general EFIC enrollment, 6 specifically stated they did not object to their enrollment (or of the patient for whom they were surrogate) but felt they could not make that judgment for others.
Table 2.
Respondents’ Acceptance of Main Trial Components (n=85)
| Strongly agree | Strongly disagree | Don’t know/refuse | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| It is important to do research to find out whether new treatments can improve care for patients with TBI. | 68 (80%) |
13 (15%) |
0 (0%) |
2 (2%) |
0 (0%) |
2 (2%) |
|
| ||||||
| I am glad that I/my family member was included in this research study. (Before explicit EFIC discussion) | 56 (66%) |
15 (18%) |
8 (9%) |
2 (2%) |
1 (1%) |
3 (4%) |
|
| ||||||
| I think it was ok for researchers to include me/my family member in the PROTECT III research study without asking for permission first. (After EFIC discussion) | 54 (64%) |
12 (14%) |
11 (13%) |
2 (2%) |
2 (2%) |
4 (5%) |
|
| ||||||
| I think that it was ok for researchers to include people in the PROTECT research study without asking them for permission first. (After EFIC discussion) | 44 (52%) |
17 (20%) |
14 (16%) |
6 (7%) |
3 (4%) |
1 (1%) |
|
| ||||||
| I think that it was acceptable for researchers to give half of the patients in the PROTECT study placebo and half of the patients Progesterone. | 55 (65%) |
13 (15%) |
9 (11%) |
2 (2%) |
1 (1%) |
5 (6%) |
|
| ||||||
| I think that it was acceptable for researchers to assign treatments at random in this study. | 63 (74%) |
15 (18%) |
5 (6%) |
0 (0%) |
1 (1%) |
1 (1%) |
Predictors of Attitudes Towards EFIC and Enrollment in ProTECT III
Female respondents (Table 3) were more likely to agree with personal (87% vs. 67%, p= 0.0253) and general EFIC enrollment (85% vs. 56%, p= 0.0038). Patients and surrogates did not have statistically significantly different views. Male patients (n=23), however, compared to male surrogates (n=16) trended toward greater acceptance of personal and general EFIC enrollment (78% vs. 50%, p= 0.0655 and 65% vs. 44%, p= 0.1836).
Table 3.
EFIC Acceptance by Demographic Characteristics
| Okay with personal enrollment under EFIC (%agree)* | Okay with EFIC usage in general in this study (%agree) † | Glad about inclusion in this study (%agree) ‡ | |||||
|---|---|---|---|---|---|---|---|
| n (%) | p-value | n (%) | p-value | n (%) | p-value | ||
| Respondent Type | Patient | 26 (84%) | 0.297 | 22 (71%) | 0.902 | 23 (74%) | 0.079 |
| Surrogate | 40 (74%) | 39 (72%) | 48 (89%) | ||||
| Gender | Male | 26 (67%) | 0.025 | 22 (56%) | 0.004 | 30 (77%) | 0.130 |
| Female | 40 (87%) | 39 (85%) | 41 (89%) | ||||
| Age | <30 | 11 (73%) | 0.750 | 11 (73%) | 0.862 | 11 (73%) | 0.038 |
| 31–50 | 25 (81%) | 21 (68%) | 23 (74%) | ||||
| 51–70 | 26 (79%) | 25 (76%) | 32 (97%) | ||||
| >70 | 3 (60%) | 3 (60%) | 4 (80%) | ||||
| Refused | 1 (100%) | 1 (100%) | 1 (100%) | ||||
| Race | White | 49 (83%) | 0.029 | 45 (76%) | 0.047 | 49 (83%) | 0.458 |
| Black | 6 (55%) | 5 (45%) | 8 (73%) | ||||
| Hispanic/Latino | 6 (100%) | 6 (100%) | 5 (83%) | ||||
| Other | 5 (56%) | 5 (56%) | 9 (100%) | ||||
| Education | ≤ High School | 32 (78%) | 0.932 | 30 (73%) | 0.781 | 31 (76%) | 0.057 |
| > High School | 34 (77%) | 31 (70%) | 40 (91%) | ||||
| Income | < $5,000 | 4 (67%) | 0.519 | 2 (33%) | 0.146 | 4 (67%) | 0.429 |
| $5,000–$20,000 | 9 (82%) | 9 (82%) | 9 (82%) | ||||
| $20,000–$40,000 | 17 (89%) | 17 (89%) | 17 (89%) | ||||
| $40,000–$60,000 | 10 (83%) | 9 (75%) | 11 (92%) | ||||
| $60,000–$80,000 | 10 (83%) | 9 (75%) | 11 (92%) | ||||
| > $80,000 | 11 (61%) | 10 (56%) | 14 (78%) | ||||
| Don’t know | 3 (75%) | 3 (75%) | 2 (50%) | ||||
| Refused | 2 (67%) | 2 (67%) | 3 (100%) | ||||
| Prior research experience | Yes | 4 (67%) | 0.612 | 4 (67%) | 1.000 § | 5 (83%) | 1.000 § |
| No | 62 (78%) | 57 (72%) | 66 (84%) | ||||
| Time from enrollment | 0–192 days (median) | 38 (88%) | 0.0163 | 33 (77%) | 0.320 | 36 (84%) | 0.962 |
| >192 days | 28 (67%) | 28 (67%) | 35 (83%) | ||||
The 6 Hispanic/Latino participants all agreed with both personal and general EFIC enrollment, but their level of acceptance was not statistically significantly different from white respondents given the small sample size. In contrast, 83% of white participants versus 55% of black participants agreed with personal EFIC enrollment (p=0.0494). Seventy-six percent of white respondents versus 45% of black respondents agreed with general EFIC enrollment (p=0.0647). There were no significant differences by race/ethnicity regarding being “glad about inclusion,” a question that did not mention EFIC. Differences regarding EFIC acceptance may thus trace specifically to EFIC.
There were no statistically significant associations observed between any form of EFIC acceptance and education or income, although extremes of income illustrated a trend toward lower acceptance. There was also no significant relationship between acceptance of EFIC and whether the respondent believed their initial enrollment (or the patient’s) was without consent. However, several respondents who believed they (or the patient) were enrolled with prospective consent said they did not agree with personal EFIC enrollment because that did not apply to them.
Proximity to trial enrollment did appear to affect acceptance of EFIC. Respondents interviewed closer to initial enrollment (prior to the median of 192 days) were more accepting of personal EFIC enrollment than those interviewed later (88% vs 67%, p=0.0163). No statistically significant relationship was observed between proximity to trial enrollment and general EFIC acceptance or general attitude toward ProTECT III inclusion.
Reasons for Views and Understanding of Study Content
Common reasons for positive and negative views are displayed in Table 4. These reasons are independent of Likert scale responses regarding EFIC acceptance. For example, some respondents had concerns about EFIC but accepted it. Concern about lack of consent was the most frequent reason for a negative view (26%). Other notable concerns included: believing that others may not be as accepting of EFIC; potential for side effects; uncertainty about risk-benefit ratio; not wanting to be experimented on; and use of placebo.
Table 4.
Positive and negative reasons for views of EFIC in ProTECT III
| Negative reasons given | N | % |
|---|---|---|
| Concerns about consent | 22 | 26% |
| Other people may not be as accepting | 14 | 16% |
| Concerned about potential side effects | 11 | 13% |
| Unsure about balance of risks/benefits in trial | 7 | 8% |
| Believes drug is experimental (guinea pig concerns) | 6 | 7% |
| Concerned about placebo being given | 6 | 7% |
| Lack of medical benefit (or any additional benefit from standard) | 5 | 6% |
| Others disagreed with enrollment (combined: family members or patient themselves had well known negative feelings) | 5 | 6% |
|
| ||
| Positive reasons given | ||
|
| ||
| Direct medical benefits | 75 | 88% |
| Unable to get consent | 33 | 39% |
| Risks of study are low/no harm done | 30 | 35% |
| Contribute to scientific knowledge/help future patients | 27 | 32% |
| Other people agreed with enrollment (combined: family members/LARs, medical personnel, patient themselves, religious figure) | 22 | 26% |
| Trust in researchers/doctors to do what is best | 20 | 24% |
| In case of an emergency, do what needs to be done | 18 | 21% |
| Patient so badly injured that it couldn’t hurt/last option | 12 | 14% |
| Research is important | 9 | 11% |
The large majority (88%) stated the presence of, or potential for, direct medical benefit as a reason for positive views about EFIC and ProTECT III. More than a third (39%) cited the inability to get consent under the clinical circumstances as a reason for accepting EFIC, and 35% felt EFIC was acceptable because study risks were low. Other common reasons included: a desire to contribute to science; reassurance from the fact that other individuals (family members, medical personnel, religious figures, or patients in the case of surrogates) agreed with enrollment; trust that researchers and doctors were doing what they consider best for patients; the belief that in emergencies, medical teams should “do whatever needs to be done;” and the belief that the patient was so badly injured that enrollment could not cause harm and might be helpful.
Placebo and Randomization
Following a description of the reasons for placebo use, respondents were asked to rate their views of the acceptability of placebo. Sixty-eight (80%) respondents either agreed or strongly agreed with the statement: “I think that it was acceptable for researchers to give half of the patients in the ProTECT III study placebo and half of the patients progesterone.” Most stated the need for adequate comparative data as a justification, though some (n=17) who accepted placebo use did express reluctance, often stating a desire to ensure or increase the likelihood of receiving active drug. Most respondents (92%) agreed with the statement: “I think that it was acceptable for researchers to assign treatments at random in this study.”
Misunderstanding of ProTECT III and study elements
Seven respondents illustrated major misconceptions about the study’s design. For example, 2 thought that each participant would receive both placebo and progesterone; 2 others thought consent constituted choosing progesterone over placebo. Ten respondents had misunderstandings about the placebo, 9 of whom believed that it was active treatment. Eleven respondents failed to understand randomization, often conflating it with doctors’ choosing a specific therapy. In total, 21 respondents (25%) illustrated at least one form of misconception regarding ProTECT III. In the PEER-RAMPART study, 62% illustrated misconceptions, typically related to randomization (6).
Other Domains
Respondents were asked about community consultation and public notification processes required by regulation. Eighty-one (95%) respondents felt community consultation is important. When asked if there were particular people or groups that researchers should engage before starting an EFIC study, 26% suggested TBI patients and families, 22% mentioned medical professionals, and only 5% thought the general public should be consulted. Other suggestions included politicians, religious leaders, and people at risk for TBI. Of 85 respondents, only 1 (a nurse) had heard of the ProTECT III study prior to enrollment.
DISCUSSION
EFIC research is essential to advancing treatment for a wide range of acute illnesses. These studies produce important societal benefits that are otherwise unattainable, but it is important to maximize the extent to which these studies respect participants’ concerns and experiences. As a placebo-controlled trial of an investigational agent in a condition with high morbidity and mortality, ProTECT III represents an ideal context for studying perceptions of EFIC-enrolled patients and surrogates. Using a design that allows both quantitative comparison of respondents’ views and contextualization of responses, this study strengthens understanding of EFIC research and carries important practical implications for research spanning a broad range of acute conditions.
Most directly, these findings demonstrate significant acceptance of EFIC among the most relevant population. With nearly 80% accepting of personal EFIC enrollment in this trial and only 4% clearly disagreeing with personal enrollment, these data suggest that enrollment was largely consistent with the preferences of most enrollees. These findings cohere with limited data from other populations (6, 10), but ProTECT III is notable in using a placebo design and an experimental agent; two features representing potential concerns among enrollees (9). Importantly, there was no relationship between placebo acceptance and EFIC acceptance, and most concerns about placebo use related to a preference for an active investigational agent that was otherwise unavailable.
Despite the low rate of objections, these findings demonstrate the predictable fact that EFIC trials unavoidably enroll some individuals who would prefer not to be enrolled. Importantly, most of the concerns expressed focused on lack of consent and general uncertainty about risk and benefit in research rather than specific features of this investigational agent. These concerns do not undermine the justification for EFIC, but the presence of individuals who would prefer not to be included reinforces the need to ensure appropriate opportunities to opt out of inclusion. In this and other studies, public disclosure efforts appear to have very low penetration, making it difficult to effectively identify those who would want to be excluded in advance. Moreover, many opt-out options (e.g. wearing a bracelet for years) place some burden on patients who wish to do so. Given these difficulties, an important area for further discussion and research is the clarification of appropriate opt-out strategies near the time of enrollment. Whether such opportunities should be offered over the telephone, for example, or in other situations where decisions are likely to be suboptimally informed is an area that warrants further discussion and clarification.
On the whole, these participants had better understanding of the parent trial than has been observed previously. Many respondents, for example, seemed to understand both the scientific necessity of a placebo and the need for EFIC in acute care research. There are several reasons this may be the case. First, ProTECT III involves a four-day infusion and long-term follow-up after discharge. These elements likely contributed to confusion about timing of consent and study initiation; however, they also allowed multiple interactions with study staff and probably contributed to increased understanding of essential features of the ProTECT III study compared to PEER-RAMPART respondents. As has been observed in other research contexts (20–22), a sizeable subgroup still did not fully understand randomization and placebo use despite a highly interactive interview methodology designed to maximize respondent engagement and understanding. We suspect that the same misunderstandings are widespread among community consultation participants as well, which is important to appreciate in interpreting community consultation feedback.
These data also inform community consultation efforts more directly. EFIC acceptance exceeded acceptance observed in a large study of community consultation participants across 12 ProTECT III sites. In that study, 71% and 54% were accepting of personal and general EFIC enrollment respectively, compared to 78% and 72% here (19). Similar patterns were found in RAMPART-related studies conducted within the same network (6, 13, 23). It is reassuring that community consultation activities do not overestimate enrollees’ acceptance, though questions remain regarding whether community consultation adequately represents substantive views or concerns of enrollees, information that may not be reflected in acceptance rates alone. It would also be problematic if community consultation efforts with lower estimates of acceptance resulted in disapproval of studies that are acceptable to and important for patients affected by the condition. Some of these concerns are echoed by these respondents’ lack of enthusiasm for community consultation efforts targeting the general public.
Findings of lower EFIC acceptance among black respondents are important and have been suggested elsewhere, though these differences were not observed in the larger ProTECT community consultation study (6, 19). Whether the high rate of acceptance among Hispanic subjects would persist in larger samples is unknown. At the very least, these data suggest a need for careful attention to the racial and ethnic composition of community consultation activities and to potential background concerns about exploitation in the black community (24, 25). They also clarify the need for larger scale data regarding the impact of demographic characteristics on participants’ views of the importance of consent and of EFIC trial enrollment. Additionally, the gender-based differences observed in this study highlight the need to assess whether these differences are really driven by the experience of being a patient versus a surrogate. These differences were not observed, for example, in the ProTECT community consultation study (19).
Several limitations are important. First, this descriptive study had limited ability to examine associations between respondent characteristics and EFIC views. Larger studies, using less in-depth methods and driven by hypotheses from this and related work, will help to assess important issues such as racial and other demographic predictors of attitudes as well as differences between patients and surrogates. Second, we cannot presently assess relationships between clinical outcomes and attitudes, because outcomes data are still being collected in the parent trial. Importantly, the few respondents who were surrogates of individuals who had died did not demonstrate significantly negative attitudes toward the study or enrollment, suggesting that poor clinical outcomes may not necessarily correlate with negative perceptions of enrollment. Third, while understanding of ProTECT III was reasonably robust, some positive views may suggest therapeutic misconception or misestimation, both commonly seen in research subjects (26, 27). We did not specifically assess therapeutic misconception or misestimation, but there was no overtly disproportionate effect in this study. While it is concerning that 25% of respondents had some level of misunderstanding of study details, this is not unusual in other research contexts as well (20, 28–30). Awareness of such prevalent misunderstanding is essential to contextualizing these and other empirical data on patients’ views of clinical research.
Several sources of potential bias are also important to recognize. While most non-responders did not overtly refuse participation, non-response may indicate a negative attitude toward the ProTECT III study. Interactive interview methods also entail a potential for interviewer bias. This was recognized and minimized by training and the use of a small number of interviewers. Moreover, this method facilitated contextualization of answers, allowed respondents to form views on unfamiliar topics, and provided insights regarding respondents’ understanding. These are critical features given the minimal data available in this area. Finally, the long amount of time between study enrollment and some interviews could have introduced recall bias. There was, for example, increased acceptance of personal EFIC enrollment among respondents interviewed closer to the time of enrollment. The extent to which individual respondents’ attitudes toward EFIC enrollment actually changed over time cannot be ascertained in this cross-sectional study. Similarly, these data do not allow for a determination of the extent to which these differences were driven by short versus long-term patient outcomes.
CONCLUSIONS
Well-designed randomized trials in the context of severe, acute illness have advanced care for important public health threats, and more research is needed to reduce morbidity and mortality in many of these conditions. This study provides reassurance that trials in acute illness using EFIC and placebo designs do not appear to conflict with most enrolled patients’ wishes. This is an important determinant of these trials’ ethical acceptability. Continued assessment of the views of enrolled patients in the context of future EFIC trials will help to confirm whether these findings persist across a range of conditions and study types. Further studies may also help to refine community consultation and opt-out efforts so that they most effectively assess and address the impact on and concerns of the most directly affected populations- enrolled patients and their families.
Supplementary Material
Acknowledgments
We are grateful to participating sites as well as to members of the NETT Human Subjects Protection Working Group for their involvement and insights regarding this study. We also thank Karen Buerkle and Lynn Pelicano at APCO Insight.
This study was funded by an award from the Greenwall Foundation (PI, Dickert). The parent study was funded by the PROTECT III™ grant from the National Institute of Neurological Disorders and Stroke (5U01NS062778). Dr. Dickert was supported by an award from the National Heart Lung and Blood Institute (F32HL095358-01A1) during a portion of the study period. Study sponsors played no role in the design or conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Financial Support: This work was supported by the Greenwall Foundation and the National Heart, Lung, and Blood Institute (F32HL095358-01A1). The parent trial was supported by the National Institute for Neurological Disorders and Stroke (5U01NS062778).
Copyright form disclosures
Dr. Dickert served as a board member for the National Heart, Lung, and Blood Institute (NHBLI) CT Surgery Clinical Trials Network Date Safety and Monitoring Board and received support for article research from the National Institutes of Health (NIH). His institution received grant support from The Greenwall Foundation (Dr. Dickert is a Greenwall Faculty Scholar and receives salary support for other projects from this funder) and from the NHBLI. Dr. Scicluna received support for article research from the NIH. Her institution received grant support from the NIH-NINDS and the NIH. Dr. Baren’s institution received grant support from the NINDS, NHLBI, and NICHD. Dr. Biros received royalties from Rosen’s textbook of E Med/Up to Date (editor of subsection of textbook). Her institution received grant support from NINDS. Dr. Jones received support for article research from the NIH. Her institution received grant support from the NIH NINDS (Dr. Jones is PI for a NIH NETT grant which supports emergency neurological research and for the ProTECT study), received support for travel from the NIH NINDS (Travel to ProTECT meetings has been paid by the ProTECT grant), and received grant support from the NIH NINDS (NEET grant). Dr. Pancioli received support for article research from the NIH. His institution received grant support (NIH funding only). Dr. Wright received support for article research from the NIH, is employed by Emory University, provided expert testimony for a Variety of Malpractice Cases, and received grant support from the NIH and DOD. Dr. Wright and his institution received royalties from Emory/BHR (BHR Licensed Progesterone for TBI from Emory. Dr. Wright is an Inventor). His institution received grant support from the NIH (NIH Grant for Parent study) and has a patent with Emory (BHR Licensed Progesterone for TBI). Dr. Pentz served as a board member for St. Jude DSMB, received support from the NIH Biobanking Working Group and the NIH Ethical and Regulatory Issues in Emergency Medicine, and received support for article research from the NIH. Her institution received grant support from NINDS and from PCORI.
Footnotes
CONFLICTS OF INTEREST
Dr. Wright is entitled to royalty through Emory University, derived from any BHR Pharma sales of progesterone for use in traumatic brain injury. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict of interest policies. The authors report no additional conflicts of interest relevant to this manuscript.
The remaining authors have disclosed that they do not have any potential conflicts of interest.
Contributor Information
Neal W Dickert, Emory University School of Medicine, Department of Medicine, Division of Cardiology, Rollins School of Public Health, Department of Epidemiology, and Atlanta VA Medical Center, Atlanta, GA.
Victoria M Scicluna, Emory University School of Medicine, Atlanta, GA.
Jill M Baren, University of Pennsylvania Perelman School of Medicine, Department of Emergency Medicine, Philadelphia, PA.
Michelle H Biros, University of Minnesota School of Medicine, Department of Emergency Medicine, Hennepin County Medical Center, Minneapolis, MN.
Ross J Fleischman, Harbor-UCLA Medical Center, Department of Emergency Medicine, Torrance, CA.
Prasanthi R Govindarajan, University of California San Francisco School of Medicine, Department of Emergency Medicine, San Francisco, CA.
Elizabeth B Jones, University of Texas Medical School at Houston, Department of Emergency Medicine, Houston, TX.
Arthur M Pancioli, University of Cincinnati School of Medicine, Department of Emergency Medicine, Cincinnati, OH.
David W Wright, Emory University School of Medicine, Department of Emergency Medicine, Atlanta, GA.
Rebecca D Pentz, Emory University School of Medicine, Winship Cancer Institute, Atlanta, GA.
References
- 1.Emanuel EJ, Wendler D, Grady C. What Makes Clinical Research Ethical? JAMA. 2000;283(20):2701–11. doi: 10.1001/jama.283.20.2701. [DOI] [PubMed] [Google Scholar]
- 2.U.S. Department of Health and Human Services. Title 45 (Code of Federal Regulations), Part 46 Protection of Human Subjects [Google Scholar]
- 3.U.S. Department of Health and Human Services and Food and Drug Administration. Guidance for Institutional Review Boards, clinical investigators, and sponsors: Exception from informed consent requirements for emergency research. Rockville, MD: U.S. Food and Drug Administration; 2013. http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm249673.pdf. Accessed July 14, 2014. [Google Scholar]
- 4.U.S. Food and Drug Administration. Title 21 (Code of Federal Regulations), Part 50.24 Protection of Human Subjects. 2004 [Google Scholar]
- 5.Largent EA, Wendler D, Emanuel E, Miller FG. Is emergency research without initial consent justified?: the consent substitute model. Arch Intern Med. 2010;170(8):668–74. doi: 10.1001/archinternmed.2010.80. [DOI] [PubMed] [Google Scholar]
- 6.Dickert NW, Mah VA, Baren JM, Biros MH, Govindarajan P, Pancioli A, et al. Enrollment in research under exception from informed consent: The Patients’ Experiences in Emergency Research (PEER) study. Resuscitation. 2013 doi: 10.1016/j.resuscitation.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potter JE, McKinley S, Delaney A. Research participants’ opinions of delayed consent for a randomised controlled trial of glucose control in intensive care. Intensive Care Med. 2013;39(3):472–80. doi: 10.1007/s00134-012-2732-8. [DOI] [PubMed] [Google Scholar]
- 8.Kasner SE, Baren JM, Le Roux PD, Nathanson PG, Lamond K, Rosenberg SL, et al. Community views on neurologic emergency treatment trials. Ann Emerg Med. 2011;57(4):346–54 e6. doi: 10.1016/j.annemergmed.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Dickert N, Kass NE. Patients’ Perceptions of Research in Emergency Settings: a Study of Survivors of Sudden Cardiac Death. Social Science and Medicine. 2009;68(1):183–91. doi: 10.1016/j.socscimed.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kämäräinen A, Silfvast T, Saarinen S, Virta J, Virkkunen I. Conduct of emergency research in patients unable to give consent—Experiences and perceptions of patients, their consent providing next of kin, and treating physicians following a prehospital resuscitation trial. Resuscitation. 2012;83(1):81–5. doi: 10.1016/j.resuscitation.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 11.Lo B. Strengthening community consultation in critical care and emergency research. Crit Care Med. 2006;34(8):2236–8. doi: 10.1097/01.CCM.0000229632.85246.3A. [DOI] [PubMed] [Google Scholar]
- 12.Bulger EM, Schmidt TA, Cook AJ, Brasel KJ, Griffiths DE, Kudenchuk PJ, et al. The random dialing survey as a tool for community consultation for research involving the emergency medicine exception from informed consent. Ann Emerg Med. 2009;53(3):341–50. 50 e1–2. doi: 10.1016/j.annemergmed.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biros MH, Sargent C, Miller K. Community attitudes towards emergency research and exception from informed consent. Resuscitation. 2009;80(12):1382–7. doi: 10.1016/j.resuscitation.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lecouturier J, Rodgers H, Ford GA, Rapley T, Stobbart L, Louw SJ, et al. Clinical Research Without Consent in Adults in the Emergency Setting: a Review of Patient and Public Views. BMC Medical Ethics. 2008;9:9. doi: 10.1186/1472-6939-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Contant C, McCullough LB, Mangus L, Robertson C, Valadka A, Brody B. Community consultation in emergency research *. Critical Care Medicine. 2006;34(8):2049–52. doi: 10.1097/01.CCM.0000227649.72651.F1. [DOI] [PubMed] [Google Scholar]
- 16.Abboud PA, Heard K, Al-Marshad AA, Lowenstein SR. What determines whether patients are willing to participate in resuscitation studies requiring exception from informed consent? J Med Ethics. 2006;32(8):468–72. doi: 10.1136/jme.2005.012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mason J. Qualitative Researching. 2nd. Thousand Oaks: SAGE Publications; 2002. [Google Scholar]
- 18.Cassell C. The SAGE Dictionary of Qualitative Management Research. SAGE Publications Ltd. London, United Kingdom: SAGE Publications Ltd; 2008. TEMPLATE ANALYSIS. [Google Scholar]
- 19.Dickert N, Mah VA, Biros MH, Harney D, Silbergleit R, Sugarman J, et al. Consulting Communities When Patients Cannot Consent: A Multicenter Study of Community Consultation for Research in Emergency Settings. Critical Care Medicine. 2014;42(2):272–80. doi: 10.1097/CCM.0b013e3182a27759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snowdon C, Garcia J, Elbourne D. Making sense of randomization; responses of parents of critically ill babies to random allocation of treatment in a clinical trial. Soc Sci Med. 1997;45(9):1337–55. doi: 10.1016/s0277-9536(97)00063-4. [DOI] [PubMed] [Google Scholar]
- 21.Featherstone K, Donovan JL. Random allocation or allocation at random? Patients’ perspectives of participation in a randomised controlled trial. BMJ. 1998;317(7167):1177–80. doi: 10.1136/bmj.317.7167.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SY, de Vries R, Wilson R, Parnami S, Frank S, Kieburtz K, et al. Research participants’ “irrational” expectations: common or commonly mismeasured? IRB. 2013;35(1):1–9. [PubMed] [Google Scholar]
- 23.Govindarajan P, Dickert NW, Meeker M, De Souza N, Harney D, Hemphill CJ, et al. Emergency research: using exception from informed consent, evaluation of community consultations. Acad Emerg Med. 2013;20(1):98–103. doi: 10.1111/acem.12039. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt TA. The legacy of the Tuskegee syphilis experiments for emergency exception from informed consent. Ann Emerg Med. 2003;41(1):79–81. doi: 10.1067/mem.2003.17. [DOI] [PubMed] [Google Scholar]
- 25.Holloway KF. Accidental Communities: Race, Emergency Medicine, and the Problem of PolyHeme®. American Journal of Bioethics. 2006;6(3):7–17. doi: 10.1080/15265160600685556. [DOI] [PubMed] [Google Scholar]
- 26.Appelbaum PS, Lidz CW, Grisso T. Therapeutic misconception in clinical research: frequency and risk factors. IRB. 2004;26(2):1–8. [PubMed] [Google Scholar]
- 27.Horng S, Grady C. Misunderstanding in clinical research: distinguishing therapeutic misconception, therapeutic misestimation, and therapeutic optimism. IRB. 2003;25(1):11–6. [PubMed] [Google Scholar]
- 28.Fallowfield L, Jenkins V, Brennan C, Sawtell M, Moynihan C, Souhami R. Attitudes of patients to randomised clinical trials of cancer therapy. European Journal of Cancer. 1998;34(10):1554–9. doi: 10.1016/s0959-8049(98)00193-2. [DOI] [PubMed] [Google Scholar]
- 29.Nishimura A, Carey J, Erwin PJ, Tilburt JC, Murad MH, McCormick JB. Improving understanding in the research informed consent process: a systematic review of 54 interventions tested in randomized control trials. BMC Med Ethics. 2013;14:28. doi: 10.1186/1472-6939-14-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Falagas ME, Korbila IP, Giannopoulou KP, Kondilis BK, Peppas G. Informed consent: how much and what do patients understand? Am J Surg. 2009;198(3):420–35. doi: 10.1016/j.amjsurg.2009.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.