

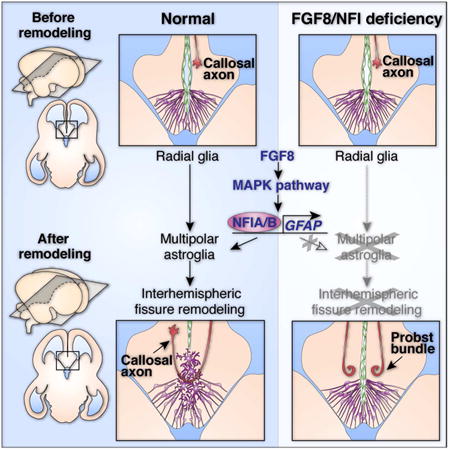
Abstract
The corpus callosum is the major axon tract that connects and integrates neural activity between the two cerebral hemispheres. Although ∼1:4000 children are born with developmental absence of the corpus callosum, the primary etiology of this condition remains unknown. Here, we demonstrate that midline crossing of callosal axons is dependent upon the prior remodeling and degradation of the intervening interhemispheric fissure. This remodeling event is initiated by astroglia either side of the interhemispheric fissure, which intercalate with one another and degrade the intervening leptomeninges. Callosal axons then preferentially extend over these specialized astroglial cells to cross the midline. A key regulatory step in interhemispheric remodeling is the differentiation of these astroglia from radial glia, which is initiated by Fgf8 signaling to downstream Nfi transcription factors. Crucially, our findings from human neuroimaging studies reveal that developmental defects in interhemispheric remodeling are likely to be a primary etiology underlying human callosal agenesis.
Keywords: astrocyte, callosal agenesis, interhemispheric fissure, Fgf8, Nfia, Nfib
Graphical abstract
Introduction
A significant milestone in the evolution of the mammalian brain was the emergence of the corpus callosum, which is exclusively present in placental mammals (Suárez et al., 2014b and references therein). This commissure forms the largest axon tract in the human brain, and is required for the integration of sensory, motor and associative processes between the two cerebral hemispheres (Gazzaniga, 2005; Paul et al., 2007). In congenital absence (or agenesis) of the corpus callosum, callosal axons often extend towards the midline but are unable to cross, affecting approximately 1:4000 live births (Hetts et al., 2006). This results in the aberrant accumulation of axons either side of the interhemispheric midline into structures known as Probst bundles (Probst, 1901). Callosal agenesis results in a wide spectrum of neurological deficits, however the primary causes underlying this major brain malformation remain unknown (Edwards et al., 2014; Paul et al., 2007).
During development, callosal connections form between the cerebral hemispheres through a midline region that is formerly separated by a deep interhemispheric fissure (IHF; Rakic and Yakovlev, 1968; Silver et al., 1993; Silver et al., 1982). This structure is primarily composed of leptomeningeal fibroblasts and extracellular matrix (Siegenthaler and Pleasure, 2011; Silver et al., 1982). Given that the cellular composition of the IHF is distinct from the surrounding neuroepithelial tissue, a key unanswered question is how callosal axons navigate across this midline territory to form a tract. Previously, it has been proposed that callosal axons cross directly through the IHF and physically interact with the leptomeningeal tissue during tract formation (Choe et al., 2012). However, this scenario is not consistent with data indicating that the substrate underlying the early callosal tract is comprised of astroglial cells rather than leptomeningeal fibroblasts (Silver et al., 1993; Silver et al., 1982). These cells express the astroglial marker Gfap and are known as the midline zipper glia (MZG; Shu et al., 2003b; Silver et al., 1993). The spatiotemporal distribution of these cells suggests that, rather than using the leptomeninges, callosal axons may instead require astroglial cells as an underlying substrate to cross the midline. Determining how callosal axons navigate the IHF during development is key to understanding the etiology of callosal agenesis, yet to date no experimental investigations have been performed to resolve this question.
Using in utero electroporation in mice, we find that pioneer callosal axons exclusively use MZG cells as a substrate to cross into the contralateral hemisphere, instead of crossing directly through the interhemispheric fissure. Further, we demonstrate that MZG cells actively remodel the interhemispheric fissure just prior to callosal tract formation, and that the differentiation of MZG cells from radial glia is a key regulatory step in this remodeling process. Crucially, we find that either delayed or precocious MZG differentiation prevents interhemispheric remodeling from occurring correctly, thus affecting subsequent callosal tract formation. In vivo gain- and loss-of-function experiments in mice further reveal that MZG differentiation is initiated by Fgf8 signaling through the Map kinase pathway to activate pro-astrogliogenic Nuclear factor one (Nfi) transcription factors. Moreover, our observations from human neuroimaging studies and analysis of human NFI mutations suggest that developmental defects in interhemispheric remodeling are likely to be a primary etiology leading to human callosal agenesis. Together, these findings reveal that remodeling of the interhemispheric midline is an astroglial-mediated process that is crucial for corpus callosum formation during placental brain development.
Results
Pioneer callosal axons do not cross through the IHF but rather use the MZG as a growth substrate to cross the midline
To investigate whether callosal axons cross directly through the IHF during development as suggested previously (Choe et al., 2012), we labeled individual pioneer callosal axons with a membrane-bound tdTomato reporter and observed their precise growth trajectory as they cross the midline. Neurons in the mouse cingulate cortex were electroporated at embryonic day (E)13 and examined at E16 in combination with pan-Laminin immunofluorescence, which labels leptomeningeal fibroblasts and the basement membrane (Halfter et al., 2002). This revealed that growing callosal axons always project around the basement membrane of the IHF, and do not pass directly through this structure (Figure 1). We then investigated whether, instead, callosal axons use astroglial cells as a growth substrate, as suggested by Silver et al. (1993; 1982). Dual labeling for the glial marker Glast and pan-Laminin revealed that callosal axons preferentially cross the midline through the MZG astroglial population, which is found at the base of the IHF (Figure 1A and 1B). Together, these observations suggest that the basement membrane and leptomeninges form a non-permissive barrier to callosal growth, and that the position of astroglia along the interhemispheric midline determines the precise site at which callosal axons cross the interhemispheric midline.
Figure 1. Pioneer callosal axons do not cross through the IHF but rather use the MZG as a growth substrate to cross the midline.
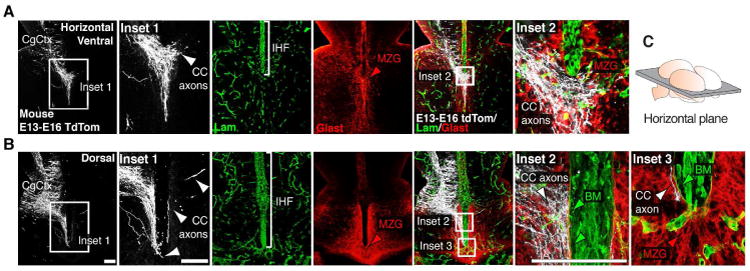
(A-B) Electroporation of the mouse cingulate cortex (CgCtx) at E13 with a myristoylated tdTomato reporter plasmid (white) labels pioneering callosal axons at E16, visualized in relation to Glast-positive MZG cells (red) and the Laminin-positive interhemispheric fissure (IHF; green) in ventral (A) and dorsal (B) horizontal sections. Callosal axons (white arrowheads) project around the basement membrane (BM) of the IHF (green arrowheads) and across the midline over MZG cells (red arrowheads). (C) Schematic showing the orientation of the horizontal plane shown in A and B. CC, corpus callosum. Scale bars, 100μm.
Progressive remodeling of the IHF occurs simultaneously with callosal tract formation and expansion
In order to further elucidate the developmental relationship between the IHF, MZG cells and the corpus callosum, we performed a spatio-temporal analysis of these structures throughout forebrain development. In mice, the IHF forms between E9 and E12 (Figure 2A). Following formation of the IHF at E12, the telencephalic hemispheres are joined at the midline only by a small hinge of tissue (defined here as the telencephalic hinge; Figure 2B). At this stage, the IHF can be delineated by pan-Laminin immunohistochemistry (Figure 2B). From E12 to E15, the IHF appears to be gradually compressed by the bilateral expansion of the septum, which culminates in fusion of the septal halves at E15 (Figure 2B and Figure S1). During this event, Nestin-positive glial fibers bridge the septal midline, and the Laminin-positive IHF retracts rostrally. Furthermore, from E16 to E17, the amount of glial fibers bridging the midline increases considerably, whilst the IHF continues to retract further rostrally (Figure 2B), suggesting that the fissure may undergo active tissue remodeling during this period. Critically, the growth of callosal axons across the midline coincides precisely with the formation of glial fibers across the interhemispheric midline (Figure 2C and 2D). This suggests that continuous expansion of the MZG population and simultaneous retraction of the fissure along the midline may facilitate the addition of fibers to the callosal tract. Quantification of IHF length throughout development revealed that the IHF is progressively eliminated from the septum along an oblique plane, beginning from the ventro-caudal base of the septum at E15 and ending at the dorso-rostral apex of the septum at approximately E18 (Figure S1). Furthermore, we observed a similar oblique progression of interhemispheric remodeling in the developing human brain (Figure S1). This suggests that developmental remodeling of the IHF may be evolutionarily conserved in placental mammals and facilitate the formation of the corpus callosum across the midline in these mammals.
Figure 2. Developmental remodeling of the IHF is associated with callosal tract formation in mice.
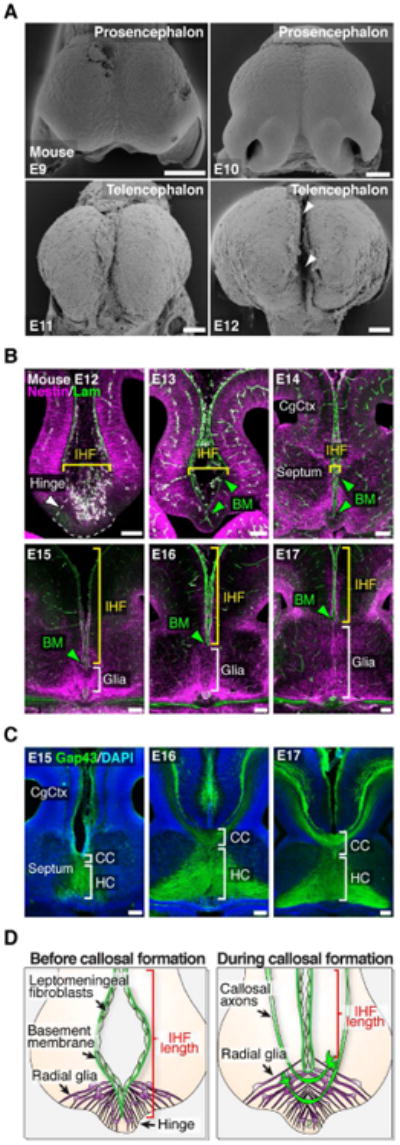
(A) Frontal view scanning electron microscopy photomicrographs show the development of the mouse IHF (arrowheads). (B) Fluorescence immunohistochemistry for Nestin-positive radial glia (magenta) and pan-Laminin-positive leptomeninges and basement membrane (BM; green) from E12 to E17. Brackets indicate the spatial extent of radial glia and IHF. Green arrowheads denote the BM. (C) Fluorescence immunohistochemistry for Gap43 labeling corpus callosum (CC) and hippocampal commissure (HC) axons from E15 to E17. (D) Schema of the midline remodeling events occurring before and during callosal tract formation. CgCtx, cingulate cortex. Scale bars, 100 μm. See also Figure S1.
The transition of radial MZG into multipolar astroglia initiates IHF remodeling
Immunohistochemistry for the MZG marker Gfap in developing human and mouse brains further revealed that, as the fissure is eliminated, MZG cells transition from two columns of radial (or bipolar) cells on either side of the fissure to a single column of tightly intercalated multipolar cells along the midline (Figure 3A). Intercalation of multipolar MZG across the fissure may therefore regulate the remodeling of this structure in humans and mice. Prior to E16, we identified radial MZG cells with Glast immunolabeling (Mori et al., 2006), as Glast is expressed before Gfap in these cells (Figure S2). Along the interhemispheric midline, Glast labeling was concentrated within the telencephalic hinge region (Figure S2), suggesting that radial MZG cells may derive from this progenitor niche. To study the progeny of cells born within the telencephalic hinge, we electroporated this region at E12 with a piggyBac Glast transposase/GFP reporter system (Chen and LoTurco, 2012). This labeled radial MZG progenitor cells undergoing somal translocation towards the surface of the IHF at E15, as well as intercalated multipolar MZG cells at the midline (Figure 3B and Figure S2). Moreover, EdU-birthdating of Glast-positive MZG progenitors within the telencephalic hinge (Figure 3C) also suggests that increased proliferation of radial MZG progenitors either side of the IHF gradually compresses this structure and primes this region for remodeling between E12 and E15.
Figure 3. The transition of radial MZG into multipolar astroglia initiates IHF remodeling and callosal tract formation.
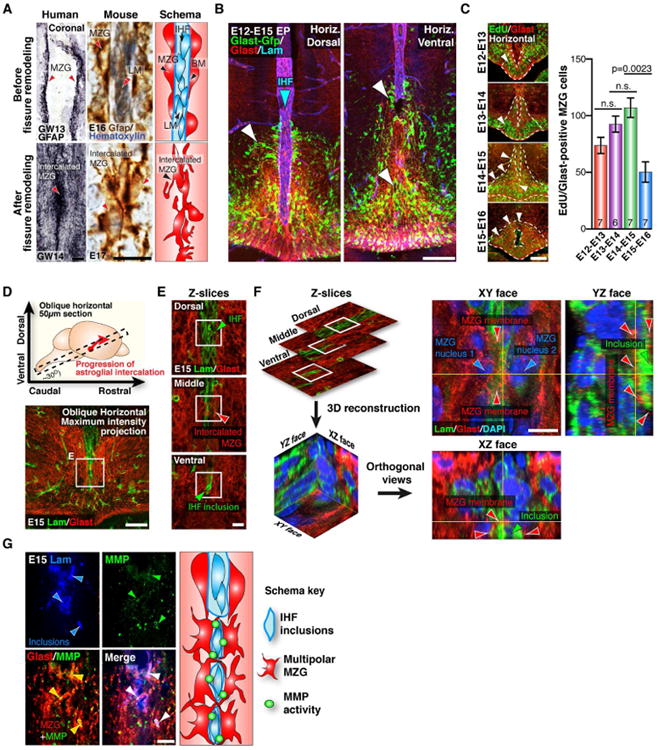
(A) Embryonic human and mouse brain sections immunostained for Gfap (black and brown, respectively), counterstained with hematoxylin (blue) in mouse sections. Gfap-positive MZG cells (arrowheads) are schematically represented far-right panel. (B) In utero electroporation (EP) of the mouse telencephalic hinge with piggyBac Glast transposase/GFP reporter plasmids (green), colabeled for Glast (red) and Laminin (blue) imunohistochemistry in dorsal and ventral sections. GFP labeling shows both MZG cells (white arrowheads) undergoing somal translocation towards the IHF (blue arrowhead) and MZG cells undergoing intercalation. (C) Glast-positive mouse MZG cells (red) were birthdated with the thymidine analogue EdU (green) every 24 hours, from E12 to E15, quantified on the right. Arrowheads show distribution of MZG progenitors within the telencephalic hinge niche (dotted lines). (D) Schema of the oblique horizontal sectioning plane (top) and confocal maximum intensity projection of an E15 oblique horizontal section (bottom) immunostained with Laminin (green) and Glast (red). (E) Optical sectioning of the region shown in (D). (F) Reconstruction of z-planes in (E) to resolve the XY, XZ and YZ orthogonal views of the MZG-IHF interface, showing MZG projections (red arrowheads) partitioning laminin-positive inclusions of the IHF (green arrowheads). (G) Pan-matrix metalloproteinase activity labeled with MMPSense (green) is associated with Glast-positive glial membranes adjacent to Laminin-positive IHF inclusions (blue), with schema (right). BM= basement membrane, LM = leptomeninges. Data are represented as means ± SEM (n-values within bars). Scale bars, 50μm (A, left), 5μm (A, right), 100 μm (B, C, and D), 10μm (E, F and G). See also Figure S2.
Next we performed oblique horizontal sectioning to analyze the entire remodeling interface in a single plane (Figure 3D). Confocal optical sectioning further revealed that Glast-positive MZG cell bodies positioned on either side of the fissure extend multiple processes that intercalate with each other through the leptomeninges (Figure 3E). These processes form a meshlike structure that partitions inclusions of the Laminin-positive leptomeninges away from the remainder of the fissure (Figure 3E and 3F). Furthermore, MZG cells express Matrix metalloproteinase 2 (Mmp2) mRNA (Figure S2), and we identified that proteolytic pan-MMP activity (Bremer et al., 2001) localizes to the membrane of MZG cells as they intercalate around leptomeningeal inclusions (Figure 3G). These findings suggest that multipolar MZG cells actively degrade the leptomeninges as they intercalate with one another.
Fgf8 signaling regulates MZG development and IHF remodeling
Previously we have shown that the expression of the morphogen Fgf8 within the septal midline is critical for the tissue patterning of this region (Moldrich et al., 2010; Storm et al., 2006). We therefore investigated whether Fgf8 may also regulate later aspects of septal midline development such as interhemispheric midline remodeling. We found that Fgf8 protein and mRNA expression is particularly enriched at the remodeling interface in radial MZG progenitors undergoing somal translocation (Figures 4A, 4B, S3 and S4).
Figure 4. Fgf8 signaling is required for MZG development and interhemispheric remodeling in mice.
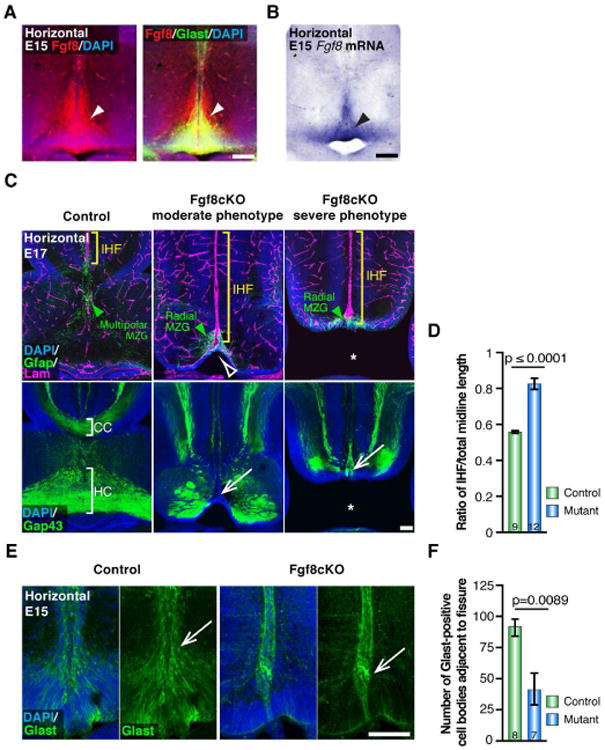
(A) Fluorescence immunohistochemistry shows expression of Fgf8 (red) in Glast-positive radial MZG (green) at E15 along the interhemispheric fissure (IHF; arrowheads). (B) In situ hybridization for Fgf8 mRNA. (C) Immunohistochemistry for Gfap (green), Laminin (magenta) and Gap43 (green) in E17 Fgf8 conditional knockout (cKO) mice and littermate controls. Note thinning of the MZG progenitor niche in moderate phenotype mutants (open arrowhead) versus complete absence of septum in severe mutants (asterisk). Green arrowheads indicate Gfap-positive MZG, yellow brackets indicate the IHF, and white arrows indicate complete agenesis of the corpus callosum and hippocampal commissure. (D) Quantification of IHF length in E17 Fgf8 cKO and control brains. (E) Immunohistochemistry for Glast (green) in E15 Fgf8cKO mutants and littermate controls, note reduction in Glast-positive MZG adjacent to the fissure pial surface in Fgf8cKO mutants (arrows). (F) Quantification of the number of Glast-positive cell bodies adjacent to the IHF in E15 Fgf8 cKO mice and littermate control brains. Data are represented as means ± SEM (n-values within bars). Scale bars, 100 μm. See also Figure S3.
To examine the functional consequence of reducing Fgf8 expression on interhemispheric remodeling, we analyzed the development of conditional Fgf8flox/Emx1Cre mutants, which exhibit loss of Fgf8 in the pallium and pallial derivatives of the septum from E10.5 (Figures 4C and S3; Moldrich et al., 2010). Homozygous Fgf8flox/Emx1Cre-positive (Fgf8cKO) mutants display moderate to severe midline defects compared to controls. While the septum is present in moderate Fgf8cKO mutants (14/25 analyzed), it is completely absent in severe Fgf8cKO mutants (11/25 analyzed), resulting in semilobar holoprosencephaly with communicating lateral ventricles (Figures 4C and S3; Moldrich et al., 2010). Notably, IHF remodeling failed to occur in either moderate or severe Fgf8cKO mutants, resulting in abnormal retention of the IHF and agenesis of the corpus callosum and hippocampal commissure in both mutant types (Figures 4C and 4D). An analysis of MZG distribution revealed that, at E15, just prior to the initiation of remodeling, there is a significant reduction in the number of Glast-positive MZG cells that have translocated to the pial surface of the IHF in Fgf8cKO mutants compared to controls (p = 0.0089, Mann-Whitney test, n ≥ 7 per condition; Figure 4E and 4F). Consistent with this observation, we found that Gfap-positive MZG cells maintain a radial morphology and fail to intercalate with one another across the fissure in E17 Fgf8cKO mutants (Figure 4C). Additionally, we observed that these aberrant radial MZG cells remain confined to the telencephalic hinge, and do not migrate rostrally (Figure 4C). These observations indicate that Fgf8 signaling is required for multiple aspects of MZG maturation, including the somal translocation of radial MZG cells to the pial surface of the IHF, as well as their subsequent transition into multipolar MZG cells.
To further investigate the requirement of Fgf8 signaling in MZG migration and maturation, we generated a gain-of-function model by electroporating an ectopic source of Fgf8 on one side of the midline at E12 (Figure 5A and Figure S4). Following analysis at E15, we observed an increase in the number of Glast-positive MZG cells translocating to the surface of the IHF, such that Glast fluorescence intensity is significantly increased on the electroporated side compared to control (p = 0.0095, Mann-Whitney test, n ≥ 4 per condition; Figure 5A and 5B). This demonstrates that Fgf8 overexpression is sufficient to elicit precocious somal translocation of radial MZG progenitors to the pial surface of the IHF.
Figure 5. Fgf8 signaling promotes astroglial maturation of the MZG in mice.
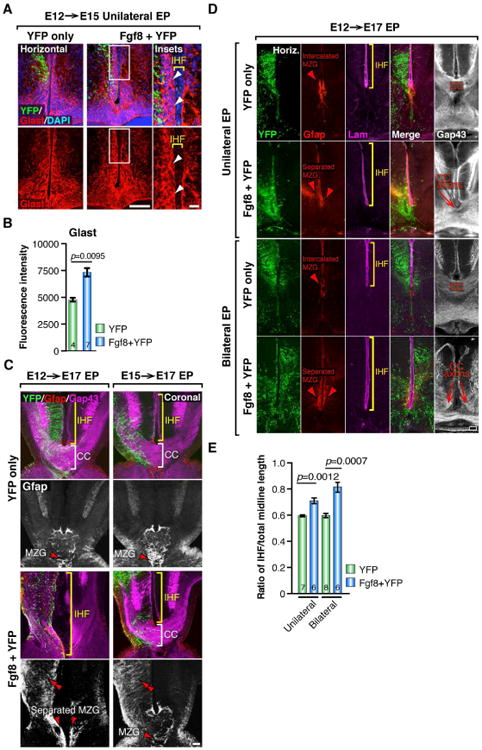
(A) Unilateral E12→E15 in utero electroporation (EP) of YFP alone or coelectroporation of Fgf8 + YFP plasmids followed by fluorescence immunohistochemistry for YFP (green) and Glast (red). Arrowheads indicate excess Glast-positive cell bodies along the pial surface of the Fgf8-electroporated hemisphere. (B) Quantification of Glast fluorescence intensity in YFP versus Fgf8+YFP electroporated hemispheres. (C) E12→E17 and E15→E17 unilateral EP of YFP alone or coelectroporation of Fgf8 + YFP plasmids followed by fluorescence immunohistochemistry for YFP (green), Gfap (red and white) and Gap43 (magenta) in coronal sections. Double red arrowheads indicate precocious Gfap expression following Fgf8 overexpression, brackets indicate the length of the IHF and the presence/absence of the corpus callosum (CC) in each condition, and single red arrowheads indicate the distribution of the MZG. (D) Unilateral or bilateral E12→E17 EP of Fgf8 + YFP followed by fluorescence immunohistochemistry for YFP, Gfap, Laminin and Gap43 in horizontal sections. Brackets indicate the IHF length in each condition, and arrowheads indicate the distribution of MZG cells. Note dysgenesis or agenesis of the CC in Fgf8 EP conditions. (E) Quantification of IHF length in unilateral and bilateral E12→E17 Fgf8 + YFP and YFP control electroporated brains. Data are represented as means ± SEM (n-values within bars). Scale bars, 100 μm (A, left; C and D), 20μm (20μm). See also Figure S4.
Next, we overexpressed Fgf8 unilaterally either before or after astroglial-remodeling of the fissure is initiated (E12 or E15, respectively), and compared the effects of each manipulation at E17, when fissure remodeling is almost complete. In both cases, we observed a dramatic increase in Gfap expression in the electroporated hemisphere (Figure 5C), demonstrating that Fgf8 is sufficient to induce precocious expression of this astroglial protein. Interestingly, however, we observed distinct midline phenotypes when Fgf8 was electroporated either before or after the initiation of fissure remodeling. Following Fgf8 overexpression at E12, we observed an increase in Gfap-positive MZG along the IHF on the electroporated side, resulting in disrupted MZG intercalation, abnormal retention of the IHF and stalling of callosal axons on either side of the fissure (Figure 5C, left; n = 6). In contrast, although overexpression of Fgf8 at E15 increased Gfap expression in the electroporated cingulate cortex, it had no effect on MZG development, IHF remodeling or callosal axon crossing (Figure 5C, right panels; n = 5). These results suggest that Fgf8 expression normally initiates somal translocation of radial MZG and the subsequent transition of these cells into multipolar Gfap-positive astroglia during the critical period of fissure remodeling, and that after MZG cells have differentiated, they are no longer responsive to this Fgf8-maturation signal.
To further investigate how precocious maturation of MZG cells may affect fissure remodeling, we examined E17 brains unilaterally electroporated with Fgf8 at E12 in horizontal sections. We found that unilateral overexpression of Fgf8 results in an asymmetric distribution of the MZG, with the electroporated hemispheres containing more Gfap-positive MZG cells compared to the unelectroporated hemispheres (Figure 5D). Interestingly, these asymmetrically distributed MZG populations fail to intercalate with one another, such that the Laminin-positive IHF is aberrantly retained in this region (11.46% increase in fissure length following unilateral Fgf8 overexpression, p = 0.0012, Mann-Whitney test, n ≥ 6 per condition; Figure 5D and 5E). Furthermore, Gap43-positive callosal axons are prevented from crossing the interhemispheric midline in this location (Figure 5D). Since maturation of the MZG normally occurs as a bilaterally symmetrical process, we then performed bilateral overexpression of Fgf8 on both sides of the midline at E12 to control for the potential effects of asymmetric MZG development. Comparison of E12 unilateral versus bilateral Fgf8 electroporations at E17 revealed that bilateral Fgf8 overexpression mirrors the effects of unilateral overexpression, resulting in excess Gfap-positive MZG cells either side of the IHF that fail to intercalate with one another (Figure 5D). Furthermore, we observed that aberrant retention of the IHF is further exacerbated following bilateral Fgf8 overexpression (21.83% increase in fissure length following bilateral Fgf8 overexpression compared to YFP controls, p = 0.0007, Mann-Whitney test, n ≥ 6 per condition; Figure 5D and 5E). Consequently, the majority of Gap43-positive callosal axons are unable to cross the IHF in these brains (Figure 5D). Moreover, consistent with our previous analysis of unilateral Fgf8 overexpression at E15 (Figure 5A), we observed a similar bilateral increase in the amount of Glast-positive MZG reaching the pial surface of the fissure at E15 following bilateral Fgf8 overexpression at E12, compared to YFP controls (p = 0.001, Mann-Whitney test, n ≥ 8 per condition; Figure S4). These results suggest that precocious MZG maturation, either unilaterally or bilaterally, prevents timely and sequential intercalation of MZG cells at the base of the IHF. Thus, we find that delayed MZG maturation in the absence of Fgf8, as well as precocious MZG maturation in the presence of excess Fgf8, both result in aberrant retention of the interhemispheric fissure and callosal agenesis. Collectively, these observations indicate that the correct level of Fgf8 expression at the midline is required at precise developmental stages in order for MZG-mediated fissure remodeling and subsequent callosal formation to occur.
As the responsiveness of MZG cells to Fgf8 signaling is crucial to subsequent midline remodeling, we investigated the mechanisms mediating the pro-astrogliogenic effects of Fgf8 on this cell population. We found that key downstream effectors of Fgf8 signaling have enriched expression in radial MZG progenitors, but are not enriched in mature intercalated MZG cells (Figure S4). These include the Fgf8 receptors, Fgfr1 and Fgfr2, as well as Mapk-Erk signaling effectors that transduce and modulate intracellular Fgf signaling (Mason, 2007), including phospho-Erk1/2, Spry1, and Spry2 (Figure S3 and S4). This suggests that the local effects of Fgf8 are limited to radial MZG progenitors, indicating that secreted Fgf8 may likely act in an autocrine fashion within this population. Furthermore, we found that Fgf8 overexpression is sufficient for significant upregulation of multiple mature astroglial-specific proteins including Gfap, Glast, S100ß, and Acsbg1, but not the radial glial marker Nestin (Figure S4), suggesting that Fgf8 may initiate multiple aspects of astroglial maturation via a pro-astrogliogenic transcriptional effector.
Nfia and Nfib, are downstream transcriptional effectors of Fgf8 and are required for remodeling of the IHF and corpus callosum formation in humans and mice
Nfia and Nfib (collectively referred to here as Nfi) are homologous transcription factors that regulate astrogliogenesis in the embryonic telencephalon (Namihira et al., 2009; Piper et al., 2010; Piper et al., 2009a). Colocalization analyses at E15 demonstrated that Nfi expression is enriched in Glast-positive radial MZG cells (Figure 6A) but is not expressed in Cxcl12-positive leptomeningeal fibroblasts during fissure remodeling (Figure S5). Unilateral electroporation of Fgf8 at E12 resulted in a significant increase of Nfi-expressing cells at the midline by E17 (Figure S5), indicating downstream involvement of both Nfia and Nfib in Fgf8-mediated MZG astrogliogenesis. As Mapk-Erk signaling appears to be activated during normal MZG somal translocation, as well as following Fgf8 overexpression (Figures S3 and S4), we hypothesized that the intracellular Mapk-Erk pathway acts as an intermediate effector of Fgf8, which then activates Nfi to induce astrogliogenesis. In line with this, disruption of the Mapk-Erk pathway with the Mek inhibitor U0126 was sufficient to decrease both Nfia- and Nfib-induced activation of a Gfap-luciferase reporter, further indicating that the Fgf8-Mapk signaling pathway promotes Nfi transcriptional activation of the astroglial gene Gfap (Figure 6B). To directly address whether Nfi signaling is required for Fgf8-mediated astrogliogenesis in vivo, we then electroporated Fgf8 into Nfia and Nfib knockout embryos at E14 and examined their brains at E17. In wildtype littermates, Fgf8 overexpression induced robust unilateral increases in Gfap and Glast expression (Figure 6C and 6D). Fgf8 overexpression also increased radial glial expression of Glast in both Nfia and Nfib knockouts (note comparable increase in the mean fold change of Glast fluorescence in the electroporated hemisphere compared to the unelectroporated hemisphere in Nfi knockouts and wildtype littermates; Figure 6D). However, Fgf8 overexpression did not elicit Gfap expression in either radial glia or multipolar astroglia in Nfi knockout brains, as compared to wildtypes (Figure 6D). These findings demonstrate that Nfia and Nfib are critical downstream effectors required for Fgf8-mediated astroglial maturation.
Figure 6. Nfia and Nfib are downstream transcriptional effectors of Fgf8 signaling required for IHF remodeling and corpus callosum formation.
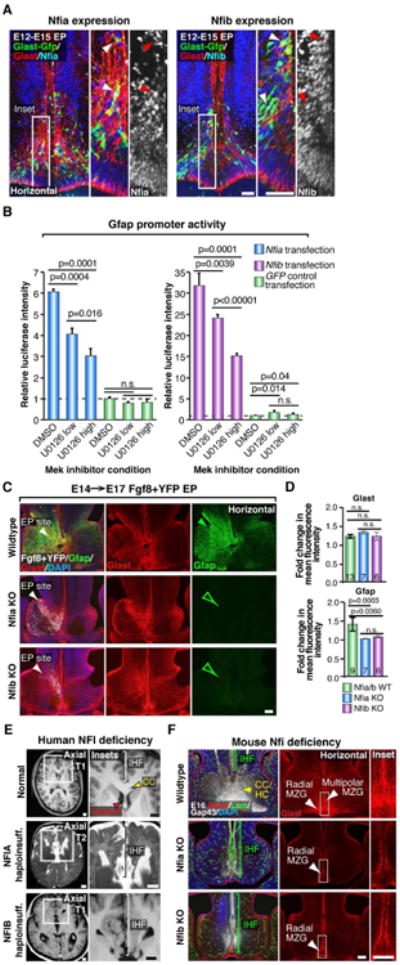
(A) E12→E15 in utero electroporation (EP) of the mouse telencephalic hinge with piggyBac Glast transposase/GFP (green) reporter plasmids, colabeled with Glast (red) and either Nfia or Nfib (blue/white) demonstrates Nfia and Nfib are both expressed by radial MZG (arrowheads). (B) Luciferase activity of U251 cells cotransfected with a Gfap promoter luciferase construct and either Nfia, Nfib or GFP control expression constructs, following addition of the Mek inhibitor U0126 (low = 5 μM; high = 20 μM), performed in triplicate. Data are represented as mean luciferase intensity ± SD, normalized to the GFP DMSO control (dotted line) and are representative of at least 2 independent experiments. Significant differences were determined with a Student's t-test. (C) Unilateral E14→E17 EP of Fgf8 + YFP plasmids into Nfia and Nfib knockout (Nfi KO) mice and their wildtype littermates, followed by fluorescence immunohistochemistry for Gfap (green) and Glast (red). Note increase in Gfap expression in wildtypes (closed green arrowhead) and absence of Gfap expression in Nfi KO mice (open green arrowheads). (D) Quantification of the fold change in Gfap and Glast fluorescence intensity. Data are represented as means ± SEM (n-values within bars, significant differences determined with a non-parametric Mann-Whitney test). (E) Coronal and axial structural T1- and T2-weighted MRI images of a normal human brain compared with NFIA- and NFIB-haploinsufficient individuals. Brackets indicate interhemispheric fissure (IHF) length and red arrowheads indicate the separated septum in NFI haploinsufficient brains. (F) Fluorescence immunohistochemistry for Glast (red), Laminin (green) and Gap43 (white) in Nfia and Nfib KO embryos and wildtype littermates at E16. Brackets indicate IHF length and white arrowheads indicate the distribution of radial versus multipolar MZG cells. Yellow arrowhead indicates the callosal (CC) and hippocampal commissural (HC) tract in wildtypes. Scale bars, 50μm (A), 100 μm (C and F) and 1 cm (E). See also Figure S5.
Since human individuals with NFIA haploinsufficiency, as well as Nfia and Nfib knockout mice, exhibit callosal malformations (Chen et al., 2011; Lu et al., 2007; Piper et al., 2009a; Sajan et al., 2013; Shu et al., 2003a; Steele-Perkins et al., 2005), we sought to investigate whether Nfi transcription factors regulate callosal development via astroglial IHF remodeling. Examination of two cases of human NFIA and NFIB haploinsufficiency using structural MRI revealed that these individuals display abnormal retention of the IHF associated with callosal agenesis (Figure 6E). Consistent with this, we found that radial MZG progenitors fail to transition into intercalated multipolar MZG in both Nfia and Nfib knockout mouse strains (Figure 6F and Figure S5). Instead, they retain an immature radial morphology along both sides of the IHF and fail to express Gfap (Figure 6F and Figure S5). Consequently, this MZG maturation defect results in abnormal retention of the IHF and callosal agenesis in these mice (Figure 6F). These observations indicate that Nfi transcription factors crucially promote IHF remodeling by regulating MZG maturation and cell polarity. Taken together, our findings elucidate Fgf8-Nfi signaling as a key astrogliogenic maturation program that initiates remodeling of the IHF and corpus callosum formation in both mice and humans.
Abnormal retention of the IHF is highly correlated with human congenital agenesis of the corpus callosum
Our cellular analyses in developing mice and humans indicate that remodeling and elimination of the IHF is a critical event preceding callosal development, and that disrupting this developmental event prevents subsequent tract formation. This suggests that defects in this remodeling event could be a significant etiological factor of agenesis of the corpus callosum. In order to quantify the co-occurrence of interhemispheric remodeling defects with callosal agenesis, we analyzed T1 or T2-weighted magnetic resonance images (MRI) from a large cohort of individuals previously diagnosed with agenesis of the corpus callosum. Individuals were selected for the analysis based upon a previous diagnosis of complete agenesis of the corpus callosum that was not associated with any other compounding brain abnormalities. We confirmed isolated complete callosal agenesis in 38 cases (Supplementary Table 1). In 100% of these 38 cases, we identified abnormal retention of the IHF, resulting in abnormal separation of the septal halves in these individuals (Figure 7A and 7B). In each of these cases, we also identified that callosal axons consistently accumulated into Probst bundles, either side of the abnormally-retained IHF (Figure 7A and 7B). These data further suggest that the IHF is non-permissive to callosal axons during human brain development, and that abnormal retention of the IHF invariably results in callosal agenesis. Thus, interhemispheric remodeling defects are a significant etiological factor in callosal agenesis disorders.
Figure 7. Characterization of human IHF phenotypes associated with agenesis of the corpus callosum based on structural MRI studies.
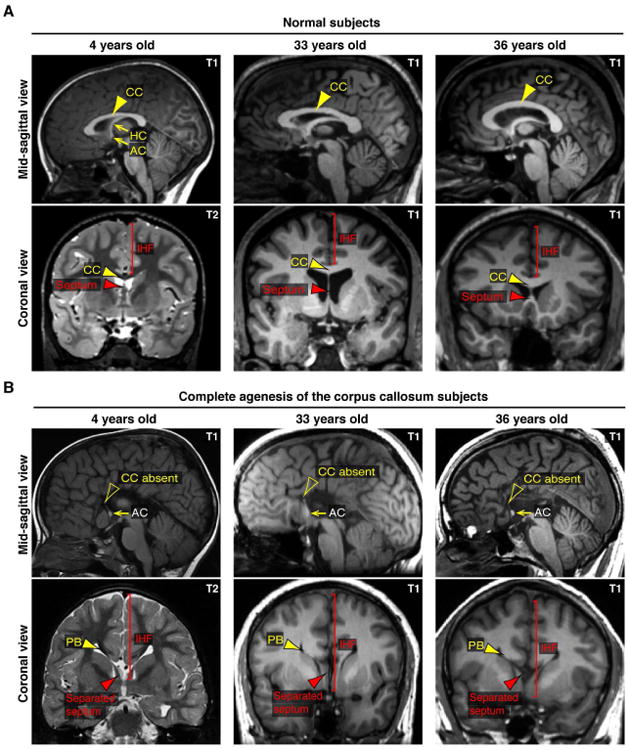
(A and B) Comparison of T1- or T2-weighted structural MRI images from 3 individuals previously diagnosed with isolated complete agenesis of the corpus callosum and age-matched controls. Note abnormal retention of the IHF and accumulation of callosal axons into Probst bundles (PB) either side of the IHF in individuals with callosal agenesis. Arrowheads indicate the morphology of the septum (red arrowheads) and corpus callosum (CC; yellow arrowheads). AC = anterior commissure, HC = hippocampal commissure. See also Table S1.
Discussion
The corpus callosum is the largest fiber tract in the human brain and is particularly important for integrating processes occurring in lateralized regions of the human neocortex. Here, we demonstrate that a critical event required for callosal tract formation in mice and humans is the remodeling of the interhemispheric midline via astroglia, and that callosal tract malformations occur if this tissue remodeling process is perturbed.
Astroglia are an essential neural cell type in the postnatal and adult brain, however their function during fetal brain development is still poorly understood. In this study, we show that a specialized population of fetal astroglia, the MZG, has a major influence on the morphology and connectivity of the adult brain. Although the MZG were identified by early histological studies (Silver et al., 1993; Silver et al., 1982), the function and molecular regulation of this cell population has remained elusive. Here, we demonstrate in fetal mouse and human brains that the MZG simultaneously remodel the IHF and provide the growth substrate for callosal axons to cross the interhemispheric midline. Previously, it has been hypothesized that callosal axons are developmentally timed to cross directly through the intervening leptomeninges via the activity of Bmp7 and Wnt3 (Choe et al., 2012). While our data do not exclude a role for Bmp7 and Wnt3 in regulating the timing of callosal midline crossing, we conclusively show that astroglia, rather than the leptomeninges, provide the growth substrate for callosal axons as they cross the midline.
We also identify that the transition of radial MZG progenitors into multipolar MZG cells is a key cellular event during remodeling of the midline that is regulated by a molecular signaling cascade initiated by the early morphogen Fgf8. Loss- and gain-of-function paradigms demonstrate that the levels and timing of Fgf8 expression at the midline are crucial for regulating the sequential chronology of MZG maturation and thus subsequently interhemispheric remodeling. Additionally, we demonstrate that Fgf8 is a crucial ligand signaling upstream of the Mapk pathway to induce astroglial differentiation via the pro-astrogliogenic transcription factors Nfia and Nfib. Taken together, our findings elucidate a key cellular mechanism that reconciles the relationship between delayed astroglial development and callosal agenesis, and are consistent with previous observations following mouse or human mutations in either Fgf signaling genes or Nfi genes (Barton et al., 1995; Chen et al., 2011; Dode and Hardelin, 2009; Dode et al., 2003; Ji et al., 2014; Koehler et al., 2010; Lajeunie et al., 1999; Li et al., 2012; Lu et al., 2007; McCabe et al., 2011; Piper et al., 2009a; Rao et al., 2014; Raybaud and Di Rocco, 2007; Sajan et al., 2013; Shu et al., 2003a; Sivasankaran et al., 1997; Smith et al., 2006; Steele-Perkins et al., 2005; Stewart et al., 2016; Tokumaru et al., 1996; Wilkie et al., 1995).
Various mutant mice that lack proteins involved in either axon guidance and/or midline guidepost cell development have been previously identified with callosal agenesis (Bagri et al., 2002; Fothergill et al., 2014; Islam et al., 2009; Keeble et al., 2006; Magnani et al., 2014; Mendes et al., 2006; Niquille et al., 2009; Piper et al., 2009b; Sanchez-Camacho et al., 2011; Smith et al., 2006). The prevailing hypothesis from these studies has therefore been that congenital callosal agenesis predominantly results from defects in axon guidance mechanisms. Although our findings do not preclude the possibility that callosal axon pathfinding defects may occur independent of interhemispheric remodeling defects, we demonstrate that appropriate elimination of the IHF is essential for subsequent callosal pathfinding across the midline. Furthermore, we observe that complete agenesis of the corpus callosum is remarkably correlated with abnormal retention of the IHF in humans. Thus, these findings suggest that the majority of isolated callosal agenesis cases associated with Probst bundles are primarily due to interhemispheric remodeling defects and aberrant retention of the IHF. Our observations further suggest that genetic abnormalities that perturb fetal astrogliogenesis may be a significant risk factor for subsequently developing a callosal tract malformation. We have identified Fgf8-Nfi signaling as an important pathway in this process, however, the prevalence of human interhemispheric remodeling defects associated with callosal agenesis suggests that multiple molecular pathways are likely to be involved. Furthermore, whether acallosal mice deficient for axon guidance and guidepost development proteins also display defects in interhemispheric midline remodeling or astrogliogenesis remains an interesting question for future research.
In summary, our findings demonstrate the cellular and molecular mechanisms required for astroglial-mediated remodeling of the interhemispheric midline, and show that this process is essential for callosal tract formation. Furthermore, clinical application of our findings may improve the future detection and diagnostic classification of human callosal malformations, as well as provide a crucial cellular target for genetic studies when investigating congenital callosal disorders.
Experimental Procedures
Human tissue
Human fetal tissue was obtained from the National Institute of Child Health and Human Development (NICHD) Brain and Tissue Bank for Developmental Disorders, at the University of Maryland, Baltimore School of Medicine, under contract N01-HD-4-3368. Collection and use of this tissue was approved by the University of Maryland Medical School human ethics institutional review board and The University of Queensland medical research ethics committee. This project complies with the provisions contained in the Australian National Statement on Ethical Conduct in Research Involving Humans and with the regulations governing experimentation on humans. Brains used for immunohistochemical analyses were immersion-fixed in 4% paraformaldehyde for 4 weeks, prior to sectioning and analysis.
Human MRI and genetic studies
Patients with disorders of the corpus callosum were enrolled in a study approved by the University of California San Francisco Committee on Human Research (UCSF CHR) organized by the Brain Development Research Program (brain.ucsf.edu). Drs. Sherr, Barkovich, Dobyns, Reardon and colleagues obtained consent for patients and families to participate, and clinical T1-weighted or T2-weighted brain MRI scans were obtained and systematically reviewed. Genetic information on two individuals with either NFIA or NFIB haploinsufficiency was obtained through both clinical and research-based array comparative genomic hybridization studies, for which the assembled cohort has been previously reported (Sajan et al., 2013; identification numbers: LR01-282 and 1127-0). For the correlative analysis of IHF remodeling defects, patients with complete agenesis of the corpus callosum, without additional cortical malformations, were selected for evaluation. Control participants were recruited under a separate UCSF CHR-approved protocol, the Simons Variation in Individuals Project led by Dr. Sherr at the University of California, San Francisco. Dr. Sherr and colleagues obtained consent for healthy volunteers to participate in a brain imaging study, which included completion of multi-echo MPRAGE T1-weighted and T2-weighted MRI scans. All MRI scans were systematically reviewed by a Board-certified neuroradiologist. All control scans showed normal morphology.
Animals
Nfia (Shu et al., 2003a) and Nfib (Steele-Perkins et al., 2005) knockout mice, Fgf8flox (Meyers et al., 1998), Emx1Cre (Iwasato et al., 2004), and ROSA26CAG-flox-STOP-flox-tdTomato reporter mice (Jackson Laboratory strain B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J;(Madisen et al., 2010)) were all maintained on a C57BL/6J background. These strains, wildtype C57BL/6J and CD1 mice were bred at the University of Queensland. All breeding and experiments were performed with approval from the University of Queensland Animal Ethics Committee. Timed-pregnant mouse females were obtained by placing male and female mice together overnight, and the following morning was designated as E0 if a vaginal plug was detected. Nfia and Nfib knockout litters and Fgf8flox/flox-STOP-flox-tdTomato/Emx1Cre litters were genotyped by PCR as previously described (Iwasato et al., 2004; Madisen et al., 2010; Meyers et al., 1998; Shu et al., 2003a; Steele-Perkins et al., 2005). Control embryos were wildtypes for Nfia and Nfib knockout analyses, and phenotypically normal homozygous Fgf8flox/heterozygous tdTomato/Cre-negative or heterozygous Fgf8flox/heterozygous tdTomato/Cre-positive animals for Fgf8 cKO mutant analyses. A minimum of 3 animals were analyzed for each separate phenotypic analysis.
In utero electroporation, in vivo procedures and tissue collection
In utero electroporation was performed as previously described (Suárez et al., 2014a), with minor modifications where 0.5-1 μL of plasmid DNA was injected either into one or both lateral ventricles and electroporated medially, or into the third ventricle and electroporated rostrally using 30-45V, depending on the embryonic stage. Plasmid expression constructs are described in the Supplemental Information. Pan-matrix metalloproteinase activity was detected by injecting 0.5nM MMPSense® 645 FAST in vivo reagent (PerkinElmer) into the lateral ventricles at E14 and collecting brains 24 hours later to allow uptake and cleavage of the fluorescent substrate. Mouse brains were fixed via transcardial perfusion or immersion fixation with 4% paraformaldehyde.
Immunohistochemistry
Brain sections were processed for either chromogenic immunohistochemistry or standard fluorescence immunohistochemistry as previously described (Moldrich et al., 2010) with minor modifications. With the exception of MMPSense-treated tissue, all sections were post-fixed in 4% paraformaldehyde, and subjected to antigen retrieval (125°C for 4 minutes at 15 psi in sodium citrate buffer) prior to incubation with primary antibodies. Primary and secondary antibodies used for immunohistochemistry are described in the Supplemental Information.
IHF measurements and fluorescence intensity quantification
Measurements of the IHF in horizontal sections were performed using ImageJ software (NIH). To account for inter-brain variability, this length was then normalized to the entire rostro-caudal length of the telencephalon along the interhemispheric midline.
For fluorescence intensity analyses, a region of interest (ROI) within each brain hemisphere was cropped from fluorescence images using consistent anatomical landmarks. The fluorescence intensity was then plotted and averaged over the width of the ROI using ImageJ.
Statistics
For in vivo phenotypic comparisons, data were first assessed for normality with a D'Agostino-Pearson omnibus normality test, and then statistical differences between two groups were determined either with a parametric Student's t-test, or a non-parametric Mann-Whitney test in Prism 6 software. P ≤ 0.05 was considered significantly different. All values are presented as mean ± standard error of the mean.
Supplementary Material
Highlights.
Developmental remodeling of the midline is required for callosal tract formation
Maturation and intercalation of astroglia mediates interhemispheric remodeling
Fgf8-Nfi signaling regulates astroglial-mediated interhemispheric remodeling
Interhemispheric remodeling defects underlie congenital callosal agenesis in humans
Acknowledgments
We are grateful to Laura Fenlon and Rowan Tweedale for critical comments on the manuscript. We thank Kathryn Green for assistance with scanning electron microscopy, Nyoman Kurniawan for assistance with MR imaging, and Jonathan Lim for assistance with luciferase assays. Microscopy was performed in the Queensland Brain Institute's Advanced Microscopy Facility. This work was supported by Australian NHMRC grants 456027 and 1048849 to LJR and 631552 to LJR and JLRR, Australian ARC LEIF grant LE130100078 and NINDS R01 NS34661 to JLRR from the United States. IG was supported by a University of Queensland Research Scholarship, LM is supported by an Australian Postgraduate Award, RS is supported by an ARC DECRA Research Fellowship and LJR is supported by an NHMRC Principal Research Fellowship.
Footnotes
Author Contributions: IG designed, performed and analyzed the experiments, and prepared all display items. LJR and IG conceived of the study and directed the project. LM, RS, JB, and JLRR helped to design, perform or analyze animal experiments and prepare display items. PB, WR, WBD, AJB, and EHS provided human imaging, genetic data and analysis of these experiments. IG, RS, LM, JB and LJR wrote the manuscript. All authors reviewed and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Laura Morcom, Email: laura.morcom@uqconnect.edu.au.
Rodrigo Suárez, Email: rsuarezsaa@gmail.com.
Jens Bunt, Email: j.bunt@uq.edu.au.
Polina Bukshpun, Email: polina.buk@gmail.com.
Willie Reardon, Email: Willie.Reardon@olchc.ie.
William B. Dobyns, Email: wbd@u.washington.edu.
John L.R. Rubenstein, Email: john.rubenstein@ucsf.edu.
A. James Barkovich, Email: James.Barkovich@uscf.edu.
Elliott H. Sherr, Email: SherrE@neuropeds.ucsf.edu.
References
- Bagri A, Marin O, Plump AS, Mak J, Pleasure SJ, Rubenstein JL, Tessier-Lavigne M. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 2002;33:233–248. doi: 10.1016/s0896-6273(02)00561-5. [DOI] [PubMed] [Google Scholar]
- Barton JS, O'Loughlin J, Howell RT, L'E Orme R. Developmental delay and dysmorphic features associated with a previously undescribed deletion on chromosome 1. Journal of medical genetics. 1995;32:636–637. doi: 10.1136/jmg.32.8.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer C, Tung CH, Weissleder R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nature medicine. 2001;7:743–748. doi: 10.1038/89126. [DOI] [PubMed] [Google Scholar]
- Chen CP, Su YN, Chen YY, Chern SR, Liu YP, Wu PC, Lee CC, Chen YT, Wang W. Chromosome 1p32-p31 deletion syndrome: prenatal diagnosis by array comparative genomic hybridization using uncultured amniocytes and association with NFIA haploinsufficiency, ventriculomegaly, corpus callosum hypogenesis, abnormal external genitalia, and intrauterine growth restriction. Taiwanese journal of obstetrics & gynecology. 2011;50:345–352. doi: 10.1016/j.tjog.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Chen F, LoTurco J. A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. J Neurosci Methods. 2012;207:172–180. doi: 10.1016/j.jneumeth.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe Y, Siegenthaler JA, Pleasure SJ. A cascade of morphogenic signaling initiated by the meninges controls corpus callosum formation. Neuron. 2012;73:698–712. doi: 10.1016/j.neuron.2011.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dode C, Hardelin JP. Kallmann syndrome. Eur J Hum Genet. 2009;17:139–146. doi: 10.1038/ejhg.2008.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain. 2014;137:1579–1613. doi: 10.1093/brain/awt358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fothergill T, Donahoo AL, Douglass A, Zalucki O, Yuan J, Shu T, Goodhill GJ, Richards LJ. Netrin-DCC signaling regulates corpus callosum formation through attraction of pioneering axons and by modulating Slit2-mediated repulsion. Cereb Cortex. 2014;24:1138–1151. doi: 10.1093/cercor/bhs395. [DOI] [PubMed] [Google Scholar]
- Gazzaniga MS. Forty-five years of split-brain research and still going strong. Nat Rev Neurosci. 2005;6:653–659. doi: 10.1038/nrn1723. [DOI] [PubMed] [Google Scholar]
- Halfter W, Dong S, Yip YP, Willem M, Mayer U. A critical function of the pial basement membrane in cortical histogenesis. J Neurosci. 2002;22:6029–6040. doi: 10.1523/JNEUROSCI.22-14-06029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetts SW, Sherr EH, Chao S, Gobuty S, Barkovich AJ. Anomalies of the corpus callosum: an MR analysis of the phenotypic spectrum of associated malformations. AJR Am J Roentgenol. 2006;187:1343–1348. doi: 10.2214/AJR.05.0146. [DOI] [PubMed] [Google Scholar]
- Islam SM, Shinmyo Y, Okafuji T, Su Y, Naser IB, Ahmed G, Zhang S, Chen S, Ohta K, Kiyonari H, et al. Draxin, a repulsive guidance protein for spinal cord and forebrain commissures. Science. 2009;323:388–393. doi: 10.1126/science.1165187. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Nomura R, Ando R, Ikeda T, Tanaka M, Itohara S. Dorsal telencephalon-specific expression of Cre recombinase in PAC transgenic mice. Genesis. 2004;38:130–138. doi: 10.1002/gene.20009. [DOI] [PubMed] [Google Scholar]
- Ji J, Salamon N, Quintero-Rivera F. Microdeletion of 1p32-p31 involving NFIA in a patient with hypoplastic corpus callosum, ventriculomegaly, seizures and urinary tract defects. European journal of medical genetics. 2014;57:267–268. doi: 10.1016/j.ejmg.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Keeble TR, Halford MM, Seaman C, Kee N, Macheda M, Anderson RB, Stacker SA, Cooper HM. The Wnt receptor Ryk is required for Wnt5a-mediated axon guidance on the contralateral side of the corpus callosum. J Neurosci. 2006;26:5840–5848. doi: 10.1523/JNEUROSCI.1175-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler U, Holinski-Feder E, Ertl-Wagner B, Kunz J, von Moers A, von Voss H, Schell-Apacik C. A novel 1p31.3p32.2 deletion involving the NFIA gene detected by array CGH in a patient with macrocephaly and hypoplasia of the corpus callosum. European journal of pediatrics. 2010;169:463–468. doi: 10.1007/s00431-009-1057-2. [DOI] [PubMed] [Google Scholar]
- Lajeunie E, Cameron R, El Ghouzzi V, de Parseval N, Journeau P, Gonzales M, Delezoide AL, Bonaventure J, Le Merrer M, Renier D. Clinical variability in patients with Apert's syndrome. J Neurosurg. 1999;90:443–447. doi: 10.3171/jns.1999.90.3.0443. [DOI] [PubMed] [Google Scholar]
- Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, Snider WD. MEK Is a key regulator of gliogenesis in the developing brain. Neuron. 2012;75:1035–1050. doi: 10.1016/j.neuron.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Quintero-Rivera F, Fan Y, Alkuraya FS, Donovan DJ, Xi Q, Turbe-Doan A, Li QG, Campbell CG, Shanske AL, et al. NFIA haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet. 2007;3:e80. doi: 10.1371/journal.pgen.0030080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani D, Hasenpusch-Theil K, Benadiba C, Yu T, Basson MA, Price DJ, Lebrand C, Theil T. Gli3 controls corpus callosum formation by positioning midline guideposts during telencephalic patterning. Cereb Cortex. 2014;24:186–198. doi: 10.1093/cercor/bhs303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason I. Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci. 2007;8:583–596. doi: 10.1038/nrn2189. [DOI] [PubMed] [Google Scholar]
- McCabe MJ, Gaston-Massuet C, Tziaferi V, Gregory LC, Alatzoglou KS, Signore M, Puelles E, Gerrelli D, Farooqi IS, Raza J, et al. Novel FGF8 mutations associated with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J Clin Endocrinol Metab. 2011;96:E1709–1718. doi: 10.1210/jc.2011-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes SW, Henkemeyer M, Liebl DJ. Multiple Eph receptors and B-class ephrins regulate midline crossing of corpus callosum fibers in the developing mouse forebrain. J Neurosci. 2006;26:882–892. doi: 10.1523/JNEUROSCI.3162-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Moldrich RX, Gobius I, Pollak T, Zhang J, Ren T, Brown L, Mori S, De Juan Romero C, Britanova O, Tarabykin V, Richards LJ. Molecular regulation of the developing commissural plate. J Comp Neurol. 2010;518:3645–3661. doi: 10.1002/cne.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Tanaka K, Buffo A, Wurst W, Kuhn R, Gotz M. Inducible gene deletion in astroglia and radial glia--a valuable tool for functional and lineage analysis. Glia. 2006;54:21–34. doi: 10.1002/glia.20350. [DOI] [PubMed] [Google Scholar]
- Namihira M, Kohyama J, Semi K, Sanosaka T, Deneen B, Taga T, Nakashima K. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell. 2009;16:245–255. doi: 10.1016/j.devcel.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Niquille M, Garel S, Mann F, Hornung JP, Otsmane B, Chevalley S, Parras C, Guillemot F, Gaspar P, Yanagawa Y, Lebrand C. Transient neuronal populations are required to guide callosal axons: a role for semaphorin 3C. PLoS Biol. 2009;7:e1000230. doi: 10.1371/journal.pbio.1000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul LK, Brown WS, Adolphs R, Tyszka JM, Richards LJ, Mukherjee P, Sherr EH. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8:287–299. doi: 10.1038/nrn2107. [DOI] [PubMed] [Google Scholar]
- Piper M, Barry G, Hawkins J, Mason S, Lindwall C, Little E, Sarkar A, Smith AG, Moldrich RX, Boyle GM, et al. NFIA controls telencephalic progenitor cell differentiation through repression of the Notch effector Hes1. J Neurosci. 2010;30:9127–9139. doi: 10.1523/JNEUROSCI.6167-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, Moldrich RX, Lindwall C, Little E, Barry G, Mason S, Sunn N, Kurniawan ND, Gronostajski RM, Richards LJ. Multiple non-cell-autonomous defects underlie neocortical callosal dysgenesis in Nfib-deficient mice. Neural Dev. 2009a;4:43. doi: 10.1186/1749-8104-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, Plachez C, Zalucki O, Fothergill T, Goudreau G, Erzurumlu R, Gu C, Richards LJ. Neuropilin 1-Sema signaling regulates crossing of cingulate pioneering axons during development of the corpus callosum. Cereb Cortex. 2009b;19(Suppl 1):i11–21. doi: 10.1093/cercor/bhp027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P, Yakovlev PI. Development of the corpus callosum and cavum septi in man. J Comp Neurol. 1968;132:45–72. doi: 10.1002/cne.901320103. [DOI] [PubMed] [Google Scholar]
- Rao A, O'Donnell S, Bain N, Meldrum C, Shorter D, Goel H. An intragenic deletion of the NFIA gene in a patient with a hypoplastic corpus callosum, craniofacial abnormalities and urinary tract defects. European journal of medical genetics. 2014;57:65–70. doi: 10.1016/j.ejmg.2013.12.011. [DOI] [PubMed] [Google Scholar]
- Raybaud C, Di Rocco C. Brain malformation in syndromic craniosynostoses, a primary disorder of white matter: a review. Childs Nerv Syst. 2007;23:1379–1388. doi: 10.1007/s00381-007-0474-7. [DOI] [PubMed] [Google Scholar]
- Sajan SA, Fernandez L, Nieh SE, Rider E, Bukshpun P, Wakahiro M, Christian SL, Riviere JB, Sullivan CT, Sudi J, et al. Both rare and de novo copy number variants are prevalent in agenesis of the corpus callosum but not in cerebellar hypoplasia or polymicrogyria. PLoS Genet. 2013;9:e1003823. doi: 10.1371/journal.pgen.1003823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Camacho C, Ortega JA, Ocana I, Alcantara S, Bovolenta P. Appropriate Bmp7 levels are required for the differentiation of midline guidepost cells involved in corpus callosum formation. Dev Neurobiol. 2011;71:337–350. doi: 10.1002/dneu.20865. [DOI] [PubMed] [Google Scholar]
- Shu T, Butz KG, Plachez C, Gronostajski RM, Richards LJ. Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice. J Neurosci. 2003a;23:203–212. doi: 10.1523/JNEUROSCI.23-01-00203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Puche AC, Richards LJ. Development of midline glial populations at the corticoseptal boundary. J Neurobiol. 2003b;57:81–94. doi: 10.1002/neu.10252. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Pleasure SJ. We have got you ‘covered’: how the meninges control brain development. Curr Opin Genet Dev. 2011;21:249–255. doi: 10.1016/j.gde.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Edwards MA, Levitt P. Immunocytochemical demonstration of early appearing astroglial structures that form boundaries and pathways along axon tracts in the fetal brain. J Comp Neurol. 1993;328:415–436. doi: 10.1002/cne.903280308. [DOI] [PubMed] [Google Scholar]
- Silver J, Lorenz SE, Wahlsten D, Coughlin J. Axonal guidance during development of the great cerebral commissures: descriptive and experimental studies, in vivo, on the role of preformed glial pathways. J Comp Neurol. 1982;210:10–29. doi: 10.1002/cne.902100103. [DOI] [PubMed] [Google Scholar]
- Sivasankaran S, Ho NK, Knight L. Annals of the Academy of Medicine. Vol. 26. Singapore: 1997. De novo interstitial deletion of chromosome 1p with absent corpus callosum--a case report; pp. 507–509. [PubMed] [Google Scholar]
- Smith KM, Ohkubo Y, Maragnoli ME, Rasin MR, Schwartz ML, Sestan N, Vaccarino FM. Midline radial glia translocation and corpus callosum formation require FGF signaling. Nat Neurosci. 2006;9:787–797. doi: 10.1038/nn1705. [DOI] [PubMed] [Google Scholar]
- Steele-Perkins G, Plachez C, Butz KG, Yang G, Bachurski CJ, Kinsman SL, Litwack ED, Richards LJ, Gronostajski RM. The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol Cell Biol. 2005;25:685–698. doi: 10.1128/MCB.25.2.685-698.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CE, Corella KM, Samberg BD, Jones PT, Linscott ML, Chung WCJ. Perinatal midline astrocyte development is impaired in fibroblast growth factor 8 hypomorphic mice. Brain Research. 2016;1646:287–296. doi: 10.1016/j.brainres.2016.06.015. [DOI] [PubMed] [Google Scholar]
- Storm EE, Garel S, Borello U, Hebert JM, Martinez S, McConnell SK, Martin GR, Rubenstein JL. Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development. 2006;133:1831–1844. doi: 10.1242/dev.02324. [DOI] [PubMed] [Google Scholar]
- Suárez R, Fenlon LR, Marek R, Avitan L, Sah P, Goodhill GJ, Richards LJ. Balanced interhemispheric cortical activity is required for correct targeting of the corpus callosum. Neuron. 2014a;82:1289–1298. doi: 10.1016/j.neuron.2014.04.040. [DOI] [PubMed] [Google Scholar]
- Suárez R, Gobius I, Richards LJ. Evolution and development of interhemispheric connections in the vertebrate forebrain. Frontiers in human neuroscience. 2014b;8:497. doi: 10.3389/fnhum.2014.00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumaru AM, Barkovich AJ, Ciricillo SF, Edwards MS. Skull base and calvarial deformities: association with intracranial changes in craniofacial syndromes. AJNR Am J Neuroradiol. 1996;17:619–630. [PMC free article] [PubMed] [Google Scholar]
- Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.