

Abstract
Background
Non-small-cell lung cancer (NSCLC) patients with activating epidermal growth factor receptor (EGFR) mutations may exhibit primary resistance to EGFR tyrosine kinase inhibitor (TKI). We aimed to examine genomic alterations associated with de novo resistance to gefitinib in a prospective study of NSCLC patients.
Methods
One-hundred and fifty two patients with activating EGFR mutations were included in this study and 136 patients' tumor sample were available for targeted sequencing of genomic alterations in 22 genes using the Colon and Lung Cancer panel (Ampliseq, Life Technologies).
Results
All 132 patients with EGFR mutation were treated with gefitinib for their treatment of advanced NSCLC. Twenty patients showed primary resistance to EGFR TKI, and were classified as non-responders. A total of 543 somatic single-nucleotide variants (498 missense, 13 nonsense) and 32 frameshift insertions/deletions, with a median of 3 mutations per sample. TP53 was most commonly mutated (47%) and mutations in SMAD4 was also common (19%), as well as DDR2 (16%), PIK3CA (15%), STK11 (14%), and BRAF (7%). Genomic mutations in the PI3K/Akt/mTOR pathway were commonly found in non-responders (45%) compared to responders (27%), and they had significantly shorter progression-free survival and overall survival compared to patients without mutations (2.1 vs. 12.8 months, P=0.04, 15.7 vs. not reached, P<0.001). FGFR 1-3 alterations, KRAS mutations and TP53 mutations were more commonly detected in non-responders compared to responders.
Conclusion
Genomic mutations in the PI3K/Akt/mTOR pathway were commonly identified in non-responders and may confer resistance to EGFR TKI. Screening lung adenocarcinoma patients with clinical cancer gene test may aid in selecting out those who show primary resistance to EGFR TKI (NCT01697163).
Keywords: primary resistance, epidermal growth factor, next-generation sequencing, tyrosine kinase inhibitor
INTRODUCTION
Lung cancer is the leading cause of cancer deaths worldwide [1]. Adenocarcinoma, which consists of more than 50% of non-small-cell lung cancer (NSCLC), is the most frequent type. Recent efforts to characterize molecular subclassifications of NSCLC have provided a marked benefit to patients whose tumors harbor specific genetic alterations [2–4], and the three major driver oncogenic mutations are epidermal growth factor receptor (EGFR) mutation, KRAS mutation, and anaplastic lymphoma kinase (ALK) rearrangement.
EGFR activating mutations are the most important predictive markers of response to EGFR tyrosine kinase inhibitor (TKI) treatment [5]. Despite the demonstrated benefits of EGFR TKIs, not all patients respond to treatment. Approximately 30% of patients with EGFR activating mutations do not show objective responses to EGFR TKI [6]. Intrinsic, de novo or primary resistance is defined as the failure to respond to EGFR-targeted therapies and little is known about the mechanisms of primary resistance. On the contrary, acquired resistance occurs in patients who initially benefited from EGFR-targeted therapies and the underlying mechanisms of acquired resistance include EGFR T790M mutation, activation of bypass signaling (such as MET amplification, HER2 upregulation or KRAS activation), and histologic transformation to small cell lung cancer or epithelial-mesenchymal transition [7].
Recent studies have revealed that both somatic mutations and germline polymorphisms may result in primary resistance to EGFR TKI. For example, mutations in phosphoinositide-3-kinase catalytic alpha (PIK3CA), the p110α catalytic subunit of phosphatidylinositol-3-kinase, are found in approximately 4% of NSCLC patients [8] and result in resistance to EGFR TKI. Loss of phosphatase and tensin homolog (PTEN) and de novo MET amplification could also be associated with resistance [9, 10]. In addition, germline polymorphisms of BIM, a pro-apoptotic protein, which result in BIM deletion may confer primary resistance [11]. SRC and MAP kinase pathways may also act as bypass pathways which confer resistance to EGFR TKIs [12]. However, other mechanisms of primary resistance remain largely unknown.
With the advancement of next-generation sequencing (NGS), it is now possible to identify oncogenic alterations that would previously been missed by conventional sequencing. Rather than sequencing the entire genome or exome, clinical cancer gene test which include genes that show frequent alterations in cancer can save the amount of tissue, time and effort to perform sequencing. These panels use PCR capture-based NGS assay that allow deep targeted sequencing of genes of interest from limited formalin-fixed, paraffin-embedded (FFPE) specimens [13]. Since incorporating NGS into routine oncologic practice requires accurate genomic profiling in a single assay, clinical cancer gene test may be appropriately used for clinical use.
In this study, we aimed to discover novel mechanisms of primary resistance to EGFR TKIs by using patient tumor samples from a large-scaled, prospective trial. We performed clinical cancer gene test of patient tissue samples which were obtained before treatment with EGFR TKIs in order to identify genetic alterations that confer primary resistance to EGFR TKIs.
RESULTS
Patient characteristics
The baseline characteristics of all patients are summarized in Table 1. The median age of all patients was 60 (range, 32-84) and there were 86 females (63.3%). The majority of patients (61%) were never-smokers and nearly all patients had adenocarcinoma histology (97.8%). At the time of their cancer diagnosis, 1 patient (0.7%) had stage IIIB disease, 119 (87.5%) had stage IV disease, and 16 (11.8%) had relapsed after surgical resection of lung cancer. EGFR mutations included exon 19 deletion (n=75), L858R mutation (n=65) and the rest included G719X, L861Q and others (n=6). Ten patients had two or more coexisting EGFR mutations (complex mutation).
Table 1. Baseline characteristics of all patients (N=136).
| Characteristic | N | % |
|---|---|---|
| Age (years) | ||
| Median | 60 | |
| Range | 32-84 | |
| Gender | ||
| Male | 50 | 36.7 |
| Female | 86 | 63.3 |
| Smoking history | ||
| Never-smoker | 83 | 61 |
| Ever smoker | 53 | 49 |
| Histologic diagnosis | ||
| Adenocarcinoma | 133 | 97.8 |
| Squamous | 1 | 0.7 |
| Adenosquamous | 2 | 1.5 |
| Clinical stage | ||
| IIIB | 1 | 0.7 |
| IV | 119 | 87.5 |
| Postoperative relapse | 16 | 11.8 |
| Type of EGFR mutation* | ||
| Exon 19 deletion | 75 | 51.4 |
| L858R | 65 | 44.5 |
| Others* | 6 | 4.1 |
10 patients had two or more coexsiting EGFR mutations
Treatment outcome of EGFR TKI
The median follow-up duration was 14 months and 101 (74.3%) patients received gefitinib as their first-line of treatment. As for best response, 87 patients (63.8%) showed partial response (PR), 33 patients (24.5%) showed SD and 6 patients (4.4%) showed PD (Table 2). Ten patients (7.3%) had not undergone response evaluation due to clinical disease progression, study withdrawal and follow-up loss. According to our prespecified definition of primary resistance to EGFR TKI, 20 patients showed PD as best response to gefitinib or PFS of less than 4 months. We classified them as non-responders to gefitinib. The median PFS was 9.1 months (95% confidence interval [CI] 7.15 – 11.05) for all patients, 13.8 months (95% CI, 12.03 – 15.57) for responders, 1.7 months (95% CI, 0.67 – 2.72) for non-responders (Figure 1A). The median OS for responders was 37.5 months (95% CI, 26.52 – 48.18), whereas it was 9.3 months (95% CI, 0.0-20.32) for non-responders (Figure 1B). When OS was compared only among patients who received gefitinib for their first line therapy, the median OS was 37.5 months (95% CI, 24.98 – 50.02) for responders and 4.5 months (95% CI, 0 – 16.19) for non-responders (Supplementary Figure S1).
Table 2. Summary of EGFR TKI treatment outcome.
| Characteristic | N | % |
|---|---|---|
| Number of previous treatment | ||
| 0 | 101 | 74.3 |
| 1 | 32 | 23.5 |
| 2 | 3 | 2.2 |
| Best response | ||
| Complete response | 0 | 0 |
| Partial response | 87 | 63.8 |
| Stable disease | 33 | 24.5 |
| Progressive disease | 6 | 4.4 |
| Not assessable | 10 | 7.3 |
| EGFR TKI | ||
| Gefitinib | 136 | 100 |
| Erlotinib | 0 | 0 |
Figure 1. Kaplan-Meier curve showing.
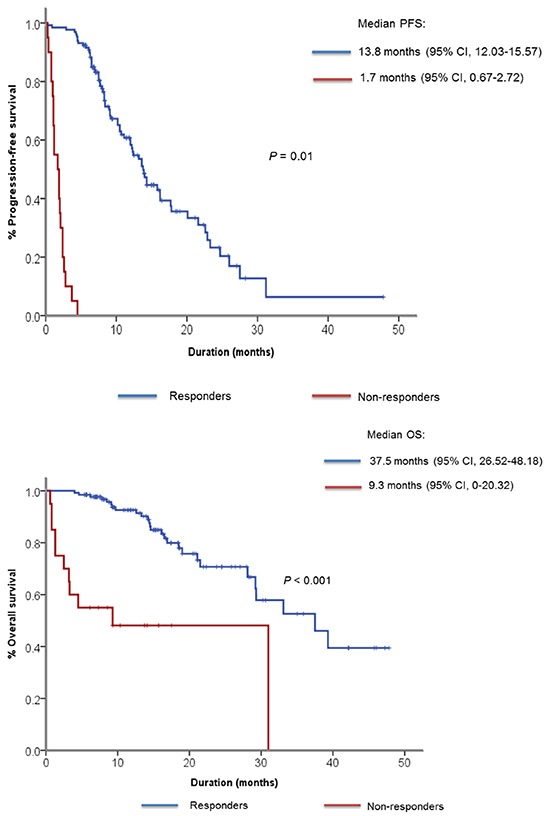
A. median progression-free survival and B. median overall survival among responders and non-responders to gefitinib.
Genomic landscape of responders and non-responders
The genomic landscape of 136 patients is depicted in Figure 2. A total of 543 somatic single-nucleotide variants (498 missense, 13 nonsense) and 32 frameshift insertions/deletions, with a median of 3 mutations per sample. All patients harbored EGFR mutations (not depicted in Figure 2), and EGFR mutations included deletions in exon 19 (n=75), L858R (n=65), 1 missense mutation (Thr751Pro) in exon 19, 1 de novo T790M gatekeeper mutation in exon 20, 1 missense mutation (L861Q) in exon 19, 2 missense mutation (G873R, K860I) in exon 21, and 1 missense mutation (N700S) in exon 18.
Figure 2. Landscape genomic profile of patients.
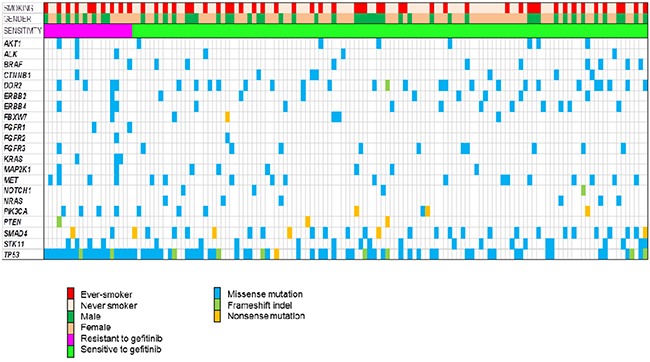
Samples are grouped by response to gefitinib.
The median number of mutations per sample in non-responders was 4, whereas the median number of mutations per sample in responders was 3. The number of mutations was significantly greater in non-responders compared to responders (13.6% vs. 10.6%, P = 0.009). There were 4 smokers who concurrently harbored 6 to 9 mutations among non-responders whereas all responders had mutations range from 1 to 4. In non-responders, all patients harbored TP53 mutations and the proportion of smokers was higher than in responders (50% vs. 37%, P = NS). Of note, KRAS mutations were identified in 3 patients among non-responders, which were previously not detected by conventional testing by direct sequencing method.
TP53 was most commonly mutated (47%) in all patients. Mutations in SMAD4 was also common (19%), as well as DDR2 (16%), PIK3CA (15%), STK11 (14%), and BRAF (7%). Recurrent mutations in AKT1, ALK, CTNNB1, ERBB2, ERBB4, FGFR1-3, MAP2K1, MET, NOTCH1, NRAS, and PTEN were also noted.
Mechanisms of primary resistance to EGFR TKI
Genomic mutations in the PI3K/Akt/mTOR pathway were commonly found in non-responders compared to responders (45% vs. 27%), although the difference was not significant. These mutations included missense mutations in AKT1, PIK3CA, STK11, nonsense mutations in PIK3CA, PTEN, and frameshift indels in PTEN. Patients with mutations in the PI3K/Akt/mTOR pathway had significantly shorter PFS and OS compared to those without (2.1 vs. 12.8 months, P=0.03, 15.7 vs. not reached, P < 0.001) (Figure 3A, 3B). FGFR 1-3 alterations were also more commonly found in non-responders compared to non-responders (20% vs. 8.3%, P = NS). All KRAS mutations were detected in non-responders (15% vs. 0%, P < 0.001), and TP53 mutations were detected in 100% of non-responders compared to 39% in responders (P < 0.001). There were no significant differences in alterations between the patients who showed upfront resistance (n=5) vs. patients with stable disease < 4 months (n=15).
Figure 3. Kaplan-Meier curve showing.
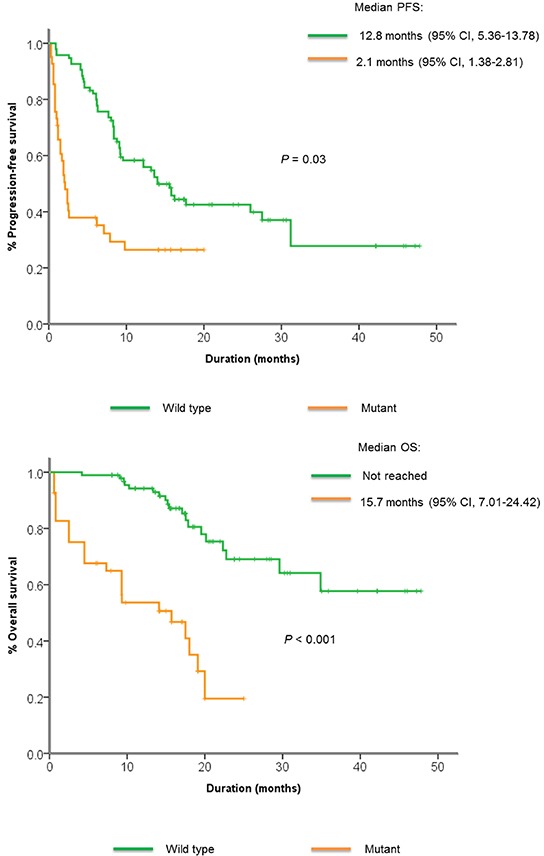
A. median progression-free survival and B. median overall survival among patients with or without PI3K/Akt/mTOR pathway alterations.
DISCUSSION
Here, we described genomic alterations identified in patient tumors that showed primary resistance to gefitinib. We suggest that PI3K/Akt/mTOR pathway alterations may confer resistance to gefitinib.
Although EGFR TKI has undoubtedly revolutionized the treatment of EGFR-mutant lung cancer patients, but more investigation is necessary to elucidate mechanisms of primary resistance to EGFR TKI. Previous studies have assessed mechanisms of primary resistance to EGFR TKIs: alterations in EGFR-downstream genes, EGFR T790M mutation, KRAS mutation, cMET amplification and HGF overexpression [19–22]. However, most studies did not comprehensively examine the genomic alterations in association with treatment outcomes to EGFR TKI.
In our study, patients with pathway alterations in PIK3CA/Akt/mTOR pathway had significantly shorter PFS to EGFR TKI compared to those without. Genetic alterations in the PIK3CA/Akt/mTOR pathway may impact the response to EGFR TKI. This finding is consistent with the previous study that reported that mutations in EGFR-downstream genes such as PIK3CA, AKT, PTEN and STK11 were associated with de novo resistance to EGFR TKI [8]. PIK3CA mutations were identified in 7% of our patients, and somatic missense mutations in helical or kinase domains of catalytic subunit encoded by the PIK3CA gene, which is known to increase the kinase activity of PIK3CA contributing to cellular transformation, were found in 2 out of 5 non-responder patients. We assume that activating PIK3CA mutations may have impaired response to EGFR TKI. Nine AKT mutations (5%) were identified in our patients and all mutations were located in pleckstrin homology domain. Of note, AKT1 E17K mutation, previously known to activate PI3K pathway, was found in 1 non-responder which could explain the non-response to gefitinib [23]. PTEN is a tumor suppressor gene which negatively regulates the PI3K/Akt/mTOR pathway, and loss of PTEN was previously identified as a poor prognostic marker [24]. While 1 frameshift deletion (p.Glu242fs) has been noted in a non-responder, its functional impact is not yet known. Mutations in STK11 were commonly identified (14%) and inactivation of STK11 has been known to promote lung tumorigenesis and associated with worse survival outcome [25, 26]. Pathogenic STK11 mutations (p.Phe354Leu) were recurrently identified in 3 non-responders whereas pathogenic mutations were not seen in responders. In patients with PI3K/Akt/mTOR pathway alterations, combination of PI3K inhibitor with gefitinib may be attempted to as a new therapeutic option.
Apart from PIK3CA/Akt/mTOR pathway alterations, KRAS mutations were identified exclusively in non-responders. KRAS mutations are known predictors of resistance to EGFR TKI [21], and interestingly, these mutations were previously not found with conventional sequencing. Possible reasons for the discordant results are low sensitivity of detection method and intratumoral heterogeneity. Genomic alterations with low allele frequencies lead to false-negative results on conventional sequencing and subclonal mutations may be heterogeneous according to biopsy sites [27, 28].
TP53 was the most commonly altered gene in our study and this is consistent with findings from previous studies [29]. Of note, all non-responders had TP53 mutations (frameshift deletion and missense mutations). Interestingly, it has been reported that TP53 may enhance sensitivity to EGFR inhibitor, and loss of TP53 may lead to resistance to EGFR inhibitor [30]. Four pathogenic mutations were identified among non-responders, and they may have led to functional loss of TP53, resulting in primary resistance to gefitinib.
Six uncommon EGFR mutations identified in our study were missense mutations in exon 18 (F712S, G721A) and 19 (A743S, P733L, L747F, N756D). Their oncogenicity and sensitivity to EGFR TKI have not been fully elucidated. Wu et al. observed an objective response rate of 48.4% and a median PFS of 5.0 months in patients with uncommon EGFR mutations [31]. In our study, two uncommon EGFR mutations were identified among non-responders and four were identified among responders. Prospective trials which examine the efficacy of EGFR inhibitors in patients with uncommon EGFR mutations are necessary.
Our study has a few limitations. The selected genes in this study may only explain a portion of mechanisms of resistance. Other genetic or epigenetic alterations that are not covered in the gene test may be missed out even if they promote resistance to EGFR TKI. However, most tumor samples were acquired from small biopsy samples and thus there were not enough tissue available for a more comprehensive sequencing. In addition, the number of patients analyzed was relatively small and 35 (25.7%) patients did not receive gefitinib as their first line of therapy, so data must be interpreted cautiously. Lastly, functional effects of resistant alterations were not assessed in vitro.
In conclusion, we note that more comprehensive genomic characterization of the tumor reveals alterations that may confer resistance to EGFR TKI in EGFR-mutant lung adenocarcinoma patients. This study highlights previously unappreciated genetic alterations, enabling further refinement in sub-classification for the improved personalization of lung cancer treatment.
MATERIALS AND METHODS
Study populations
A total of 152 patients with NSCLC harboring activating EGFR mutations were enrolled in a prospective trial of gefitinib between 2012 and 2015 at institutions in Korean Lung Cancer Consortium. Activating EGFR mutations were defined as mutations known to be associated with EGFR TKI sensitivity, including exon 19 deletion and L858R [14]. Patients with available archival tissue and those with measurable lesions at baseline were enrolled. This study was approved by the Institutional Review Board of Severance Hospital and the ethics committee. All patients provided written informed consent for study participation and genetic analysis.
Data collection
Medical records and radiologic images of all patients were reviewed to evaluate demographic and clinicopathologic parameters, tumor response and progression-free survival (PFS) and overall survival (OS) using a predesigned data collection format. PFS was measured from the first day of treatment with EGFR TKI to tumor progression or death. OS was measured from the first date of treatment with EGFR TKI until the date of death. Patients were censored on December 7, 2015 if alive and progression-free. Never-smokers were defined as those with a lifetime smoking-dose less than 100 cigarettes.
Tumor assessment
Response to gefitinib was assessed by a computed tomography scan performed at 4 weeks and then every 8 weeks thereafter in accordance with the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 [15]. Primary resistance to EGFR TKIs was defined as followings: (1) the best response to EGFR TKI is progressive disease (PD) and (2) PFS to EGFR TKI is less than 4 months with stable disease (SD) as best response [8, 16].
Genetic analysis
Tumor samples from 136 patients were available for genetic alterations. DNA was extracted from FFPE tumor samples using the QIAamp FFPE tissue kit (Qiagen, Antwerp, Belgium). The DNA obtained was quantified using the Qubit® fluorometer in combination with the Qubit dsDNA HS assay kit (Life Technologies, Gent, Belgium). For library construction, 10ng of DNA was amplified using the Colon and Lung Cancer panel (Ampliseq, Life Technologies). An amplicon library was generated for sequencing 1825 hotspot mutations in 22 genes including AKT1 (NM_05163), ALK (NM_004304), BRAF (NM_004333), CTNNB1 (NM_001904), DDR2 (NM_001014796), EGFR (NM_005228), ERBB2 (NM_004448), ERBB4 (NM_005235), FBXW7 (NM_033632), FGFR1 (NM_023110), FGFR2 (NM_022970), FGFR3 (NM_000142), KRAS (NM_033360), MAP2K1 (NM_002755), MET (NM_001127500), NOTCH1 (NM_017617), NRAS (NM_002524), PIK3CA (NM_006218), PTEN (NM_000314), SMAD4 (NM_005359), STK11 (NM_000455), TP53 (NM_000546). The amplicons were then digested, barcoded and amplified using the Ion Ampliseq™ Library kit 2.0 and Ion Xpress™ barcode adapters kit (Life technologies) according to the manufacturer's instructions. The library was quantified using the Qubit1 fluorometer and the Qubit1 dsDNA HS assay kit (Life technologies). 8pM of each library was multiplexed and clonally amplified on Ion sphere™ particles (ISP) by emulsion PCR performed on the Ion One Touch 2 instrument with the Ion PGM™ template OT2 200 kit (Life technologies) according to the manufacturer's instructions. Quality control was performed using the Ion Sphere™ Quality Control kit (Life Technologies) to ensure that 10–30% of template positive ISP were generated in the emulsion PCR. Finally, the template ISP were enriched, loaded on an Ion 316™ or on an Ion 318™ chip and sequenced on a PGM™ sequencer with the Ion PGM™ sequencing 200 kit v2 according to the manufacturer's instructions.
Data analysis
The raw data were analyzed using the torrent suite software v3.6.2 (Life technologies). The coverage analysis was performed using the coverage analysis plug-in v3.6. Cases for which the number of mapped reads was <100000 and/or the average base coverage was <500x were considered as non-informative. Mutations were detected using the Variant Caller plug-in v3.6 with low stringency settings (Life Technologies). In the variant list obtained, each mutation was verified in the Integrative genome viewer (IGV) from the Broad Institute (http://www.broadinstitute.org/igv/). Only mutations reported in the COSMIC (Sanger Institute Catalogue of Somatic Mutations in Cancer) database (http://www.sanger.ac.uk/cosmic) were taken into account and silent or intronic mutations were not reported. Locis were further analyzed for functional prediction of amino acid changes using two different prediction algorithms (Provean and SIFT) [17, 18].
Statistical analysis
OS and PFS were estimated using the Kaplan–Meier method. Differences between groups were compared by the log-rank test. Two-sided P-values < 0.05 were considered significant. All analyses were carried out using SPSS 20.0 (IBM SPSS Statistics, IBM Corp., Somers, NY).
SUPPLEMENTARY FIGURES
Acknowledgments
This study was supported partly by Astra Zeneca Korea Ltd. This research was supported in part by a grant of the Research Driven Hospital R&D project, funded by the CHA Bundang Medical Center (BDCHA R&D 2015-34 to S.M. Lim), a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI13C1948 to H.R. Kim), and by Korea Ministry of Environment (MOE) as “the Environmental Health Action Program” (2015001350002 to H.R. Kim).
Footnotes
CONFLICTS OF INTEREST
The authors have no potential conflicts of interest to disclose.
REFERENCES
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Herbst RS, Heymach JV, Lippman SM. Lung cancer. The New England journal of medicine. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee CK, Brown C, Gralla RJ, Hirsh V, Thongprasert S, Tsai CM, Tan EH, Ho JC, Chu da T, Zaatar A, Osorio Sanchez JA, Vu VV, Au JS, Inoue A, Lee SM, Gebski V, et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: a meta-analysis. Journal of the National Cancer Institute. 2013;105:595–605. doi: 10.1093/jnci/djt072. [DOI] [PubMed] [Google Scholar]
- 6.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. The New England journal of medicine. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 7.Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nature medicine. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HR, Cho BC, Shim HS, Lim SM, Kim SK, Chang J, Kim DJ, Kim JH. Prediction for response duration to epidermal growth factor receptor-tyrosine kinase inhibitors in EGFR mutated never smoker lung adenocarcinoma. Lung cancer. 2014;83:374–382. doi: 10.1016/j.lungcan.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Cappuzzo F, Janne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, Zucali PA, Terracciano L, Toschi L, Roncalli M, Destro A, Incarbone M, Alloisio M, Santoro A, Varella-Garcia M. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Annals of oncology. 2009;20:298–304. doi: 10.1093/annonc/mdn635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, Girard L, Peyton M, Gazdar AF, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer research. 2009;69:3256–3261. doi: 10.1158/0008-5472.CAN-08-4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, Ariyaratne PN, Takahashi N, Sawada K, Fei Y, Soh S, Lee WH, Huang JW, Allen JC, Jr, Woo XY, Nagarajan N, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nature medicine. 2012;18:521–528. doi: 10.1038/nm.2713. [DOI] [PubMed] [Google Scholar]
- 12.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, Lee D, Kalsy A, Gomez-Caraballo M, Elamine L, Howe E, Hur W, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–1486. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, Iwanik K, Maillet A, Buell J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature biotechnology. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng L, Li Y, Zhang SB, Teng XD. Molecular pathology of lung cancer: key to personalized medicine [Article in Chinese] Zhonghua Bing Li Xue Za Zhi. 2012;41:715–720. doi: 10.3760/cma.j.issn.0529-5807.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 15.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1. 1) European journal of cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA, Lynch T, Johnson BE, Miller VA. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Journal of clinical oncology. 2010;28:357–360. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 19.Benedettini E, Sholl LM, Peyton M, Reilly J, Ware C, Davis L, Vena N, Bailey D, Yeap BY, Fiorentino M, Ligon AH, Pan BS, Richon V, Minna JD, Gazdar AF, Draetta G, et al. Met activation in non-small cell lung cancer is associated with de novo resistance to EGFR inhibitors and the development of brain metastasis. The American journal of pathology. 2010;177:415–423. doi: 10.2353/ajpath.2010.090863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, Mitsudomi T, Tanaka H, Kimura T, Kudoh S, Nokihara H, Ohe Y, Yokota J, Uramoto H, Yasumoto K, Kiura K, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. Journal of thoracic oncology. 2011;6:2011–2017. doi: 10.1097/JTO.0b013e31823ab0dd. [DOI] [PubMed] [Google Scholar]
- 21.Ludovini V, Bianconi F, Pistola L, Chiari R, Minotti V, Colella R, Giuffrida D, Tofanetti FR, Siggillino A, Flacco A, Baldelli E, Iacono D, Mameli MG, Cavaliere A, Crino L. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. Journal of thoracic oncology. 2011;6:707–715. doi: 10.1097/JTO.0b013e31820a3a6b. [DOI] [PubMed] [Google Scholar]
- 22.Lee SY, Kim MJ, Jin G, Yoo SS, Park JY, Choi JE, Jeon HS, Cho S, Lee EB, Cha SI, Park TI, Kim CH, Jung TH, Park JY. Somatic mutations in epidermal growth factor receptor signaling pathway genes in non-small cell lung cancers. Journal of thoracic oncology. 2010;5:1734–1740. doi: 10.1097/JTO.0b013e3181f0beca. [DOI] [PubMed] [Google Scholar]
- 23.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 24.Tang JM, He QY, Guo RX, Chang XJ. Phosphorylated Akt overexpression and loss of PTEN expression in non-small cell lung cancer confers poor prognosis. Lung cancer. 2006;51:181–191. doi: 10.1016/j.lungcan.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 25.Pecuchet N, Laurent-Puig P, Mansuet-Lupo A, Legras A, Alifano M, Pallier K, Didelot A, Gibault L, Danel C, Just PA, Riquet M, Le Pimpec-Barthes F, Damotte D, Fabre E, Blons H. Different prognostic impact of STK11 mutations in non-squamous non-small-cell lung cancer. Oncotarget. 2015 doi: 10.18632/oncotarget.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, Buttarazzi MS, Sanofsky BA, McNamara KL, Brandstetter KA, Walton ZE, Gu TL, Silva JC, Crosby K, Shapiro GI, Maira SM, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer cell. 2010;17:547–559. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han HS, Lim SN, An JY, Lee KM, Choe KH, Lee KH, Kim ST, Son SM, Choi SY, Lee HC, Lee OJ. Detection of EGFR mutation status in lung adenocarcinoma specimens with different proportions of tumor cells using two methods of differential sensitivity. Journal of thoracic oncology. 2012;7:355–364. doi: 10.1097/JTO.0b013e31823c4c1b. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, Gold KA, Kalhor N, Little L, Mahadeshwar H, Moran C, Protopopov A, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256–259. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. The Lancet Oncology. 2015;16:e342–351. doi: 10.1016/S1470-2045(15)00077-7. [DOI] [PubMed] [Google Scholar]
- 30.Huang S, Benavente S, Armstrong EA, Li C, Wheeler DL, Harari PM. p53 modulates acquired resistance to EGFR inhibitors and radiation. Cancer research. 2011;71:7071–7079. doi: 10.1158/0008-5472.CAN-11-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu JY, Yu CJ, Chang YC, Yang CH, Shih JY, Yang PC. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clinical cancer research. 2011;17:3812–3821. doi: 10.1158/1078-0432.CCR-10-3408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.