

Abstract
Chondrosarcoma is the second most frequently occurring type of bone malignancy that is characterized by the distant metastasis propensity. Vascular endothelial growth factor-C (VEGF-C) is the major lymphangiogenic factor, and makes crucial contributions to tumor lymphangiogenesis and lymphatic metastasis. Chemokine CCL5 has been reported to facilitate angiogenesis and metastasis in chondrosarcoma. However, the effect of chemokine CCL5 on VEGF-C regulation and lymphangiogenesis in chondrosarcoma has largely remained a mystery. In this study, we showed a clinical correlation between CCL5 and VEGF-C as well as tumor stage in human chondrosarcoma tissues. We further demonstrated that CCL5 promoted VEGF-C expression and secretion in human chondrosarcoma cells. The conditioned medium (CM) from CCL5-overexpressed cells significantly induced tube formation of human lymphatic endothelial cells (LECs). Mechanistic investigations showed that CCL5 activated VEGF-C-dependent lymphangiogenesis by down-regulating miR-507. Moreover, inhibiting CCL5 dramatically reduced VEGF-C and lymphangiogenesis in the chondrosarcoma xenograft animal model. Collectively, we document for the first time that CCL5 induces tumor lymphangiogenesis by the induction of VEGF-C in human cancer cells. Our present study reveals miR-507/VEGF-C signaling as a novel mechanism in CCL5-mediated tumor lymphangiogenesis. Targeting both CCL5 and VEGF-C pathways might serve as the potential therapeutic strategy to block cancer progression and metastasis in chondrosarcoma.
Keywords: CCL5, VEGF-C, lymphangiogenesis, miR-507
INTRODUCTION
Human chondrosarcoma is the second most common sarcoma arising in bone malignancy which chiefly occurs in adults over 40 years of age [1]. Chondrosarcoma has been characterized as the aggressive and pathologically diverse malignant tumor with poor disease progression [2]. To date, surgical resection remains the primary treatment for chondrosarcoma, since conventional radiotherapy and chemotherapy are mostly ineffective. The relapse ususally occurs after surgical resection because of the potential for metastatic propensity. The need for a specific targeted therapy to impede the metastasis of chondrosarcoma remains urgent [3].
Metastasis is the leading cause of cancer mortality worldwide. The metastatic spread of tumor cells to sentinel lymph nodes represents the first step of tumor dissemination in most human cancers [4]. Lymphangiogenesis, the formation of new lymphatic vessels from preexisting ones, is crucial for the development of tumor metastasis [5]. Tumors can actively promote the formation of lymphatic vessels through secretion of lymphangiogenic factors, and that tumor lymphangiogenesis has been implicated in the correlation with lymph node metastasis in many types of human cancer [6]. Vascular endothelial growth factor-C (VEGF-C) is the best-characterized lymphangiogenic factor, acting predominantly via VEGF receptor-3 (VEGFR-3) that is specifically expressed by lymphatic endothelial cells (LECs). The activation of VEGF-C/VEGFR-3 axis is responsible for LECs survial, proliferation, migration and tube formation during lymphangiogenic process [7]. Recent studies have revealed that VEGF-C production by tumor cells is recognized as the chief promoter of tumor-associated lymphangiogenesis and lymphatic metastasis [5]. Moreover, clinical evidences suggest the existence of a relationship between tumor expressing VEGF-C and the disease progression of cancer in various tumor types, including melanoma, pancreatic, breast, colorectal and lung cancer [8–12]. Blockade of tumor-induced lymphangiogenesis has been shown to markedly suppress cancer metastasis. Therefore, the identification of mechanisms underlying VEGF-C-mediated lymphangiogenesis is essential for developing novel prognostic and therapeutic strategies in the treatment of cancer [13, 14].
MicroRNAs (miRNAs) are small non-protein-coding RNAs that function as endogenous negative gene regulators by interfering with the translation or stability of target transcripts [15]. They control gene expression by binding to the 3′untranslated region (3′UTR) of mRNA through complementary base pairing. Dysfunctions of miRNAs are frequently found in malignancies, including chondrosarcoma [16]. Increasing studies have focused on the role of miRNAs in cancer progression and metastasis. miRNAs have been proposed to intervene numerous functions of cancer cell, including survival, apoptosis, autophagy, migration, invasion, angiogenesis and lymphangiogenesis [17]. Several investigations demonstrate that miRNAs inhibit lymphangiogenesis and tumor dissemination through the dysregulation of miR/VEGF-C signaling [18, 19]. miR-128 has been reported to inhibit lymphangiogenesis in human non-small cell lung cancer by directly suppressing VEGF-C expression [20]. miR-206 also abrogates the expression and secretion of VEGF-C, and subsequently inhibits tumor lymphangiogenesis in pancreatic cancer [21]. Furthermore, miR-101 has been documented to suppress migration and invasion via negatively regulating VEGF-C expression in bladder cancer and cholangiocarcinoma cells, respectively [22]. However, the role of miRNA in regulating VEGF-C expression in human chondrosarcoma cells is poorly understood.
CCL5 is widely established as an inflammatory chemokine secreted by many cell types including activated T cell, macrophage, endothelial cell, stromal cells and cancer cells [23, 24]. CCL5 has been demonstrated to play the important role in many chronic inflammatory diseases. Compelling evidences indicate that CCL5 is associated with tumourigenesis and metastasis in several types of cancer [25]. We previously reported that CCL5 enchances cell migration through activation of matrix metalloproteinase-3 (MMP-3) and promotes VEGF-A-dependent tumor angiogenesis in human chondrosarcoma [26–28], implying that CCL5 is involved in the metastasis of chondrosarcoma. However, it is still not well-recognized whether CCL5 increases VEGF-C expression to facilitate tumor-associated lymphangiogenesis in human chondrosarcoma. In this study, we investigated the relationship of CCL5 with VEGF-C-mediated lymphangiogenesis, and evaluated the involvement of miRNA in human chondrosarcoma cells.
RESULTS
Clinical significance of CCL5 and VEGF-C expression in specimens from patients with chondrosarcoma
Our previous studies have shown that chemokine CCL5 facilitates tumor angiogenesis in human chondrosarcoma [27, 28]. We indicated that the expression of CCL5 is highly correlated with tumor stage according the IHC analysis of human chondrosarcoma tissues. To characterize the role of CCL5 in tumor lymphangiogenesis of chondrosarcoma, we first analyzed the expression profile of VEGF-C in specimens of chondrosarcoma patients. As shown in Figure 1A, the expression of VEGF-C was higher in tumor specimens than in normal tissues. Accordingly, the high level of VEGF-C expression correlated significantly with tumor stage (Figure 1B). The quantitative data also showed the high positive relationship between the expression of CCL5 and VEGF-C in human chondrosarcoma patients (Figure 1C). In addition, we examined the mRNA expression of CCL5, CCL5 receptors and VEGF-C between normal caritlage and chondrosarcoma. The expression of CCL5 and VEGF-C in chondrosarcoma patients was obvioustly higher than that in normal cartilage (Supplementary Figure 1A&1B). The mRNA levels of CCR5 and GPR75 were higher in tumor specimens than in normal tissues (Supplementary Figure 1C&1D). These results suggest that chemokine CCL5 and its receptors are strongly associated with VEGF-C expression and tumor progression of human chondrosarcoma.
Figure 1. Clinical significance of CCL5 and VEGF-C expression in specimens of chondrosarcoma patients.
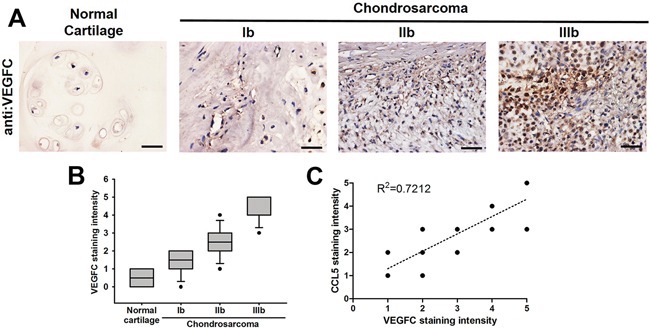
A. Immunohistochemistry of VEGF-C expression in human normal cartilage and chondrosarcoma tissues. B&C. The quantitative data and correlation between CCL5 and VEGF-C as well as tumor stage. The representative specimens were shown, and the staining intensity was ranked into five groups (1-5) according to the histologic scoring. The correlation analyses were performed by Spearman rank correlation test (R2 > 0.5).
CCL5 promotes VEGF-C-dependent lymphangiogenesis in human chondrosarcoma cells
To clarify direct relationship between CCL5 and VEGF-C in lymphangiogenic process, CCL5 stable transfectant in human chondrosarcoma cells were established (Supplementary Figure 2). After proper selection by antibiotics, CCL5-overexpressed clone (JJ012/CCL5 and SW1353/CCL5), CCL5-knockdowned clone (JJ012/shCCL5 and SW1353/shCCL5) and vector control (JJ012/vector and SW1353/vector) cells were generated to investigate the effect of CCL5 on VEGF-C expression and lymphangiogenesis. The results showed that CCL5 overexpression manifestly increased VEGF-C mRNA and protein levels, and the secretion of VEGF-C (Figure 2A-2C). To elucidate the interaction of CCL5 with it specific receptor CCR5, the selective CCR5 antagonists (maraviroc and DAPTA) were used in CCL5-overexpressed cells. Maraviroc and DAPTA markedly attenuated CCL5-induced VEGF-C mRNA expression, implying that CCL5 promotes VEGF-C expression through chemokine receptor CCR5 in an autocrine manner (Supplementary Figure 3). We then examined whether CCL5-dependent VEGF-C expression induced lymphangiogenesis using an in vitro LECs model. Incubation of human LECs with conditioned medium (CM) from CCL5-overexpressed cells significantly promoted migration and tube formation of LECs (Figure 2D&2E). The human recombinant VEGF-C was used as the positive control to induce tube formation of LECs. In contract, shRNA-mediated knockdown of CCL5 notably inhibited VEGF-C expression and secretion in human chondrosarcoma cells, and subsequently suppressed migration and tube formation in human LECs (Figure 2). These results indicate that CCL5/CCR5 axis enhances VEGF-C production and lymphangiogenesis in human chondrosarcoma.
Figure 2. CCL5 promotes lymphangiogenesis by VEGF-C production in human chondrosarcoma cells.
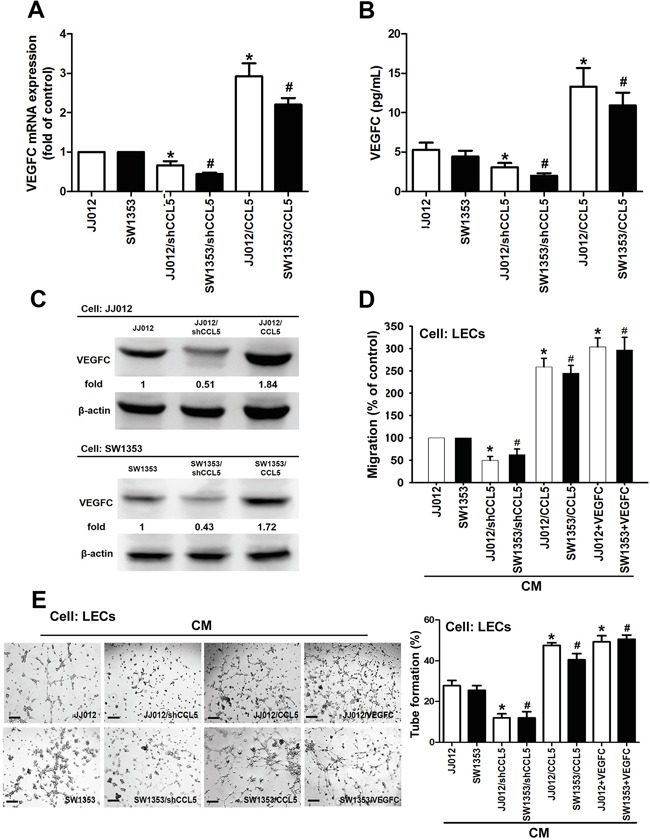
A-C. The JJ012/vector, JJ012/shCCL5, JJ012/CCL5, SW1353/vector, SW1353/shCCL5 and SW1353/CCL5 cells were cultured for 24 h, and VEGF-C expression was examined by qPCR, ELISA, and Western blot (n = 4-6). D&E. The medium was collected as CM, then applied to LECs for 6 h treatment. Then, cell migration and capillary-like structure formation in human LECs were examined by Transwell migration and tube formation assay. The CM of JJ012/vector and SW1353/vector cells were treated with VEGF-C (25 ng/ml), then used to induce LECs migration and tube formation (n = 4-6). Results are expressed as the mean ± SE. *, p < 0.05 compared with JJ012 control; #, p < 0.05 compared with SW1353 control.
CCL5 enhances VEGF-C production and lymphangiogenesis by down-regulating miR-507
Emerging studies have indicated that miRNAs are important regulators of VEGF-C expression and tumor lymphangiogenesis in cancer progression [17, 18]. We next performed miRNome microRNA Profilers QuantiMir™ kit to elucidate miRNA differential expression in CCL5-overexpressed chondrosarcoma cells. This kit contained 384 human miRNA primer, we analyzed 33 metastatic miRNA and found the miR-507 was the most downregulated in response to CCL5 overexpression (Supplementary Figure 4). We further integrated the miRNA screening results with open source software (www.TargetScan.org and www.microrna.org), and found that the 3′-UTR of VEGF-C mRNA harbors potential binding sites for miR-507. We confirmed miR-507 expression using quantitative real-time PCR (qPCR) in both CCL5-overexpressed and CCL5-knockdowned cells. As shown in Figure 3A, the miR-507 was downregulated in JJ012/CCL5 and SW1353/CCL5 cells, and upregulated in JJ012/shCCL5 and SW1353/shCCL5 cells. To explore the role of miR-507 in CCL5-induced VEGF-C production and lymphangiogenesis, we transiently transfected the miR-507 mimic or miR-507 inhibitor into CCL5-overexpressed cells and CCL5-knockdowned cells, respectively. The data showed that VEGF-C production and LECs tube formation were obviously reduced by the miR-507 mimic in JJ012/shCCL5 and SW1353/shCCL5 cells. In addition, the miR-507 mimic clearly diminished the CCL5-induced VEGF-C production and LECs tube formation in JJ012/CCL5 and SW1353/CCL5 cells (Figure 3B-3E).
Figure 3. CCL5 promotes VEGF-C-dependent lymphangiogenesis by suppressing miR-507.
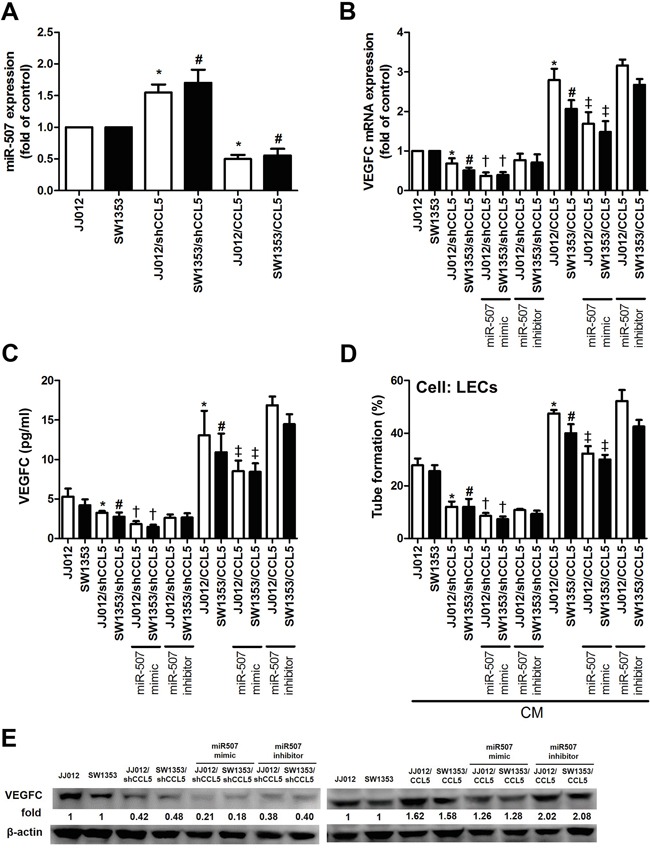
The JJ012/vector, JJ012/shCCL5, JJ012/CCL5, SW1353/vector, SW1353/shCCL5 and SW1353/CCL5 cells cultured for 24 h, and miR-507 expression was examined by qPCR A. In addition, cells were transfected with miR-507 mimic or inhibitor for 24 h, and VEGF-C expression was examined by qPCR B. ELISA C. and Western blot E. (n = 5-7). The medium was collected as CM and then applied to LECs for tube formation assay D. (n = 5). Results are expressed as the mean ± SE. *, p < 0.05 compared with JJ012 control; #, p < 0.05 compared with SW1353 control; †, p < 0.05 compared with CCL5-knockdowned group; ‡, p < 0.05 compared with CCL5-overexpressed group.
In order to verify whether miR-507 regulates the 3′UTR of VEGF-C, we constructed luciferase reporter vectors harboring wildtype 3′UTR of VEGF-C mRNA (wt-VEGFC-3′UTR) and vector containing mismatches in the predicted miR-507 binding site (mt-VEGFC-3′UTR) and transfected these vectors into CCL5-overexpressed cells, CCL5-knockdowned cells and control cells. We found that cotransfection with the miR-507 mimic inhibited luciferase activity in the wt-VEGFC-3′UTR plasmid but not in the mt-VEGFA-3′UTR plasmid (Figure 4). Taken together, these findings demonstrate that miR-507 directly represses VEGF-C expression via binding to 3′UTR of the human VEGF-C gene.
Figure 4. The miR-507 inhibits VEGF-C expression through binding to the 3′UTR of human VEGF-C.
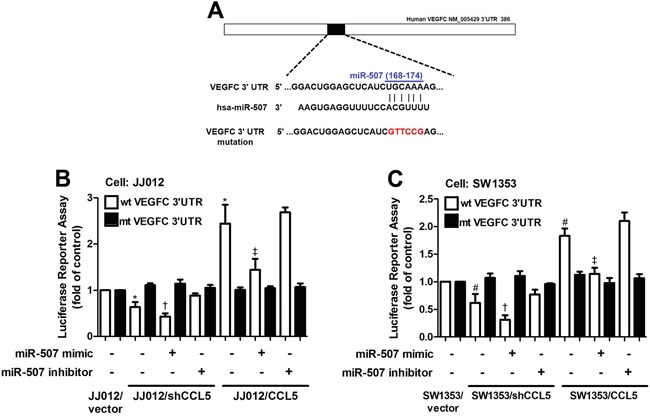
A. Schematic representation of the 3′UTR of the human VEGF-C containing a miR-507 binding site. B&C. Cells were cotransfected with miR-507 mimic or inhibitor and wt-VEGFC-3′UTR or mt-VEGFC-3′UTR plasmid for 24 h, and the relative luciferase/renilla activities were measured by luciferase reporter assay (n = 5). Results are expressed as the mean ± SE. *, p < 0.05 compared with JJ012 control; #, p < 0.05 compared with SW1353 control; †, p < 0.05 compared with CCL5-knockdowned group; ‡, p < 0.05 compared with CCL5-overexpressed group.
CCL5 boosts tumor-associated lymphangiogenesis by down-regulating miR-507
Herein, we investigated whether CCL5 promoted tumor lymphangiogenesis in vivo. To validate the effect and targeting miRNA of CCL5 on tumor lymphangiogenesis, we analyzed the expression of CCL5, VEGF-C and miR-507 in tumor tissues which were excised from chondrosarcoma xenograft mice. Immunohistochemical analysis revealed that overexpression of CCL5 increased the expression of CCL5 and VEGF-C, whereas knockdown of CCL5 decreased the expression of CCL5 and VEGF-C in mouse chondrosarcoma tissues (Figure 5A). The quantitative data exhibited the positive correlation between CCL5 and VEGF-C, and the negative correlation between CCL5 and miR-507 using qPCR analysis in tumor specimens form animals (Figure 5B&5C). Furthermore, LYVE-1 is the specific marker of lymphatic vessels in tumor lymphangiogenesis. As shown in Figure 5A, the number of lymphatic vessels, stained with anti-LYVE-1, was reduced by knockdown of CCL5, and promoted by overexpression of CCL5. Therefore, we suggest that CCL5 promotes VEGF-C-dependent tumor-associated lymphangiogenesis by down-regulating miR-507 in vivo.
Figure 5. CCL5 promotes tumor lymphangiogenesis by suppressing miR-507 in vivo.
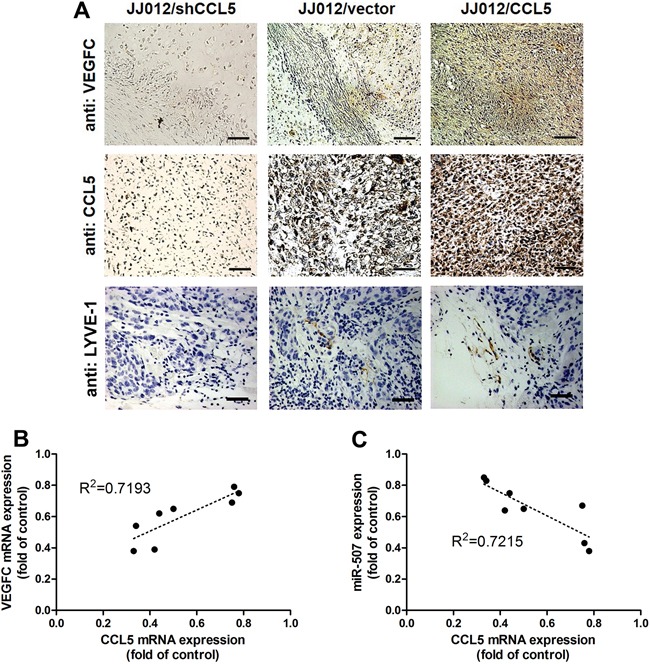
The JJ012/vector, JJ012/shCCL5 and JJ012/CCL5 cells were mixed with Matrigel and injected into flank sites of mice (n = 8-10) for six weeks. Then, the xenografted tumors were excised and stained with CCL5, VEGF-C and LYVE-1 by IHC A. The correlation between CCL5/VEGF-C B. and CCL5/miR-507 C. in tumor tissues was analyzed by qPCR and Spearman rank correlation test (n = 8) (R2 > 0.5).
DISCUSSION
Although chondrosarcoma is a relatively rare human cancer, the notorious aggressiveness of chondrosarcoma is due to its metastatic potential and poor prognosis [3]. Lymphngiogenesis is an indispensable step for cancer metastasis, facilitating cancer development by the generation of new lymphatic vessels that serve as conduits for tumor dissemination to lymph nodes and beyond [4]. Accumulating evidences demonstrate that increased level of VEGF-C promotes tumor relapse and poor prognosis, and thus VEGF-C represents a potential target for preventing lymphatic metastasis [5, 13]. In the current study, we indicate the clinical significance of CCL5 and VEGF-C expression in specimens of chondrosarcoma patients. We reveal that CCL5 increases the production of lymphangiogenic factor VEGF-C by down-regulating miR-507 in chondrosarcoma cells, and thereby promotes tumor lymphangiogenesis in chondrosarcoma microenvironment (Figure 6). Recent studies have shown that chemokine/chemokine receptor system is directly or indirectly involved in promoting tumor lymphangiogenesis [5]. CXC chemokine ligand 12 (SDF-1) has been indicated to directly promote lymphangiogenesis in human LECs through the chemokine receptor CXCR4 [29]. Addtionally, CCL21/CCR7 chemokine axis regulates the expression and secretion of VEGF-C in human breast cancer cells, and contributes to LECs lymphangiogenesis and breast cancer-induced lymphangiogenesis [30]. Herein, we point out that CCL5/CCR5 axis induces VEGF-C-dependent lymphangiogenesis in human chondrosarcoma. Our study is the first to report that chemokine CCL5 induces lymphangiogenesis by the induction of VEGF-C in human cancer cells.
Figure 6. Schematic diagram summarizes the mechanism of CCL5-mediated tumor lymphangiogenesis in human chondrosarcoma.
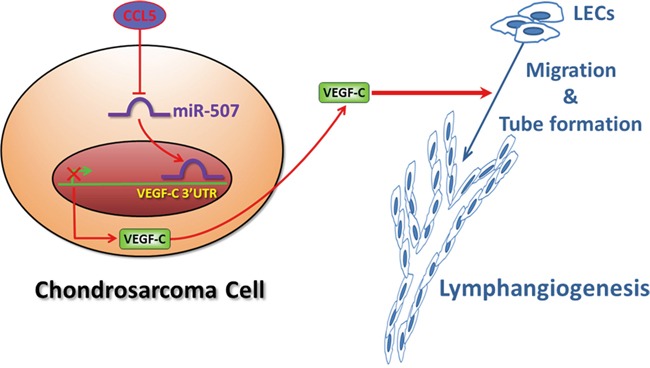
CCL5 promotes VEGF-C production by downregulating miR-507 in human chondrosarcoma cells, and subsequently induces lymphangiogenesis of human LECs.
Chemokines can be produced by tumor cells and/or by nonmalignant stromal cells in the microenvironment, and dictate tumor development by stimulating angiogenesis and lymphangiogenesis, which are the important routes for cancer metastasis [31]. Here, we investigated the effect of chemokine CCL5 on VEGF-C production and tumor lymphangiogenesis both in vitro and in vivo. We found that CCL5 increased the expression and secretion of VEGF-C in human chondrosarcoma cells, and subsequently induced migration and tube formation of human LECs. Otherwise, CCL5 knockdown dramatically suppressed VEGF-C-induced lymphangiogenesis. Moreover, we showed that CCL5 and VEGF-C display a positive correlation in the chondrosarcoma xenograft model. CCL5 knockdown diminished the expression of lymphangiogenesis marker LYVE-1 and VEGF-C in vivo. CCL5 serves as a potent positive-regulator of tumor lymphangiogenesis in human chondrosarcoma. Previously, we have demonstrated that CCL5 promotes tumor angiogenesis by VEGF-A production in human chondrosarcoma [27, 28]. These findings support the rationale that impeding CCL5 signal may inhibit both tumor angiogenesis and lymphangiogenesis, leading to impair cancer metastasis and the formation of an immunosuppressive microenvironment. Targeting CCL5 and CCR5 with antagonists will have potential therapeutic usage to restrict the disease progression of many cancers, particularly chondrosarcoma. CCR5 antagonists have been successfully used in patients with HIV infection [32]. Maraviroc and vicriviroc are CCR5 antagonists that exert promising efficacy against chemokine function and HIV entry. There are several lines of evidence suggesting possible clinical applications of CCR5 antagonists in cancer treatment [33, 34]. Importantly, inhibition of CCL5 production by cancer cell or by the tumor microenvironment may represent an additional strategy to block cancer progression [25]. The development of novel therapeutic molecules (antibody, peptide, small molecule compound, etc.) aimed to repress the expression or secretion of CCL5 is needed to proceed in the future.
The lymphatic system is the primary pathway of metastasis for most human cancers [6]. The lymphatic endothelium, which comprises LECs, is a specialized endothelium and is distinct from the vascular endothelium [35]. Tumor lymphatic vessels serve as a pivotal route for metastatic cancer cells, due to their leaky nature and secretion of tumor-recruiting factors [7]. More understanding of molecular mechanisms underlying tumor lymphangiogenesis will provide new insights in the process of metastasis. However, the study of lymphangiogenesis has been impeded by the difficulties in the isolation and propagation of LECs from different organs [36, 37]. To overcome the above limitations, we used a “conditionally immortalized” line of human LECs, which transformed with the human telomerase reverse transcriptase (hTERT), and maintained their ‘lymphatic’ endothelial characteristics after repeated passages. This immortalized human LECs keep the ability to sprout, elongate, migrate and reorganize to form the capillary-like tube structure within 4–8 h, a process called tube formation, and this function of LECs represents the major process of lymphangiogenesis. In this study, we found that CM from CCL5-overexpressed cells profoundly stimulated tube formation of human LECs. On the contrary, knockdown of CCL5 suppressed CM-induced LECs tube formation. These results provide evidences that CCL5-mediated VEGF-C production induces lymphangiogenesis in vitro. Furthermore, we found that levels of CCL5 and VEGF-C in clinical specimens from patients with chondrosarcoma were correlated with tumor stage, implying that CCL5 may be a potential prognostic indicator for disease progression of human chondrosarcoma.
Small non-coding miRNAs, the average length of approximately 18 to 22 nucleotides, negatively regulate gene expression by either translational repression or mRNA cleavage through binding to complementary 3′UTR sequences of target mRNA [15, 17]. Deregulated biogenesis of miRNAs has been widely observed in human cancer [18]. Accumulating evidences further indicate that numerous miRNAs can impede cancer progression via direct suppression of VEGF-C. miR-27b, miR-101, miR-128, miR-206 and miR-1826 have been reported to inhibit tumor growth, lymphangiogenesis and metastasis by targeting VEGF-C in a variety of human cancer cells [20–22, 38–40]. Current study showed that CCL5 markedly repressed miR-507 expression in human chondrosarcoma cells in vitro and in vivo. Cotransfection of cells with miR-507 mimic abolished CCL5-induced VEGF-C production and LECs tube formation. Strikingly, we revealed that miR-507 directly inhibited VEGF-C protein expression through binding to the 3′UTR of the human VEGF-C gene, thereby negatively regulating VEGF-C-mediated lymphangiogenesis. miR-507 has been shown to suppress the NF-E2-related factor 2 (NRF2)-mediated oncogenic pathway by directly targeting NRF2 [41]. Moreover, administration of miR-507 alone or in combination with cisplatin significantly inhibits tumor growth in esophageal squamous cell carcinoma. To our knowledge, this study is first time to indicate that miR-507 negatively regulates VEGF-C expression. Importantly, we further demonstrate that CCL5 promotes VEGF-C-mediated tumor lymphangiogenesis by suppressing miR-507 in human chondrosarcoma. Thus, these findings provide information on the potential miRNA-based molecular diagnosis and treatment for VEGF-C-dependent tumor-associated lymphangiogenesis.
MATERIALS AND METHODS
Materials
Anti-mouse and anti-rabbit IgG-conjugated horseradish peroxidase, rabbit polyclonal antibody specific for β-actin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). CCL5 and VEGF-C antibodies were purchased from Abcam (Cambridge, MA, USA). Recombinant human VEGF-C was purchased from R&D Systems (Minneapolis, MN, USA). Recombinant human CCL5 and the VEGF-C ELISA kit were purchased from PerpoTech (Rocky Hill, NJ, USA). The miR-507 mimic, miR-507 inhibitor, Lipofectamine 2000 and Trizol were purchased from Life Technologies (Carlsbad, CA, USA). The human miRNome microRNA Profilers QuantiMir™ kit was purchased from SBI (Mountain View, CA, USA). Dulbecco's modified Eagle's medium (DMEM), α-minimum essential medium (MEM), fetal bovine serum (FBS) and all other cell culture reagents were purchased from Gibco-BRL Life technologies (Grand Island, NY, USA). The dual-luciferase reporter assay kit was purchased from Promega (Madison, WI, USA). All other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA).
Cell culture
The human chondrosarcoma cell line (JJ012) was kindly provided by the laboratory of Dr. Sean P. Scully (University of Miami School of Medicine, Miami, FL) and originated from Dr. Joel Block (Rush University Medical Center, Chicago, Illinois). The human chondrosarcoma cell line (SW1353) was purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in DMEM/α-MEM supplemented with 10% FBS and 100 units/ml penicillin/streptomycin at 37°C with 5% CO2. The basal level of CCL5 in JJ012 cells is higher than in SW1351 cells (Supplementary Figure 5).
The human telomerase-immortalized human dermal lymphatic endothelial cells (hTERT-HDLECs), an immortalized human LEC line, was purchased from Lonza (Walkersville, MD, USA). These immortalized human LECs represent CD31 positive/podoplanin positive, and retain their ability to uptake acetylated LDL and induce tube formation. The human LECs were grown in EGM-2MV BulletKit Medium consisting of EBM-2 basal medium plus SingleQuots kit (Lonza). Cells were seeded onto 1% gelatin-coated plastic ware and cultured at 37°C with 5% CO2. We obtained the cryopreserved human LECs line from Lonza as passage 1, and maintained these cells according to manufacturer's instructions as well as used between passages 5 and 10 for experiments described herein.
Generation of stable cell lines
Cells were seeded on plates and infected with pLKO_AS2.puro-vector, pLKO_AS2.puro-CCL5 or pLKO_1.puro-CCL5 shRNA by prepared lentivirus as described previously [28]. At 24 h after infection, stable clones were selected with puromycin. Thereafter, the selection medium was replaced every 2 days. After 2 weeks of selection, resistant clones were established.
Preparation of conditioned medium
In the series of experiments, stable cell lines were incubated alone or transfected with miR-507 mimic or inhibitor for 24 h. After treatment, cells were washed and changed to serum-free medium. Conditioned medium (CM) was then collected 2 days after the change of medium and stored at −80°C until use.
ELISA assay
The CM was used to determine the VEGF-C level in the medium of chondrosarcoma cells using VEGF-C ELISA kit (PerpoTech) according to the procedure described by the manufacturer.
Transwell migration assay
Transwell inserts (8-μm pore size; Costar, NY, USA) in 24-well plates were used. The CM was collected from chondrosarcoma cells. LECs were seeded in the upper transwell chamber and 300 μL of CM was placed in the lower chamber. After 20 h, migrated cells were stained with crystal violet and counted under a microscope.
Tube formation assay
Matrigel (BD Biosciences; Bedford, MA, USA) was dissolved at 4°C overnight, and 48-well plates were prepared with 150 μL Matrigel in each well after coating and incubating at 37°C for 30 min. LECs were resuspended at a density of 5 × 104/100 μL in culture medium (50% EGM-2MV BulletKit Medium and 50% chondrosarcoma cell CM) and added to the wells. After 6 h of incubation at 37°C, LECs tube formation was assessed by microscopy, and each well was photographed. The number of tube branches and total tube length were calculated using the MacBiophotonics Image J software.
Western blot analysis
The cellular lysates were prepared according our previous instruction [42]. The proteins were resolved using SDS-PAGE, then transferred to Immobilon polyvinyldifluoride (PVDF) membranes. Blots were blocked with 4% BSA for 1 h at room temperature, then probed with primary antibodies against VEGF-C (1:1000) and β-actin (1:2000) for 1 h at room temperature. After three washes, blots were subsequently incubated with the horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature and visualized by enhanced chemiluminescence, using an Imagequant LAS 4000 (GE Healthcare, Pewaukee, WI, USA).
Quantitative real-time PCR (qPCR) of mRNA and miRNA
Total RNA was extracted from cells as described previously [43]. The qPCR analysis was carried out using Taqman® one-step PCR Master Mix (Applied Biosystems, CA, USA); 100 ng of total cDNA were added per 25 μl reaction with sequence-specific primers and Taqman® probes. Sequences for all target gene primers and probes were purchased commercially (GAPDH was used as an internal control) (Applied Biosystems). qPCR assays were carried out in triplicate on a StepOnePlus sequence detection system. The cycling conditions consisted of 10-min polymerase activation at 95°C followed by 40 cycles at 95°C for 15 s and 60°C for 60 s. The threshold was set above the non-template control background and within the linear phase of the target gene amplification to calculate the cycle number at which the transcript was detected (denoted CT).
For the miRNA assay, cDNA was synthesized from total RNA (100 ng) using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). Then, miRNome microRNA Profilers QuantiMir™ kit which contained 384 human miRNA primer was used to elucidate miRNA differential expression. The reactions were incubated first at 16°C for 30 min and then at 42°C for 30 min followed by inactivation at 85°C for 5 min, then incubated in a 96-well plate at 50°C for 2 min, 95°C for 10 min, followed by 30 cycles of 95°C for 15 s and 60°C for 60 s using the StepOnePlus sequence detection system. Relative gene expression was quantified using an endogenous control gene (U6). The threshold cycle (CT) was defined as the fractional cycle number at which fluorescence passed a fixed threshold, and relative expression was calculated using the comparative CT method.
Plasmid constructs
The 3′-UTR-luciferase reporter constructs containing the 3′-UTR regions of VEGF-C with wild-type and mutant binding sites for miR-507 were amplified using the PCR method. The cDNAs were obtained from H293T cells. The PCR products were cloned into a pmirGLO reporter vector (Promega) between the NheI and XhoI restriction sites, instantly downstream of the luciferase reporter gene. The mutant 3′-UTRs were constructed by introducing seven mismatched mutations into putative seed regions of VEGF-C. All the constructs containing 3′-UTR inserts were sequenced and verified.
Luciferase reporter assay
Cells were seeded on 6-well plates, then transiently transfected with VEGF-C 3′-UTR luciferase plasmids using Lipofectamine 2000 as described previously [44]. Cells collected were lysed with reporter lysis buffer 24 h after transfection, and the luciferase and renilla activities in the cellular extracts were determined by the dual-luciferase reporter assay kit. The relative luciferase activity was calculated by the ratio of luciferase/renilla activity, and normalized to that of control cells.
Immunohistochemistry (IHC)
The human chondrosarcoma tissue array was purchased from CtbridTM (Rockville, MD, USA). The 9 cases for normal cartilage, 8 cases for type Ib chondrosarcoma (Grade I), 9 cases for type IIb chondrosarcoma (Grade II), and 12 cases for type IIIb chondrosarcoma (Grade III). The tissue slides were incubated and rehydrated in 3% hydrogen peroxide to block the endogenous peroxidase activity. After trypsinization, sections were blocked by incubation in 3% FBS in PBS. The primary antibody anti-human CCL5, VEGF-C or LYVE-1 was applied to the slides at a dilution from 1:50 to 1:150 and incubated at 4°C overnight [45]. After the slides were washed 3 times with PBS, the samples were treated with the secondary antibody at a dilution of 1:100. Bound antibodies were detected with an ABC kit (Vector Laboratories, Burlingame, CA). The samples were stained with chromogen diaminobenzidine, washed with PBS, counter-stained with Delafield's hematoxylin and dehydrated. Finally, the samples were treated with xylene and mounted. The intensity of staining was blindly evaluated by pathologist as 0, 1+, 2+, 3+, 4+, and 5+ for no staining, very weak staining, weak staining, moderate staining, strong staining, and very strong respectively. IHC score was determined as the sum of the intensity score.
Tumor xenograft in vivo study
Four weeks old male nude mice were randomly divided into 3 groups (10 mice per group). For experimental cells growing exponentially, each implanted into mice by subcutaneous injection of 1 × 106 human chondrosarcoma cells (JJ012/vector, JJ012/shCCL5 and JJ012/CCL5) were resuspended and mixed with Matrigel. After six weeks, mice were sacrificed and tumors were excised for IHC and q-PCR analysis, respectively. All procedures involving animal experiment were approved by the Institutional Animal Care and Use Committee at School of Medicine, China Medical University.
Statistics
Data are presented as mean ± standard error of the mean (SEM). Statistical analyses of pairs of samples were performed using the Student's t-test. Statistical comparisons of more than two groups were performed using one-way analysis of variance (ANOVA) with Bonferroni's post-hoc test. In all cases, p < 0.05 was considered significant.
SUPPLEMENTARY FIGURES
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology of Taiwan (MOST 101-2320-B-715-002-MY3, MOST 103-2628-B-039-002-MY3, MOST 104-2320-B-715-002); Mackay Medical College (MMC-1031B02, MMC-1041B10); Mackay Memorial Hospital (MMH-101101, MMH-103-44, MMH-MM-10303); Taipei City Hospital (TCH 10301-62-047) and Dongyang People's Hospital (2015-YB002).
Footnotes
CONFLICTS OF INTEREST
All authors have no financial or personal relationships with other people or organizations that could inappropriately influence our work.
REFERENCES
- 1.Terek RM, Schwartz GK, Devaney K, Glantz L, Mak S, Healey JH, Albino AP. Chemotherapy and P-glycoprotein expression in chondrosarcoma. J Orthop Res. 1998;16:585–590. doi: 10.1002/jor.1100160510. [DOI] [PubMed] [Google Scholar]
- 2.Chang L, Shrestha S, LaChaud G, Scott MA, James AW. Review of microRNA in osteosarcoma and chondrosarcoma. Med Oncol. 2015;32:613. doi: 10.1007/s12032-015-0613-z. [DOI] [PubMed] [Google Scholar]
- 3.Chen JC, Fong YC, Tang CH. Novel strategies for the treatment of chondrosarcomas: targeting integrins. Biomed Res Int. 2013;2013:396839. doi: 10.1155/2013/396839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Trappen PO, Pepper MS. Lymphatic dissemination of tumour cells and the formation of micrometastases. Lancet Oncol. 2002;3:44–52. doi: 10.1016/s1470-2045(01)00621-0. [DOI] [PubMed] [Google Scholar]
- 5.Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer. 2014;14:159–172. doi: 10.1038/nrc3677. [DOI] [PubMed] [Google Scholar]
- 6.Van der Auwera I, Cao Y, Tille JC, Pepper MS, Jackson DG, Fox SB, Harris AL, Dirix LY, Vermeulen PB. First international consensus on the methodology of lymphangiogenesis quantification in solid human tumours. Br J Cancer. 2006;95:1611–1625. doi: 10.1038/sj.bjc.6603445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wissmann C, Detmar M. Pathways targeting tumor lymphangiogenesis. Clin Cancer Res. 2006;12:6865–6868. doi: 10.1158/1078-0432.CCR-06-1800. [DOI] [PubMed] [Google Scholar]
- 8.Streit M, Detmar M. Angiogenesis, lymphangiogenesis, and melanoma metastasis. Oncogene. 2003;22:3172–3179. doi: 10.1038/sj.onc.1206457. [DOI] [PubMed] [Google Scholar]
- 9.Rubbia-Brandt L, Terris B, Giostra E, Dousset B, Morel P, Pepper MS. Lymphatic vessel density and vascular endothelial growth factor-C expression correlate with malignant behavior in human pancreatic endocrine tumors. Clin Cancer Res. 2004;10:6919–6928. doi: 10.1158/1078-0432.CCR-04-0397. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura Y, Yasuoka H, Tsujimoto M, Imabun S, Nakahara M, Nakao K, Nakamura M, Mori I, Kakudo K. Lymph vessel density correlates with nodal status, VEGF-C expression, and prognosis in breast cancer. Breast Cancer Res Treat. 2005;91:125–132. doi: 10.1007/s10549-004-5783-x. [DOI] [PubMed] [Google Scholar]
- 11.Tacconi C, Correale C, Gandelli A, Spinelli A, Dejana E, D'Alessio S, Danese S. Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology. 2015;148:1438–1451. doi: 10.1053/j.gastro.2015.03.005. e1438. [DOI] [PubMed] [Google Scholar]
- 12.Su JL, Shih JY, Yen ML, Jeng YM, Chang CC, Hsieh CY, Wei LH, Yang PC, Kuo ML. Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular endothelial growth factor-C up-regulation: a novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer research. 2004;64:554–564. doi: 10.1158/0008-5472.can-03-1301. [DOI] [PubMed] [Google Scholar]
- 13.Dieterich LC, Seidel CD, Detmar M. Lymphatic vessels: new targets for the treatment of inflammatory diseases. Angiogenesis. 2014;17:359–371. doi: 10.1007/s10456-013-9406-1. [DOI] [PubMed] [Google Scholar]
- 14.Lee E, Fertig EJ, Jin K, Sukumar S, Pandey NB, Popel AS. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat Commun. 2014;5:4715. doi: 10.1038/ncomms5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Croce CM. Oncogenes and cancer. N Engl J Med. 2008;358:502–511. doi: 10.1056/NEJMra072367. [DOI] [PubMed] [Google Scholar]
- 16.Yoshitaka T, Kawai A, Miyaki S, Numoto K, Kikuta K, Ozaki T, Lotz M, Asahara H. Analysis of microRNAs expressions in chondrosarcoma. J Orthop Res. 2013;31:1992–1998. doi: 10.1002/jor.22457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 18.Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. 2009;4:199–227. doi: 10.1146/annurev.pathol.4.110807.092222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA. MicroRNAs—the micro steering wheel of tumour metastases. Nat Rev Cancer. 2009;9:293–302. doi: 10.1038/nrc2619. [DOI] [PubMed] [Google Scholar]
- 20.Hu J, Cheng Y, Li Y, Jin Z, Pan Y, Liu G, Fu S, Zhang Y, Feng K, Feng Y. microRNA-128 plays a critical role in human non-small cell lung cancer tumourigenesis, angiogenesis and lymphangiogenesis by directly targeting vascular endothelial growth factor-C. Eur J Cancer. 2014;50:2336–2350. doi: 10.1016/j.ejca.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Keklikoglou I, Hosaka K, Bender C, Bott A, Koerner C, Mitra D, Will R, Woerner A, Muenstermann E, Wilhelm H, Cao Y, Wiemann S. MicroRNA-206 functions as a pleiotropic modulator of cell proliferation, invasion and lymphangiogenesis in pancreatic adenocarcinoma by targeting ANXA2 and KRAS genes. Oncogene. 2015;34:4867–4878. doi: 10.1038/onc.2014.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng G, Teng Y, Huang F, Nie W, Zhu L, Huang W, Xu H. MicroRNA-101 inhibits the migration and invasion of intrahepatic cholangiocarcinoma cells via direct suppression of vascular endothelial growth factor-C. Mol Med Rep. 2015;12:7079–7085. doi: 10.3892/mmr.2015.4239. [DOI] [PubMed] [Google Scholar]
- 23.Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347:669–671. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- 24.Borsig L, Wolf MJ, Roblek M, Lorentzen A, Heikenwalder M. Inflammatory chemokines and metastasis—tracing the accessory. Oncogene. 2014;33:3217–3224. doi: 10.1038/onc.2013.272. [DOI] [PubMed] [Google Scholar]
- 25.Aldinucci D, Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators of inflammation. 2014;2014:292376. doi: 10.1155/2014/292376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang CH, Yamamoto A, Lin YT, Fong YC, Tan TW. Involvement of matrix metalloproteinase-3 in CCL5/CCR5 pathway of chondrosarcomas metastasis. Biochem Pharmacol. 2010;79:209–217. doi: 10.1016/j.bcp.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Liu GT, Huang YL, Tzeng HE, Tsai CH, Wang SW, Tang CH. CCL5 promotes vascular endothelial growth factor expression and induces angiogenesis by down-regulating miR-199a in human chondrosarcoma cells. Cancer Lett. 2015;357:476–487. doi: 10.1016/j.canlet.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 28.Liu GT, Chen HT, Tsou HK, Tan TW, Fong YC, Chen PC, Yang WH, Wang SW, Chen JC, Tang CH. CCL5 promotes VEGF-dependent angiogenesis by down-regulating miR-200b through PI3K/Akt signaling pathway in human chondrosarcoma cells. Oncotarget. 2014;5:10718–10731. doi: 10.18632/oncotarget.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhuo W, Jia L, Song N, Lu XA, Ding Y, Wang X, Song X, Fu Y, Luo Y. The CXCL12-CXCR4 chemokine pathway: a novel axis regulates lymphangiogenesis. Clin Cancer Res. 2012;18:5387–5398. doi: 10.1158/1078-0432.CCR-12-0708. [DOI] [PubMed] [Google Scholar]
- 30.Tutunea-Fatan E, Majumder M, Xin X, Lala PK. The role of CCL21/CCR7 chemokine axis in breast cancer-induced lymphangiogenesis. Mol Cancer. 2015;14:35. doi: 10.1186/s12943-015-0306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazennec G, Richmond A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol Med. 2010;16:133–144. doi: 10.1016/j.molmed.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkin TJ, Gulick RM. CCR5 antagonism in HIV infection: current concepts and future opportunities. Annual review of medicine. 2012;63:81–93. doi: 10.1146/annurev-med-052010-145454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weitzenfeld P, Ben-Baruch A. The chemokine system, and its CCR5 and CXCR4 receptors, as potential targets for personalized therapy in cancer. Cancer Lett. 2014;352:36–53. doi: 10.1016/j.canlet.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Velasco-Velazquez M, Jiao X, De La Fuente M, Pestell TG, Ertel A, Lisanti MP, Pestell RG. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer research. 2012;72:3839–3850. doi: 10.1158/0008-5472.CAN-11-3917. [DOI] [PubMed] [Google Scholar]
- 35.Pepper MS, Skobe M. Lymphatic endothelium: morphological, molecular and functional properties. J Cell Biol. 2003;163:209–213. doi: 10.1083/jcb.200308082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nisato RE, Harrison JA, Buser R, Orci L, Rinsch C, Montesano R, Dupraz P, Pepper MS. Generation and characterization of telomerase-transfected human lymphatic endothelial cells with an extended life span. Am J Pathol. 2004;165:11–24. doi: 10.1016/S0002-9440(10)63271-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruyere F, Noel A. Lymphangiogenesis: in vitro and in vivo models. FASEB J. 2010;24:8–21. doi: 10.1096/fj.09-132852. [DOI] [PubMed] [Google Scholar]
- 38.Ye J, Wu X, Wu D, Wu P, Ni C, Zhang Z, Chen Z, Qiu F, Xu J, Huang J. miRNA-27b targets vascular endothelial growth factor C to inhibit tumor progression and angiogenesis in colorectal cancer. PloS one. 2013;8:e60687. doi: 10.1371/journal.pone.0060687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirata H, Hinoda Y, Ueno K, Shahryari V, Tabatabai ZL, Dahiya R. MicroRNA-1826 targets VEGFC, beta-catenin (CTNNB1) and MEK1 (MAP2K1) in human bladder cancer. Carcinogenesis. 2012;33:41–48. doi: 10.1093/carcin/bgr239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu HT, Xing AY, Chen X, Ma RR, Wang YW, Shi DB, Zhang H, Li P, Chen HF, Li YH, Gao P. MicroRNA-27b, microRNA-101 and microRNA-128 inhibit angiogenesis by down-regulating vascular endothelial growth factor C expression in gastric cancers. Oncotarget. 2015;6:37458–37470. doi: 10.18632/oncotarget.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto S, Inoue J, Kawano T, Kozaki K, Omura K, Inazawa J. The impact of miRNA-based molecular diagnostics and treatment of NRF2-stabilized tumors. Mol Cancer Res. 2014;12:58–68. doi: 10.1158/1541-7786.MCR-13-0246-T. [DOI] [PubMed] [Google Scholar]
- 42.Liu SC, Chen C, Chung CH, Wang PC, Wu NL, Cheng JK, Lai YW, Sun HL, Peng CY, Tang CH, Wang SW. Inhibitory effects of butein on cancer metastasis and bioenergetic modulation. Journal of agricultural and food chemistry. 2014;62:9109–9117. doi: 10.1021/jf502370c. [DOI] [PubMed] [Google Scholar]
- 43.Wang SW, Liu SC, Sun HL, Huang TY, Chan CH, Yang CY, Yeh HI, Huang YL, Chou WY, Lin YM, Tang CH. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis. 2015;36:104–114. doi: 10.1093/carcin/bgu218. [DOI] [PubMed] [Google Scholar]
- 44.Tai HC, Chang AC, Yu HJ, Huang CY, Tsai YC, Lai YW, Sun HL, Tang CH, Wang SW. Osteoblast-derived WNT-induced secreted protein 1 increases VCAM-1 expression and enhances prostate cancer metastasis by down-regulating miR-126. Oncotarget. 2014;5:7589–7598. doi: 10.18632/oncotarget.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin CC, Chen PC, Lein MY, Tsao CW, Huang CC, Wang SW, Tang CH, Tung KC. WISP-1 promotes VEGF-C-dependent lymphangiogenesis by inhibiting miR-300 in human oral squamous cell carcinoma cells. Oncotarget. 2016;7:9993–10005. doi: 10.18632/oncotarget.7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.