

Abstract
Pituitary spindle cell oncocytoma (SCO) is an uncommon primary pituitary neoplasm that presents with mass effect on adjacent neurovascular structures, similar to non-hormone-producing pituitary adenomas. To determine the molecular etiology of SCO, we performed exome sequencing on four SCO cases, with matched normal controls, to assess somatic mutations and copy number alterations. Our analysis revealed a low mutation rate and a copy-neutral profile, consistent with the low-grade nature of this tumor. However, we identified a co-occurring somatic HRAS (p.Q61R) activating point mutation and MEN1 frameshift mutation (p.L117fs) present in a primary and recurrent tumor from one patient. Other SCOs demonstrated mutations in SND1 and FAT1, which are associated with MAPK pathway activation. Immunohistochemistry across the SCO cohort demonstrated robust MAPK activity in all cases (n=4), as evidenced by strong phospho-ERK staining, while phospho-AKT levels suggested only basal levels of PI3K pathway activation. Taken together, this identifies the MAPK signaling pathway as a novel therapeutic target for spindle cell oncocytoma, which may offer a powerful adjunct for aggressive tumors refractory to surgical resection.
Keywords: spindle cell oncocytoma, pituitary, MAPK, HRAS, genomics
INTRODUCTION
Spindle cell oncocytoma (SCO) is a rare non-endocrine neoplasm of the hypophysis, which exhibits WHO grade I histology [1]. SCO presents similarly to non-functioning pituitary adenomas, clinically demonstrating pituitary hypofunction, visual field deficits, and potential headache and nausea, due to mass effect. Although they were initially regarded as benign, several subsequent recurrent and locally aggressive SCO cases have been reported [2–6]. SCO is primarily treated with surgical resection, while radiation therapy has been reported for patients with recurrence [2]. Invasion of nearby structures, including the cavernous sinus, suprasellar space, and sphenoid sinus, can challenge traditional therapeutic strategies.
Histologically, SCOs show an interlacing fascicular pattern of spindled to epithelioid cells, with eosinophilic and variably oncocytic cytoplasm. Nuclear atypia is generally minimal and mitotic indices are low [2]. Ultrastructural features of abundant mitochondria and a paucity of secretory granules help distinguish SCOs from non-functioning adenomas [1, 2]. Expression of S-100, vimentin, galectin-3, and epithelial membrane antigen (EMA) is typical of SCOs, which lack expression of pituitary adenoma markers such as synaptophysin, chromogranin, and pituitary hormones [1]. SCOs generally do not express glial fibrillary acidic protein (GFAP), distinguishing them from pituicytoma, a tumor derived from neurohypophyseal glial pituicytes [1, 2, 7]. SCOs also do not express cytokeratins, smooth muscle actin (SMA), CD34, or CD68.
The cell of origin for SCOs remains unclear. They have been postulated to derive from folliculostellate cells of the adenohypophysis, based on shared expression of S-100, vimentin, galectin-3, and EMA, as well as desmosomes and intermediate junctions found using electron microscopy [1]. However, the pituicyte has also been proposed as a potential cell of origin, on the basis of shared expression of thyroid transcription factor 1 (TTF-1), prompting a potential classification of SCOs as oncocytic pituicytomas [8].
Little is known about the genetic drivers of proliferation and infiltration in SCO. A recent report on seven cases found no BRAF V600E mutations, BRAF-KIAA fusions, or IDH R132H mutation-specific immunoreactivity [8]. One case report observed mild-to-moderate expression of phospho-AKT, phospho-mTOR, and GLI2, suggesting some degree of activation of mammalian target of rapamycin (mTOR) and sonic hedgehog (SHH) pathways [9].
In order to further examine the molecular drivers of oncogenesis in spindle cell oncocytoma, we performed whole exome sequencing and signal pathway profiling on four cases of SCO. Here we report novel genetic mutations that may provide additional insights into the future treatment of this disease.
RESULTS
Mutational profile of SCO
We identified all cases of SCO resected at Brigham and Women's Hospital since its first report at this institution in 2002, yielding four cases from three patients (Table 1). Patient three manifested with recurrent/residual tumor less than a year after initial resection, and therefore two separate samples were available for study (cases 3A and 3B). Each SCO case was reviewed and the diagnosis confirmed on the basis of histologic appearance and immunohistochemical profile (Table 2). Figure 1 illustrates typical histologic and immunohistochemical features. In concordance with a recent report [8], we found strong nuclear TTF-1 expression in each case of SCO.
Table 1. Clinical profiles of spindle cell oncocytoma cases.
| Case | Age (Yr) | Imaging characteristics |
|---|---|---|
| 1 | 66 | 2.4 cm sellar mass, abutting cavernous sinuses and third ventricle, and extending into sphenoid sinus and posterior to optic chiasm |
| 2 | 50 | 1.4 cm sellar mass, partially surrounding bilateral internal carotid arteries and abutting optic nerves |
| 3A | 63 | 2.7 cm sellar mass, extending into sphenoid sinus, partially encasing bilateral internal carotid arteries, and displacing optic chiasm and optic nerves |
| 3B | 63 | 1.7 cm residual/recurrent enhancing sellar mass, partially encasing left internal carotid artery, with displacement of optic chiasm and optic nerves |
Table 2. Immunohistochemical profiles of spindle cell oncocytoma cases.
| Case | EMA | S100 | Galectin-3 | GFAP | Chromogranin | TTF-1 | MIB-1 |
|---|---|---|---|---|---|---|---|
| 1 | + | + | + | - | - | + | 5% |
| 2 | + | + | + | - | - | + | 2% |
| 3A | - | + | + | - | - | + | 5% |
| 3B | + (focal) | + | + | - | - | + | 5% |
Figure 1. Histologic and Immunohistochemical Features of Spindle Cell Oncocytoma.
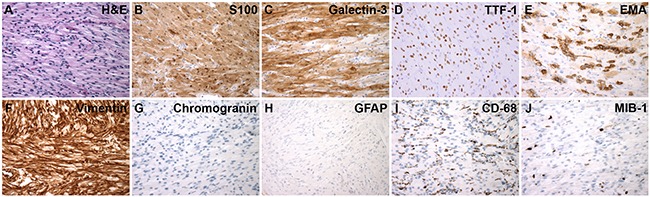
A. H&E stain. Immunohistochemistry for: B. S100, C. Galectin-3, D. TTF-1, E. EMA, F. Vimentin, G. Chromogranin, H. GFAP, I. CD68 and J. MIB-1 (Ki-67). (A-J, 600X magnification; A-I, case 1; J, case 2)
We performed whole exome next-generation sequencing on each SCO case, using matched DNA as control. Sequencing revealed 43 nonsynonymous somatic mutations, insertions, or deletions (Table 3). Among the mutations present, samples 3A and 3B both showed a Q61R mutation in the Harvey rat sarcoma viral oncogene homolog (HRAS) gene on chromosome 11, a specific variant that has been previously reported in multiple cancers [10–13]. Cases 3A and 3B also both showed two frameshift mutations in the multiple endocrine neoplasia type 1 (MEN1) gene, also on chromosome 11; these variants have been previously reported as both germline and sporadic mutations in tumors of the pituitary and other sites [14].
Table 3. Mutations identified by whole exome sequencing of spindle cell oncocytoma cases.
| Gene | Chr. | Variant | Protein Change | Allele Frequency | Case | Functional Prediction Score (SIFT) |
|---|---|---|---|---|---|---|
| NAV1 | 1 | Missense Mutation | L85V | 0.31, 0.15 | 3A, 3B | 0.00 |
| TCEB3 | 1 | Frameshift Deletion | K451fs | 0.16 | 3A | — |
| ADD2 | 2 | Missense Mutation | E480K | 0.11 | 1 | 0.00 |
| C2orf16 | 2 | Nonsense Mutation | Q788* | 0.39, 0.20 | 3A, 3B | — |
| FZD7 | 2 | Missense Mutation | D3N | 0.41, 0.27 | 3A, 3B | 0.30 |
| SH3BP4 | 2 | Missense Mutation | D922G | 0.14 | 1 | 0.00 |
| STAT4 | 2 | Missense Mutation | E388D | 0.44, 0.25 | 3A, 3B | 0.04 |
| ZNF717 | 3 | Frameshift Insertion | T45fs | 0.4 | 2 | — |
| FAT1 | 4 | Missense Mutation | N109H | 0.25 | 1 | 0.00 |
| FAT4 | 4 | Missense Mutation | N3706S | 0.25 | 3B | 0.10 |
| ANKH | 5 | Missense Mutation | E43D | 0.39, 0.09 | 3A, 3B | 0.08 |
| ASCC3 | 6 | Missense Mutation | S221Y | 0.12 | 3A | 1.00 |
| EXOC2 | 6 | Missense Mutation | H736Q | 0.33, 0.21 | 3A, 3B | 0.34 |
| GPR115 | 6 | Missense Mutation | N230S | 0.13 | 2 | 0.74 |
| GTPBP2 | 6 | Missense Mutation | R335Q | 0.11 | 1 | — |
| PIK3CG | 7 | Missense Mutation | E1073K | 0.38, 0.15 | 3A, 3B | 0.47 |
| SND1 | 7 | Missense Mutation | S578N | 0.18 | 2 | 0.17 |
| TRPV5 | 7 | Missense Mutation | M440T | 0.13 | 3A | 0.25 |
| AGO2 | 8 | Missense Mutation | P430R | 0.13 | 2 | 0.00 |
| C8orf76 | 8 | Frameshift Deletion | PERR21fs | 0.47 | 3A | — |
| TRPM6 | 9 | Missense Mutation | L333V | 0.12, 0.14 | 3A, 3B | 0.14 |
| CALHM1 | 10 | Missense Mutation | R178C | 0.40, 0.13 | 3A, 3B | 0.02 |
| CBL | 11 | Missense Mutation | R280Q | 0.41, 0.21 | 3A, 3B | 0.01 |
| HRAS | 11 | Missense Mutation | Q61R | 0.42, 0.19 | 3A, 3B | 0.04 |
| IPO7 | 11 | Nonsense Mutation | Y689* | 0.21 | 3B | — |
| MEN1 | 11 | Frameshift Deletion | K459fs | 0.36, 0.17 | 3A, 3B | — |
| MEN1 | 11 | Frameshift Deletion | LV117fs | 0.36, 0.18 | 3A, 3B | — |
| OR1S2 | 11 | Missense Mutation | P300S | 0.42, 0.20 | 3A, 3B | 0.00 |
| XRRA1 | 11 | Missense Mutation | R76Q | 0.12 | 1 | 0.77 |
| CCER1 | 12 | Nonsense Mutation | R40* | 0.13 | 2 | — |
| ITGA7 | 12 | Missense Mutation | D312A | 0.42, 0.13 | 3A, 3B | 0.43 |
| TAOK3 | 12 | Missense Mutation | E496D | 0.39, 0.14 | 3A, 3B | 0.11 |
| CCNK | 14 | Missense Mutation | G53A | 0.26 | 3B | 0.27 |
| KLHDC1 | 14 | Frameshift Deletion | W28fs | 0.36 | 1 | — |
| LTB4R | 14 | Nonsense Mutation | Y172* | 0.35, 0.14 | 3A, 3B | — |
| RIN3 | 14 | Nonsense Mutation | W275* | 0.06, 0.21 | 3A, 3B | — |
| CHD2 | 15 | Missense Mutation | R550S | 0.19 | 1 | 0.00 |
| DENND4A | 15 | Missense Mutation | R865C | 0.1 | 1 | 0.00 |
| LTK | 15 | Missense Mutation | A480T | 0.13 | 1 | 0.11 |
| TYRO3 | 15 | Missense Mutation | A48V | 0.18 | 2 | 0.08 |
| SBK1 | 16 | Frameshift Deletion | G304fs | 0.33 | 3B | — |
| CD300A | 17 | Missense Mutation | W49L | 0.21 | 3B | 0.00 |
| FMNL1 | 17 | Missense Mutation | P49R | 0.27, 0.10 | 3A, 3B | 0.00 |
Other tumor-associated genes found to be mutated as single events in individual tumors in the SCOs within our cohort include FAT atypical cadherin 1 (FAT1) [15], staphylococcal nuclease domain-containing protein 1 (SND1) [16, 17], Cbl proto-oncogene E3 ubiquitin protein ligase (CBL), frizzled class receptor 7 (FZD7), phosphatidylinositol-4,5-bisphosphate 3-kinase subunit gamma (PIK3CG), and SH3 domain binding kinase 1 (SBK1).
Copy number profile of SCO
We found no significant recurrent copy number changes or aneuploidy across the examined cohort of SCO cases. We observed a loss of chromosome 13 in case 3A, which was not detected in the recurrent tumor, case 3B (Supplementary Figure S1). However, this may be attributable to a lower tumor cell fraction in case 3B, limiting its detection.
Immunohistochemical assessment of MAPK and PI3K signaling pathways
Prompted by the HRAS mutation identified in cases 3A and 3B, we examined activation of its canonical intracellular signaling cascade, the mitogen-activated protein kinase (MAPK) pathway. Ras signals activate Raf, resulting in phosphorylation of downstream MEK and of ERK. This leads to multiple cellular responses, including phosphorylation of ribosomal protein S6, which regulates protein translation and activates cell cycle regulators. We found robust expression (>90% positivity) of downstream pathway effectors, phosphorylated ERK (p-ERK) and S6 (p-S6), in all four SCO cases, using immunohistochemistry (Figure 2). In contrast, IHC for phosphorylated protein kinase B (p-AKT) showed only a weak signal, indicating basal activation of the phosphoinositide 3-kinase (PI3K) pathway.
Figure 2. MAPK and PI3K Pathway Signaling in Spindle Cell Oncocytoma Cases.
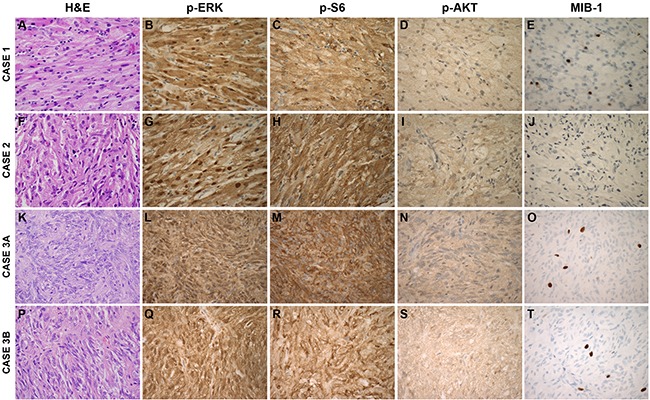
Tissue sections were stained with H&E or immunohistochemistry for MIB-1, phosphorylated ERK (p-ERK), phosphorylated AKT (p-AKT), and phosphorylated S6 (p-S6) proteins. (600X magnification)
DISCUSSION
Strong evidence of activated downstream effectors of the MAPK pathway in each pituitary SCO tumor in this study suggests a perturbation that may drive cellular proliferation. In cases 3A and 3B, we identified an HRAS Q61R mutation by whole exome sequencing, which is associated with multiple other cancers and may have caused MAPK pathway activation. Case 2 contained a mutation in SND1, which has been reported to be involved in glioblastoma and carcinomas of the colon, prostate, and liver [16, 18–20]. SND1 is a component of the RNA-induced silencing complex (RISC) and has been reported to activate the MAP kinase ERK [17]. Case 1 contained a mutation in the tumor suppressor FAT1 atypical cadherin gene, which has been implicated in glioblastoma, colorectal adenocarcinoma, and head and neck squamous cell carcinoma [15]. While FAT1 is best known for promoting Wnt signaling, FAT1 expression has also been associated with ERK activation [21]. Therefore, mutations in HRAS, SND1, and FAT1 may constitute separate genetic drivers that underlie the common MAPK activation observed in each SCO.
While our immunohistochemical and exome sequencing findings point to MAPK pathway activation in SCOs, the finding of two MEN1 mutations in cases 3A and 3B suggests that biallelic inactivation of MEN1 may be a second mechanism underlying neoplasia in SCO. Inactivation of both MEN1 alleles has been found in multiple endocrine tumors, including parathyroid adenoma, insulinoma, and a small subset of pituitary adenomas [22].
HRAS mutations have been previously associated with increased aggressiveness in pituitary adenomas [23, 24]. Given this, it is noteworthy that cases 3A and 3B, which displayed rapid recurrence leading to repeat resections, demonstrated a pathogenic HRAS mutation. As such, HRAS mutation may be an indicator of more aggressive behavior in SCO.
The recurrent tumor of case 3B may also be related to the acquisition of new somatic mutations not present in the initial tumor, case 3A. Newly mutated genes identified in case 3B include FAT atypical cadherin 4 (FAT4), Importin 7 (IPO7), Cyclin K (CCNK), SH3 domain-binding kinase 1 (SBK1), and CD300A. Of these, FAT4, CCNK, and SBK1 have been previously linked to neoplasia [25–27] and may contribute to the aggressive behavior demonstrated by case 3.
Interestingly, the similarities in presentation between SCOs and pituitary adenomas are reflected in their genetic profiles as well. Various MEN1 mutations have been implicated in pituitary adenoma [14], and, as mentioned earlier, pituitary adenomas with HRAS mutations show increased aggressiveness. The genetic similarity between SCO case 3 reported here and pituitary adenoma raises the question of diagnostic overlap. However, the immunohistochemical profile, including the absence of neuroendocrine markers and the presence of S100, strongly suggest that case 3 is indeed a spindle cell oncocytoma, rather than a pituitary adenoma [1, 28–30].
Pituitary adenomas have been associated with mutations in numerous other genes, including succinate dehydrogenase (SDH) [31], ubiquitin-specific peptidase 8 (USP8) [32, 33], cyclin-dependent kinase inhibitor 1B (CDKN1B) [34], aryl hydrocarbon receptor interacting protein (AIP) [35], and cAMP-dependent protein kinase type 1-alpha regulatory subunit (PRKAR1A) [36]. These mutations were not identified in our whole exome sequences of spindle cell oncocytoma. Cytogenetic studies of pituitary adenoma have shown scattered chromosome gains and losses, without a significant recurrent signature [37, 38]. The minimal chromosomal abnormalities we observed in our SCOs are consistent with the copy number profiles of some non-functional pituitary adenomas.
Scarce genetic information on pituicytoma is available for comparison with SCO. Comparative genomic hybridization (CGH) performed on one case of pituicytoma showed multiple copy number imbalances, with losses on 1p, 14q, and 22q and a gain on 5p [39]. This pattern appears distinct from our findings for SCO. Overall, we did not identify any significant copy number profile changes in SCO that have been reported in pituitary adenoma or pituicytoma.
In this report, we present four cases of SCO, using whole exome sequencing to reveal abnormal MAPK pathway signaling, suggesting it may be a common mechanism underlying oncogenesis as a shared phenotypic endpoint of various driver mutations. Inhibition of MAPK pathway signal transducers, or downstream nodes such as MEK, is under active clinical investigation in multiple other cancers [40–42]. Consequently, targeted inhibition of MAPK pathway signaling may offer an opportunity for treatment of spindle cell oncocytomas that cannot be controlled by surgical resection alone. Mutational profiling of many other tumor types has opened up successful personalized targeted medical treatments, and our findings suggest spindle cell oncocytomas may also be amenable to this approach.
MATERIALS AND METHODS
Sample selection
Analysis of data generated from tumor specimens and clinical information was conducted under a Dana-Farber/Brigham and Women's Cancer Center (DF/BWCC) Institutional Review Board (IRB)-approved protocol. Histologic diagnosis was confirmed on all samples by a board-certified neuropathologist (S.H.R.) and representative paraffin-embedded tissue with average estimated purity >70% was selected. Tumor DNA was extracted from 1 mm cores and normal DNA was prepared from patient salivary samples using standard techniques (Oragene kit, DNA Genotek, Kanata, Ontario, Canada; and Qiagen, Valencia, CA). The tumor-normal pairs were confirmed by mass spectrometric genotyping with an established 48-SNP panel (Sequenom, San Diego, CA) [43].
Whole exome sequencing, mutation analysis, and copy number analysis
Whole exome sequencing was performed as previously described [44]. DNA was sonicated to 150 bp fragments, size selected with Agencourt AMPure XP beads, and ligated to specific barcoded adapors (Illumina TruSeq; Illumina Inc., San Diego, CA) for multiplexed analysis. Exome hybrid capture was performed using the Agilent SureSelect hybrid capture kit (Whole Exome v4; Agilent Technologies, Santa Clara, CA) and sequenced on a HiSeq 2500 system (Illumina Inc., San Diego, CA). All samples achieved at least 80X depth of coverage across exons.
Read pairs were aligned to the hg19 reference sequence using the Burrows-Wheeler Aligner [45], and sample reads sorted and duplicate-marked using SAMtools and Picard. Bias in base quality score assignments due to flowcell, lane, dinucleotide context, and machine cycle were analyzed and recalibrated, and local realignment around insertions or deletions (indels) was achieved using the Genome Analysis Toolkit (GATK) [46, 47].
Somatic mutations and short indels were called and post-filtered using MuTect [48] and IndelLocator [49, 50]. These were annotated to genes and compared to events in the Catalogue of Somatic Mutations in Cancer (COSMIC) using Oncotator and also manually verified in the sequence output through visualization in the Integrated Genome Viewer (IGV). To analyze somatic copy number alterations from whole exome data, we used an allelic copy number pipeline, consisting of the ReCapSeg, Allelic Capseg and ABSOLUTE algorithms. ReCapseg detects total copy ratios from whole-exome sequencing data and performs a tangent normalization against a panel of normal exomes. Allelic capseg takes the output of ReCapseg and splits total copy ratios into homologue-specific copy ratios (HSCRs) from segmental estimates of multipotent allelic copy-ratios at heterozygous loci incorporating the statistical phasing software (BEAGLE) and population haplotype panels (HAPMAP3) [51–53]. Allele-specific somatic copy number alterations and tumor ploidy status were assessed with the ABSOLUTE algorithm [53].
Prediction of possible functional effect of the identified mutations was performed using the SIFT (Sorting Intolerant from Tolerant) Human Protein algorithm (J. Craig Venter Institute) [54]. The SIFT prediction score ranges from 0 to 1, and is the scaled probability of an amino acid substitution being tolerated. Amino acid substitutions with scores that fall below 0.05 are predicted to affect protein function. Notably, such prediction algorithms may be more useful for loss of function of tumor suppressor genes than for predicting gain of function for proto-oncogenes.
Immunohistochemistry
Diaminobenzidine (DAB) brightfield staining was performed according to standard protocols on 5 μm paraffin sections [55]. Antigens were retrieved using heat and 10 mM sodium citrate buffer (pH 6.0). The following primary antibodies were utilized: S100 (DAKO, 1:1000 dilution), vimentin (DAKO, 1:400), EMA (DAKO, 1:200), galectin-3 (Fitzgerald Industries, 1:100), chromogranin (Thermo Scientific, 1:4000), GFAP (DAKO, 1:2,000), TTF-1 (DAKO, 1:300), CD68 (DAKO, 1:1000), p-ERK (Cell Signaling, 1:200), p-AKT (Cell Signaling, 1:50), p-S6 (Cell Signaling, 1:50), and MIB-1 (Ki-67) (Leica, 1:200). Counterstaining for nuclei was performed using Mayer's hematoxylin stain, and cover slips were mounted using Permount (Fisher Scientific).
SUPPLEMENTARY FIGURE
Footnotes
CONFLICTS OF INTEREST
All authors declare no conflict of interest.
GRANT SUPPORT
This work is supported by NINDS K08NS087118 (SHR) award.
REFERENCES
- 1.Roncaroli F, Scheithauer BW, Cenacchi G, Horvath E, Kovacs K, Lloyd RV, Abell-Aleff P, Santi M, Yates AJ. ‘Spindle cell oncocytoma’ of the adenohypophysis: a tumor of folliculostellate cells? The American journal of surgical pathology. 2002;26:1048–1055. doi: 10.1097/00000478-200208000-00010. [DOI] [PubMed] [Google Scholar]
- 2.Kloub O, Perry A, Tu PH, Lipper M, Lopes MB. Spindle cell oncocytoma of the adenohypophysis: report of two recurrent cases. The American journal of surgical pathology. 2005;29:247–253. doi: 10.1097/01.pas.0000147400.87767.f6. [DOI] [PubMed] [Google Scholar]
- 3.Borges MT, Lillehei KO, Kleinschmidt-DeMasters BK. Spindle cell oncocytoma with late recurrence and unique neuroimaging characteristics due to recurrent subclinical intratumoral bleeding. Journal of neuro-oncology. 2011;101:145–154. doi: 10.1007/s11060-010-0229-2. [DOI] [PubMed] [Google Scholar]
- 4.Borota OC, Scheithauer BW, Fougner SL, Hald JK, Ramm-Pettersen J, Bollerslev J. Spindle cell oncocytoma of the adenohypophysis: report of a case with marked cellular atypia and recurrence despite adjuvant treatment. Clinical neuropathology. 2009;28:91–95. doi: 10.5414/npp28091. [DOI] [PubMed] [Google Scholar]
- 5.Coire CI, Horvath E, Smyth HS, Kovacs K. Rapidly recurring folliculostellate cell tumor of the adenohypophysis with the morphology of a spindle cell oncocytoma: case report with electron microscopic studies. Clinical neuropathology. 2009;28:303–308. doi: 10.5414/npp28303. [DOI] [PubMed] [Google Scholar]
- 6.Demssie YN, Joseph J, Dawson T, Roberts G, de Carpentier J, Howell S. Recurrent spindle cell oncocytoma of the pituitary, a case report and review of literature. Pituitary. 2011;14:367–370. doi: 10.1007/s11102-009-0170-6. [DOI] [PubMed] [Google Scholar]
- 7.Brat DJ, Scheithauer BW, Staugaitis SM, Holtzman RN, Morgello S, Burger PC. Pituicytoma: a distinctive low-grade glioma of the neurohypophysis. The American journal of surgical pathology. 2000;24:362–368. doi: 10.1097/00000478-200003000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Mete O, Lopes MB, Asa SL. Spindle cell oncocytomas and granular cell tumors of the pituitary are variants of pituicytoma. The American journal of surgical pathology. 2013;37:1694–1699. doi: 10.1097/PAS.0b013e31829723e7. [DOI] [PubMed] [Google Scholar]
- 9.Alexandrescu S, Brown RE, Tandon N, Bhattacharjee MB. Neuron precursor features of spindle cell oncocytoma of adenohypophysis. Annals of clinical and laboratory science. 2012;42:123–129. [PubMed] [Google Scholar]
- 10.Jebar AH, Hurst CD, Tomlinson DC, Johnston C, Taylor CF, Knowles MA. FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene. 2005;24:5218–5225. doi: 10.1038/sj.onc.1208705. [DOI] [PubMed] [Google Scholar]
- 11.Rivera M, Ricarte-Filho J, Knauf J, Shaha A, Tuttle M, Fagin JA, Ghossein RA. Molecular genotyping of papillary thyroid carcinoma follicular variant according to its histological subtypes (encapsulated vs infiltrative) reveals distinct BRAF and RAS mutation patterns. Modern pathology. 2010;23:1191–1200. doi: 10.1038/modpathol.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crona J, Delgado Verdugo A, Maharjan R, Stalberg P, Granberg D, Hellman P, Bjorklund P. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. The Journal of clinical endocrinology and metabolism. 2013;98:E1266–1271. doi: 10.1210/jc.2012-4257. [DOI] [PubMed] [Google Scholar]
- 13.Chiosea SI, Miller M, Seethala RR. HRAS mutations in epithelial-myoepithelial carcinoma. Head and neck pathology. 2014;8:146–150. doi: 10.1007/s12105-013-0506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) Best practice & research Clinical endocrinology & metabolism. 2010;24:355–370. doi: 10.1016/j.beem.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan S, Eng S, Kannan K, Zou Y, Peng L, Banuchi VE, Paty P, Zeng Z, Vakiani E, Solit D, Singh B, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nature genetics. 2013;45:253–261. doi: 10.1038/ng.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuchiya N, Ochiai M, Nakashima K, Ubagai T, Sugimura T, Nakagama H. SND1, a component of RNA-induced silencing complex, is up-regulated in human colon cancers and implicated in early stage colon carcinogenesis. Cancer research. 2007;67:9568–9576. doi: 10.1158/0008-5472.CAN-06-2707. [DOI] [PubMed] [Google Scholar]
- 17.Santhekadur PK, Akiel M, Emdad L, Gredler R, Srivastava J, Rajasekaran D, Robertson CL, Mukhopadhyay ND, Fisher PB, Sarkar D. Staphylococcal nuclease domain containing-1 (SND1) promotes migration and invasion via angiotensin II type 1 receptor (AT1R) and TGFbeta signaling. FEBS open bio. 2014;4:353–361. doi: 10.1016/j.fob.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emdad L, Janjic A, Alzubi MA, Hu B, Santhekadur PK, Menezes ME, Shen XN, Das SK, Sarkar D, Fisher PB. Suppression of miR-184 in malignant gliomas upregulates SND1 and promotes tumor aggressiveness. Neuro-oncology. 2014 doi: 10.1093/neuonc/nou220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuruma H, Kamata Y, Takahashi H, Igarashi K, Kimura T, Miki K, Miki J, Sasaki H, Hayashi N, Egawa S. Staphylococcal nuclease domain-containing protein 1 as a potential tissue marker for prostate cancer. The American journal of pathology. 2009;174:2044–2050. doi: 10.2353/ajpath.2009.080776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoo BK, Santhekadur PK, Gredler R, Chen D, Emdad L, Bhutia S, Pannell L, Fisher PB, Sarkar D. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology. 2011;53:1538–1548. doi: 10.1002/hep.24216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruder-Nascimento T, Chinnasamy P, Riascos-Bernal DF, Cau SB, Callera GE, Touyz RM, Tostes RC, Sibinga NE. Angiotensin II induces Fat1 expression/activation and vascular smooth muscle cell migration via Nox1-dependent reactive oxygen species generation. Journal of molecular and cellular cardiology. 2014;66:18–26. doi: 10.1016/j.yjmcc.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agarwal SK, Ozawa A, Mateo CM, Marx SJ. The MEN1 gene and pituitary tumours. Hormone research. 2009;71(Suppl 2):131–138. doi: 10.1159/000192450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karga HJ, Alexander JM, Hedley-Whyte ET, Klibanski A, Jameson JL. Ras mutations in human pituitary tumors. The Journal of clinical endocrinology and metabolism. 1992;74:914–919. doi: 10.1210/jcem.74.4.1312542. [DOI] [PubMed] [Google Scholar]
- 24.Pei L, Melmed S, Scheithauer B, Kovacs K, Prager D. H-ras mutations in human pituitary carcinoma metastases. The Journal of clinical endocrinology and metabolism. 1994;78:842–846. doi: 10.1210/jcem.78.4.8157709. [DOI] [PubMed] [Google Scholar]
- 25.Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, Lim KH, Ong CK, Huang D, Chin SY, Tan IB, Ng CC, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nature genetics. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 26.Marsaud V, Tchakarska G, Andrieux G, Liu JM, Dembele D, Jost B, Wdzieczak-Bakala J, Renoir JM, Sola B. Cyclin K and cyclin D1b are oncogenic in myeloma cells. Molecular cancer. 2010;9:103. doi: 10.1186/1476-4598-9-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang P, Guo J, Wang F, Shi T, Ma D. Human SBK1 is dysregulated in multiple cancers and promotes survival of ovary cancer SK-OV-3 cells. Molecular biology reports. 2011;38:3551–3559. doi: 10.1007/s11033-010-0465-8. [DOI] [PubMed] [Google Scholar]
- 28.DeStephano DB, Lloyd RV, Pike AM, Wilson BS. Pituitary adenomas. An immunohistochemical study of hormone production and chromogranin localization. The American journal of pathology. 1984;116:464–472. [PMC free article] [PubMed] [Google Scholar]
- 29.Lloyd RV, Scheithauer BW, Kovacs K, Roche PC. The Immunophenotype of Pituitary Adenomas. Endocrine pathology. 1996;7:145–150. doi: 10.1007/BF02739974. [DOI] [PubMed] [Google Scholar]
- 30.Turpin G, Heshmati HM, Kujas M, Gremain J, Jacque CM, Racadot J. Immunocytochemical study of S-100 protein in human pituitary adenomas. Virchows Archiv B, Cell pathology including molecular pathology. 1988;55:107–109. doi: 10.1007/BF02896566. [DOI] [PubMed] [Google Scholar]
- 31.Xekouki P, Szarek E, Bullova P, Giubellino A, Quezado M, Mastroyannis SA, Mastorakos P, Wassif CA, Raygada M, Rentia N, Dye L, Cougnoux A, Koziol D, de La Luz Sierra M, Lyssikatos C, Belyavskaya E, et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in human and mice. The Journal of clinical endocrinology and metabolism. 2015:jc20144297. doi: 10.1210/jc.2014-4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, Meitinger T, Mizuno-Yamasaki E, Kawaguchi K, Saeki Y, Tanaka K, Wieland T, Graf E, Saeger W, Ronchi CL, Allolio B, et al. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nature genetics. 2015;47:31–38. doi: 10.1038/ng.3166. [DOI] [PubMed] [Google Scholar]
- 33.Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, Mao Y, Li YM, Hu RG, Zhang ZY, Ye HY, Shen M, Shou XF, Li ZQ, Peng H, Wang QZ, et al. Recurrent gain-of-function USP8 mutations in Cushing's disease. Cell research. 2015;25:306–317. doi: 10.1038/cr.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, Vierimaa O, Makinen MJ, Tuppurainen K, Paschke R, Gimm O, Koch CA, Gundogdu S, Lucassen A, Tischkowitz M, Izatt L, et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. The Journal of clinical endocrinology and metabolism. 2007;92:3321–3325. doi: 10.1210/jc.2006-2843. [DOI] [PubMed] [Google Scholar]
- 35.Georgitsi M, Raitila A, Karhu A, Tuppurainen K, Makinen MJ, Vierimaa O, Paschke R, Saeger W, van der Luijt RB, Sane T, Robledo M, De Menis E, Weil RJ, Wasik A, Zielinski G, Lucewicz O, et al. Molecular diagnosis of pituitary adenoma predisposition caused by aryl hydrocarbon receptor-interacting protein gene mutations. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4101–4105. doi: 10.1073/pnas.0700004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stergiopoulos SG, Abu-Asab MS, Tsokos M, Stratakis CA. Pituitary pathology in Carney complex patients. Pituitary. 2004;7:73–82. doi: 10.1007/s11102-005-5348-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harada K, Nishizaki T, Ozaki S, Kubota H, Harada K, Okamura T, Ito H, Sasaki K. Cytogenetic alterations in pituitary adenomas detected by comparative genomic hybridization. Cancer genetics and cytogenetics. 1999;112:38–41. doi: 10.1016/s0165-4608(98)00235-0. [DOI] [PubMed] [Google Scholar]
- 38.Trautmann K, Thakker RV, Ellison DW, Ibrahim A, Lees PD, Harding B, Fischer C, Popp S, Bartram CR, Jauch A. Chromosomal aberrations in sporadic pituitary tumors. International journal of cancer. 2001;91:809–814. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1127>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 39.Phillips JJ, Misra A, Feuerstein BG, Kunwar S, Tihan T. Pituicytoma: characterization of a unique neoplasm by histology, immunohistochemistry, ultrastructure, and array-based comparative genomic hybridization. Archives of pathology & laboratory medicine. 2010;134:1063–1069. doi: 10.1043/2009-0167-CR.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120:3446–3456. doi: 10.1002/cncr.28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chapman PB, Solit DB, Rosen N. Combination of RAF and MEK inhibition for the treatment of BRAF-mutated melanoma: feedback is not encouraged. Cancer cell. 2014;26:603–604. doi: 10.1016/j.ccell.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 42.Joshi M, Rice SJ, Liu X, Miller B, Belani CP. Trametinib with or without Vemurafenib in BRAF Mutated Non-Small Cell Lung Cancer. PloS one. 2015;10:e0118210. doi: 10.1371/journal.pone.0118210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demichelis F, Greulich H, Macoska JA, Beroukhim R, Sellers WR, Garraway L, Rubin MA. SNP panel identification assay (SPIA): a genetic-based assay for the identification of cell lines. Nucleic acids research. 2008;36:2446–2456. doi: 10.1093/nar/gkn089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, Ligon KL, Palescandolo E, Van Hummelen P, Ducar MD, Raza A, Sunkavalli A, Macconaill LE, Stemmer-Rachamimov AO, Louis DN, Hahn WC, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nature genetics. 2013;45:285–289. doi: 10.1038/ng.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, Anderson KC, Ardlie KG, Auclair D, Baker A, Bergsagel PL, Bernstein BE, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, Onofrio R, Carter SL, Park K, Habegger L, Ambrogio L, Fennell T, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.International HapMap C. Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Peltonen L, Dermitzakis E, Bonnen PE, Altshuler DM, Gibbs RA, de Bakker PI, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Browning BL, Yu Z. Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. American journal of human genetics. 2009;85:847–861. doi: 10.1016/j.ajhg.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, Lander ES, Meyerson M, Getz G. Absolute quantification of somatic DNA alterations in human cancer. Nature biotechnology. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 55.Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, Louis DN, Stiles CD, Rowitch DH. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. Journal of neuropathology and experimental neurology. 2004;63:499–509. doi: 10.1093/jnen/63.5.499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.