

Abstract
Context:
To date, all the familial hyperaldosteronism type III (FH-III) patients reported presenting with typical primary aldosteronism (PA), without showing other adrenal hormone abnormalities.
Objective:
This study characterized a novel phenotype of FH-III and explored the possible pathogenesis.
Patients and Methods:
A male patient presented with severe hypertension and hypokalemia at the age of 2 years and developed Cushing's syndrome at 20 years. He was diagnosed with PA and Cushing's syndrome on the basis of typical biochemical findings. He had massive bilateral adrenal hyperplasia and underwent left adrenalectomy. KCNJ5 was sequenced, and secretion of aldosterone and cortisol were observed both in vivo and in vitro.
Results:
A heterozygous germline p.Glu145Gln mutation of KCNJ5 was identified. ARMC5, PRKAR1A, PDE8B, PDE11A, and PRKACA genes and β-catenin, P53 immunoactivity were normal in the adrenal. CYP11B2 was highly expressed, whereas mRNA expression of CYP11B1, CYP17A1, and STAR was relatively low in the hyperplastic adrenal, compared with normal adrenal cortex and other adrenal diseases. In the primary cell culture of the resected hyperplastic adrenal, verapamil and nifedipine, two calcium channel blockers, markedly inhibited the secretion of both aldosterone and cortisol and the mRNA expression of CYP11B1, CYP11B2, CYP17A1, and STAR.
Conclusions:
We presented the first FH-III patient who had both severe PA and Cushing's syndrome. Hypersecretion of cortisol might be ascribed to overly large size of the hyperplastic adrenal because CYP11B1 expression was relatively low in his adrenal. Like aldosterone, synthesis and secretion of cortisol in the mutant adrenal may be mediated by voltage-gated Ca2+ channels.
This study presented the first FH-III patient who had both severe primary aldosteronism and Cushing's syndrome. Hypersecretion of cortisol might be ascribed to large size of the hyperplastic adrenal.
Primary aldosteronism (PA) is the most common cause of secondary hypertension. Familial hyperaldosteronism (FH) has been well documented in recent years. To date, four forms of FH have been identified and are attributed to a chimeric CYP11B1/CYP11B2 gene,, a change in chromosomal region 7p22, KCNJ5 mutation (FH-III), and CACNA1H mutation, respectively. FH-III is an autosomal-dominant hereditary disease caused by the heterozygous mutations of the KCNJ5 gene, which codes the G protein-activated inward rectifier K+ channel 4 (Kir3.4) (1). So far, 10 FH-III families, involving six different KCNJ5 mutations, have been reported (1–9). All the FH-III patients reportedly had typical clinical features and biochemical findings of PA, without showing other abnormalities of adrenal hormones. Presented in this report was a novel phenotype of FH-III characterized by concomitant presence of both severe PA and typical Cushing's syndrome. In addition, the possible mechanism of cosecretion of aldosterone and cortisol was also investigated.
Subjects and Methods
Patient
The index case was a Chinese boy who presented with severe hypertension and hypokalemia at the age of 2 years. His blood pressure (BP) was 120–140/80–90 mm Hg, and his serum potassium was 2.0 mmol/L. He visited a children's hospital and was found to have elevated serum aldosterone and suppressed plasma renin activity. He was diagnosed with PA, and a dexamethasone suppression test excluded glucocorticoid-remediable aldosteronism and hypercortisolism. The patient did not receive a computed tomography (CT) scan, and ultrasound failed to detect an enlarged adrenal or mass at that time. He was treated with spironolactone at 60 mg/d, chloride potassium 2.8 g/d, with BP at 115/75 mm Hg and serum potassium at 3.6 mmol/L. His BP rose gradually. At 13 years, his BP was 150–160/80–90 mm Hg, and serum potassium was 2.4–2.6 mmol/L under the treatment of spironolactone at 60 mg/d, chloride potassium at 8 g/d and captopril at 75 mg/d. During that time, the circadian rhythms of serum cortisol were found to be normal. A CT scan showed enlarged bilateral adrenals, with the left adrenal being 4.9X1.3 cm and the right one at 3.8X1.3 cm. His growth and development were normal. The growth chart is shown in Supplemental Figure 1.
At the age of 20 years, he gained 5 kg and had a moon face, with accompanying purple abdominal striae, thin skin, easy bruising, and generalized weakness. His BP rose to 160–180/95–130 mm Hg, and his serum potassium was 2.8–3.0 mmol/L, even when he had been put on 60 mg/d of spironolactone, 20 g/d of potassium supplementation, and 75 mg/d of captopril.
At the age of 23 years, he was referred to our hospital. Examinations showed that his serum potassium was 3.0 mmol/L; serum creatinine was 180 μmol/L (normal range 59–104 μmol/L), and his urine protein to creatinine ratio was 290 mg/gCr (normal range 0–30 mg/gCr). The diagnosis of PA was made on the basis of elevated serum aldosterone and suppressed plasma renin activity. In light of increased cortisol level and lowered plasma ACTH concentration, the diagnosis of Cushing's syndrome was also established. Serum progesterone was elevated, but 17α-hydroxyprogesterone, testosterone, and estradiol levels were normal. Dehydroepiandrosterone sulfate dropped to the lower limit level (Table 1). These data excluded the abnormality of adrenal sex hormone.
Table 1.
Serum and Urinary Steroid Profiles Before and After Operation
| Index | Before Operation | After Operation (3 mo) | Normal Range |
|---|---|---|---|
| Aldosterone, ng/dL | 46.4 | 40.8 | 6.5–15.0 |
| Plasma renin activity, ng/mL·h | 0.01 | 0.16 | 0.93–6.56 |
| Cortisol basal, μg/dL | 20.8 | 14.6 | 4.0–22.3 |
| Cortisol after 1 mg overnight LDST, μg/dL | 26.1 | ND | <1.8 |
| ACTH, pg/mL | <5.0 | <5.0 | 10.0–46.0 |
| Urinary free cortisol basal, μg/da | 1611.5 | 247.1 | 12.3–103.5 |
| Urinary free cortisol after HDST, μg/db | 1039.5 | ND | |
| Testosterone, ng/mL | 3.1 | 2.1 | 1.8–3.8 |
| Estradiol, pg/mL | 24.0 | 12.0 | <47.0 |
| LH, mU/mL | 5.3 | 3.0 | 1.2–8.6 |
| FSH, mU/mL | 4.6 | 5.2 | 1.3–19.3 |
| Progesterone, ng/mL | 5.0 | 2.6 | 0.1–0.8 |
| 17-α hydroxyprogesterone, ng/mL | 1.9 | ND | 0.3–2.2 |
| Dehydroepiandrosterone sulfate, μg/dL | 103.0 | 112.0 | 85–690 |
| Urinary norepinephrine, μg/d | 36.8 | 18.0 | 16.7–40.7 |
| Urinary epinephrine, μg/d | 4.6 | 3.6 | 1.7–6.4 |
| Urinary dopamine, μg/d | 211.9 | 150.3 | 121.0–330.6 |
Abbreviations: HDST, high-dose (8 mg/d) dexamethasone suppression test; LDST, low-dose dexamethasone suppression test; ND, not determined.
Greater than 103.5 μg/d for urinary free cortisol excretion suggests hypercortisolism.
Less than 50% reduction in urinary free cortisol during HDST suggests an adrenal disorder or an ectopic ACTH syndrome.
An enhanced CT scan (Figure 1A) revealed massive bilateral adrenal hyperplasia. He underwent complete left adrenalectomy. Pathological examination showed that his adrenal gland was oval shaped, weighing 242.8 g and measuring 12.5 × 7.5 × 5.5 cm (Figure 1B). The cut surface was homogeneous and of a golden yellow color (Figure 1C). The specimen was pathohistologically diagnosed as diffuse adrenocortical hyperplasia.
Figure 1.

A, Enhanced CT scan showed huge bilateral adrenal mass. B, Left adrenal mass. C, The cut surface of the left adrenal. D, Pedigree of kindred with germline KCNJ5 mutation. E, Sequencing of peripheral blood and adrenal DNA showed the KCNJ5 c.433G>C substitution leading to p.Glu145Gln (p.E145Q) mutation in the index case. Sequencing of peripheral blood DNA of his parents did not show an abnormality.
The patient was followed up 3 months after the operation. His urinary free cortisol and serum progesterone decreased dramatically, and serum aldosterone was decreased slightly (Table 1). He lost 4 kg, with his moon face and purple striae being partially ameliorated. The antihypertensive treatment (60 mg/d nifedipine) and potassium supplementation (10 g/d) were scaled down postoperatively, and his BP (150/90 mm Hg) and serum potassium level (3.5 mmol/L) were better controlled. He is now awaiting the removal of his right adrenal gland. The study was approved by the local ethical committee, and informed consent was obtained from the patient and his family members.
Amplification and sequencing of KCNJ5, ARMC5, PRKAR1A, PDE8B, PDE11A, and PRKACA genes
Genomic DNA was isolated from peripheral blood lymphocytes and hyperplastic adrenal tissue by using QIAamp DNA minikit from QIAGEN. All the coding sequences and the intro-exon junctions of KCNJ5, ARMC5, PRKAR1A, PDE8B, PDE11A, and hot spot regions of PRKACA were amplified and bidirectionally sequenced on an ABI3730 DNA analyzer (Applied Biosystems). The primers used for the PCR amplification are listed in Supplemental Table 1. All the reagents for amplification were purchased from QIAGEN.
Tissue homogenization
Surgically removed hyperplastic adrenal tissue and four pieces of normal adrenal cortex tissues (each 30 mg wet weight) were collected. Each tissue fragment was put in 1 mL of 0.9% sodium chloride and homogenized by using a homogenizer. The homogenate was collected and centrifuged for detection of aldosterone and cortisol by a RIA and chemiluminescence immunoassay, respectively.
Immunohistochemistry
Immunohistochemical detection was performed using EnVision detection kit (Dako). The antibodies and dilutions were as follows: Ki-67 (1:500; OriGene), β-catenin (1:300; OriGene), P53 (1:400; Leica Biosystems), CYP11B1 (1:200), and CYP11B2 (1:500). A mouse monoclonal antibody against CYP11B2 and rat monoclonal antibody against CYP11B1 were kindly provided by Dr Celso E. Gomez-Sanchez (Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi) (10).
Primary cell culture of the hyperplastic adrenal
Surgically removed adrenal tissue (1 g wet weight) was washed two times with DMEM, cut into small pieces, and dispersed by incubation in DMEM containing 2% collagenase I for 20 minutes at 37°C. Dispersed cells were harvested by centrifugation and washed once. Then the cells were resuspended in culture medium (DMEM containing 15% fatal calf serum, 25 mM HEPES, 100 μg/mL streptomycin, and 100 μg/mL penicillin). The cells were plated into 12-well plates at a density of about 1 × 105 cells/well in 1 mL medium and incubated at 37°C in a 5% CO2 atmosphere. On day 4, when the cells grew 80% confluent, cells were treated with verapamil (10 μM; Sigma-Aldrich) and nifedipine (10 μM; Sigma-Aldrich) or with the vehicle for 24 hours. Culture medium was collected for the measurement of aldosterone and cortisol, and the cells were harvested for RNA assay.
RNA isolation and real-time quantitative PCR
Four aldosterone-producing adenomas (APAs) with somatic mutation KCNJG151R, four APAs without somatic mutation of KCNJ5, ATP1A1, ATP2B3, and CACNA1D, four cortisol-producing adenomas, four specimens of ACTH-independent macronodular adrenal hyperplasia, four nonfunctional adenomas, and four specimens of normal adrenal cortex were compared against the hyperplastic adrenal of the index patient. Cells and tissues were lysed with Trizol reagent (Sigma-Aldrich), and total RNA was isolated by following the standard protocol provided by the manufacturer. The cDNA was synthesized from 1 μg of total RNA by using the high-capacity cDNA reverse transcription kit (Applied Biosystems). PCR amplification for CYP11B1, CYP11B2, CYP17A1, and STAR genes was conducted by using the LightCycler 480 SYBR Green I Master (Roche Applied Science) on a LightCycler 480 PCR system. The relative gene expression levels were calculated by using the cycle threshold value and were normalized against the expression of the human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. The primers used are listed in Supplemental Table 2. All real-time PCR was performed in triplicate for each sample.
Statistical analysis
The data were expressed as mean ± SD for the in vitro study. To identify whether calcium channel blockers could affect the hormone production, the Student t test was used to compare the secretion of aldosterone and cortisol and mRNA expression of their synthetic enzymes between calcium channel blocker-treated group and control group. P < .05 was considered to be statistically significant. The statistical analyses were performed by using the SPSS version 13.0 software package.
Results
Sequencing of the KCNJ5, ARMC5, PRKAR1A, PDE8B, PDE11A, and PRKACA genes
Sequencing of the KCNJ5 mutation from peripheral blood DNA of the patient identified a heterozygous c.433G>C (p.Glu145Gln) mutation (Figure 1E). The mutation was not found in his parents (Figure 1, D and E). The hyperplastic adrenal of the patient also had the same KCNJ5 mutation (Figure 1E). Sequencing of the ARMC5, PRKAR1A, PDE8B, PDE11A, and PRKACA genes from the adrenal DNA did not detect any mutations.
Hormone levels in tissue homogenization
Aldosterone and cortisol were found to be 112.5 ng/dL and 13.0 μg/dL, respectively, in the tissue homogenate of the index hyperplastic adrenal and 19.3 ± 3.1 ng/dL and 23.0 ± 6.1 μg/dL, respectively, in the normal adrenal cortex tissues.
Immunohistochemistry
Histologically, the hyperplastic adrenal was predominantly composed of clear cells (Figure 2A). Immunohistochemical analysis of the adrenal tissue revealed cytoplasm immunoreactivity for CYP11B1 and CYP11B2, membrane immunoreactivity for β-catenin, and sporadic nuclear immunoreactivity for P53. The overall positive ratio for the Ki-67 index was about 2%, and at the foci relatively more cells were positively stained (Figure 2B-F).
Figure 2.
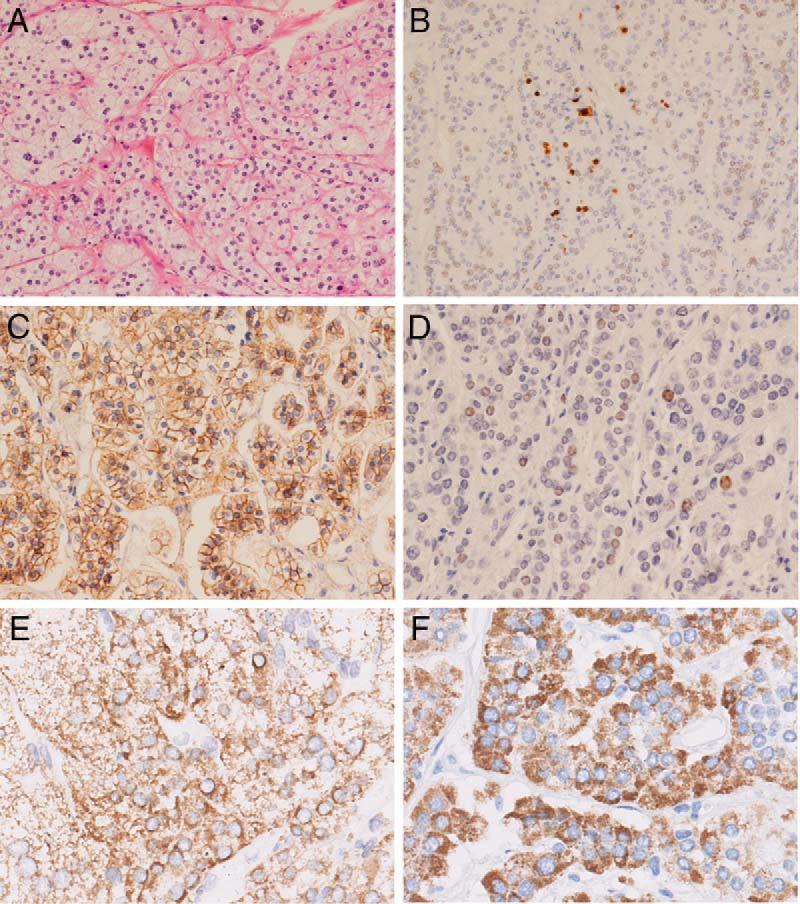
Histopathological findings in the adrenal gland of the patient. A, Hematoxylin-eosin staining (×200); B–F, Immunostaining. B, Ki-67 (×100); C, β-catenin (×200); D, P53 (X200); E, CYP11B1 (×400); F, CYP11B2 (×400)
Expression of CYP11B1, CYP11B2, CYP17A1, and STAR genes
Because clinically the patient presented with both PA and hypercortisolism, to clarify whether the hyperplastic adrenal overexpressed steroidogenic enzymes for the synthesis of aldosterone and cortisol, we examined the mRNA expression of steroidogenic enzyme CYP11B1, CYP11B2, CYP17A1, and STAR in the hyperplastic adrenal and other adrenal diseases. We found that CYP11B2 was highly expressed in APAs and the hyperplastic adrenal, whereas the expression of CYP11B1, CYP17A1, and STAR was relatively low in the hyperplastic adrenal, compared with other adrenal diseases and normal adrenal cortex (Figure 3).
Figure 3.

A and B, Serum aldosterone level and 24-hour urinary free cortisol excretion in the index patient and patients with other adrenocortical diseases. C–F, The mRNA expression of steroidogenic enzymes CYP11B1, CYP11B2, CYP17A1, and STAR in the adrenal of the index patient, specimens of other adrenocortical diseases, and normal adrenal cortex. Each bar represents the mean ± SEM of four specimens of the adrenocortical diseases. AIMAH, ACTH-independent macronodular adrenal hyperplasia; APA-m, APA with the somatic mutation KCNJG151R; APA-wt, APA without the somatic mutation of KCNJ5, ATP1A1, ATP2B3, and CACNA1D; CPA, cortisol-producing adenomas; FH-III, the index case with FH-III; NC, normal adrenal cortex; NFA, nonfunctional adenomas.
Calcium channel involved in the secretion of aldosterone and cortisol
In the primary cell culture of the resected hyperplastic adrenal, calcium channel blockers, verapamil and nifedipine, were found to markedly inhibit the secretion of both aldosterone and cortisol and the mRNA expression of their synthetic enzymes CYP11B1, CYP11B2, CYP17A1, and STAR (Figure 4).
Figure 4.

Involvement of the calcium channel in the synthesis and secretion of aldosterone and cortisol in the primary cell culture of the mutant adrenal. A and B, Effects of verapamil and nifedipine on the secretion of aldosterone and cortisol. C–F, Effects of verapamil and nifedipine on the mRNA expression of CYP11B1, CYP11B2, CYP17A1, and STAR. *, P < .05 compared with the control.
Discussion
Mutation in the KCNJ5 gene has been demonstrated to be the cause of both sporadic APA and FH-III. Kir3.4 dysfunction due to KCNJ5 mutation leads to a loss in K+ selectivity and an increased influx of Na+, resulting in depolarization of the plasma membrane, accumulation of intracellular Ca2+, and eventually increases synthesis of steroidogenic enzymes and aldosterone production (1, 2).
So far, 10 FH-III families, involving six different KCNJ5 mutations, have been reported, and among them, amino acid substitution at position 151 was the most frequent mutation (1–9). Germline mutation p.Glu145Gln (E145Q) identified in our patient had been reported recently to be responsible for severe hyperaldosteronism and massive bilateral adrenal hyperplasia in a Caucasian girl (2). This mutation was found to cause Na+-dependent cell membrane depolarization, increase intracellular Ca2+ level, and up-regulate CYP11B2 expression in adrenal cells (2).
Interestingly, our case of the germline KCNJ5 mutation presented not only severe PA but also typical Cushing's syndrome, which had not been reported in FH-III patients previously (2–9). The serum aldosterone level and urinary free cortisol in our patient were seemingly substantially higher than those in patients with other adrenal diseases. Therefore, in any patient who had an early-onset PA and extremely elevated serum aldosterone level, FH-III should be suspected. In this index patient, cosecretion of aldosterone and cortisol in adrenal has been confirmed by immunohistochemical staining and detection of hormones in tissue homogenate. Concomitant secretion of multiple adrenocortical hormones has been reported mostly in adrenocortical carcinoma (ACC). With ACCs, simultaneous hypersecretion of more than one hormone mostly involved cortisol and androgens, whereas the hypersecretion of both aldosterone and cortisol was rare (11, 12). In recent years, APA patients with accompanying cortisol hypersecretion have been reported mostly in Japanese individuals (13–15). Most previously reported cases had subclinical cortisol hypersecretion, whereas concomitant overt Cushing's syndrome has been rarely found. Yamada et al (14) even reported a case of PA and overt Cushing's syndrome harboring somatic mutation of the KCNJ5 gene. The exact mechanism of autonomous cortisol secretion in APAs remains poorly understood. Recently, by using novel monoclonal antibodies, Nakamura et al (16) found that APAs contained not only a mix of CYP11B2- and CYP11B1-positive cells but also cells expressing both CYP11B enzymes. Presumably, an APA patient might clinically have hypercortisolism if APAs contain a large number of cortisol-producing cells or if the tumor is of large size (16).
The index patient had an early-onset PA and developed Cushing's syndrome almost 20 years later. Sequencing of most genes believed to be associated with adrenal Cushing's syndrome, including ARMC5, PRKAR1A, PDE8B, PDE11A, and PRKACA (17), failed to find mutations in the hyperplastic adrenal of the patient. The mechanism of the patient developing cortisol hypersecretion is unknown, and possible causes might be as follows. First, in this patient, transcriptional expression of steroidogenic enzymes associated with cortisol synthesis (CYP11B1, CYP17A1) was relatively low in the hyperplastic adrenal, compared with those in the normal adrenal cortex and other adrenocortical disease. Although the expression of cortisol synthetase CYP11B1 was low in the case, the overall secretion of cortisol was high due to relatively larger size of the hyperplastic adrenal. Second, steroidogenic enzymes, previously promoting the synthesis of aldosterone alone, in the adrenocortical cells of the hyperplastic adrenal might experience certain changes to promote synthesis of both aldosterone and cortisol. Barzon et al (18) reported a case of ACC, who initially presented severe PA, developed overt Cushing's syndrome later when the tumor recurred, and had an accompanying change in the expression of adrenal steroidogenic enzymes in the tumor. This case suggests that adrenocortical tumors can change their hormone production pattern and reverse their differentiation program when the condition progresses (18).
Notably, the adrenals of our patient were extremely huge, thereby raising a question of whether they grew fast. Wnt/β-catenin pathway activation was found to be involved in the pathogenesis of APAs and cortisol-producing adenomas (19). β-Catenin can stimulate proliferation of adrenocortical cells during embryonic development and plays a role in cell renewal in adult adrenal cortex (19). But we failed to observe an abnormally high expression of β-catenin in the huge adrenal, suggesting the adrenal growth did not involve Wnt/β-catenin pathway. Moreover, aberrant P53 expression, which is related to malignant transformation of the adrenocortical cell, was not detected in this hyperplastic adrenal. Adrenal cells proliferated quickly at foci, as shown by Ki-67 staining, indicating that future malignant transformation of the hyperplastic adrenal could not be completely excluded.
The increased cytosolic Ca2+ has been demonstrated to be a critical event in the pathogenesis of APAs with mutant KCNJ5. Moreover, Ca2+ channel blocker can reduce CYP11B2 expression and aldosterone secretion in H295R cells expressing mutant KCNJ5 (2, 20). In the present study, by primarily culturing adrenocortical cells with mutant KCNJ5E145Q, we found that L-type Ca2+ channel blocker verapamil and nifedipine reduced the synthesis and secretion of not only aldosterone but also cortisol. In fasciculata cells, ACTH is the most important regulator for cortisol secretion. ACTH binds to melanocortin 2 receptor, leading to an increase in cAMP by activating adenylate cyclase. The increased cAMP level and the subsequent activation of protein kinase A stimulate both cortisol production and cell proliferation due to up-regulated cAMP response element-binding protein phosphorylation. On the other hand, ACTH can increase intracellular Ca2+ by triggering Ca2+ influx through voltage-gated Ca2+ channels, thereby inducing the expression of steroidogenic enzymes involved in the cortisol synthesis. ACTH-stimulated Ca2+ influx and consequent cortisol secretion can be inhibited by blockers of voltage-gated Ca2+ channels (21). In this study, Ca2+ channel blockers substantially inhibited both cortisol secretion and expression of its synthetases, suggesting that hypersecretion of cortisol in the mutant cells might be mediated by voltage-gated Ca2+ channels.
In conclusion, in this report, we presented the first FH-III patient who had both severe PA and Cushing's syndrome. Hypersecretion of cortisol might be ascribed to the overly large size of the hyperplastic adrenal because CYP11B1 expression was relatively low in his adrenal. One possible explanation might be that the FH-III patient had severe KCNJ5 mutation-related PA and bilateral adrenal hyperplasia in his childhood and gradually developed hypercortisolism when adrenal hyperplasia deteriorated. Synthesis and secretion of cortisol, like aldosterone, in the mutant adrenal may be mediated by voltage-gated Ca2+ channels.
Acknowledgments
We are grateful to Dr Celso E. Gomez-Sanchez (Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi), who kindly provided us antibodies against CYP11B1 and CYP11B2.
This work was supported by research grants from the National Key Program of Clinical Science of China (Grant WBYZ2011–873) and the National Basic Research Program (Grant 2014CB542302).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ACC
- adrenocortical carcinoma
- APA
- aldosterone-producing adenoma
- BP
- blood pressure
- CT
- computed tomography
- FH
- familial hyperaldosteronism
- FH-III
- familial hyperaldosteronism type III
- PA
- primary aldosteronism.
References
- 1. Korah HE, Scholl UI. An update on familial hyperaldosteronism. Horm Metab Res. 2015;47:941–946. [DOI] [PubMed] [Google Scholar]
- 2. Monticone S, Bandulik S, Stindl J, et al. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J Clin Endocrinol Metab. 2015;100:E114–E118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Monticone S, Hattangady NG, Penton D, et al. a Novel Y152C KCNJ5 mutation responsible for familial hyperaldosteronism type III. J Clin Endocrinol Metab. 2013;98:E1861–E1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mulatero P, Tauber P, Zennaro MC, et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension. 2012;59:235–240. [DOI] [PubMed] [Google Scholar]
- 5. Scholl UI, Nelson-Williams C, Yue P, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci USA. 2012;109:2533–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Adachi M, Muroya K, Asakura Y, Sugiyama K, Homma K, Hasegawa T. Discordant genotype-phenotype correlation in familial hyperaldosteronism type III with KCNJ5 gene mutation: a patient report and review of the literature. Horm Res Paediatr. 2014;82:138–142. [DOI] [PubMed] [Google Scholar]
- 7. Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. 2008;93:3117–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Charmandari E, Sertedaki A, Kino T, et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J Clin Endocrinol Metab. 2012;97:E1532–E1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mussa A, Camilla R, Monticone S, et al. Polyuric-polydipsic syndrome in a pediatric case of non-glucocorticoid remediable familial hyperaldosteronism. Endocr J. 2012;59:497–502. [DOI] [PubMed] [Google Scholar]
- 10. Gomez-Sanchez CE, Qi X, Velarde-Miranda C, et al. Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol 2014;383:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peppa M, Pikounis V, Papaxoinis G, et al. Adrenocortical carcinoma secreting cortisol, androgens and aldosterone: a case report. Cases J. 2009;2:8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kurtulmus N, Yarman S, Azizlerli H, Kapran Y. Co-secretion of aldosterone and cortisol by an adrenocortical carcinoma. Horm Res. 2004;62:67–70. [DOI] [PubMed] [Google Scholar]
- 13. Spath M, Korovkin S, Antke C, Anlauf M, Willenberg HS. Aldosterone- and cortisol-co-secreting adrenal tumors: the lost subtype of primary aldosteronism. Eur J Endocrinol. 2011;164:447–455. [DOI] [PubMed] [Google Scholar]
- 14. Yamada M, Nakajima Y, Taguchi R, et al. KCNJ5 mutations in aldosterone- and cortisol-co-secreting adrenal adenomas. Endocr J. 2012;59:735–741. [DOI] [PubMed] [Google Scholar]
- 15. Willenberg HS, Spath M, Maser-Gluth C, et al. Sporadic solitary aldosterone- and cortisol-co-secreting adenomas: endocrine, histological and genetic findings in a subtype of primary aldosteronism. Hypertens Res. 2010;33:467–472. [DOI] [PubMed] [Google Scholar]
- 16. Nakamura Y, Maekawa T, Felizola SJ, et al. Adrenal CYP11B1/2 expression in primary aldosteronism: immunohistochemical analysis using novel monoclonal antibodies. Mol Cell Endocrinol. 2014;392:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Faillot S, Assie G. Endocrine tumours: the genomics of adrenocortical tumors. Eur J Endocrinol. 2016;174(6):R249–R265. [DOI] [PubMed] [Google Scholar]
- 18. Barzon L, Masi G, Fincati K, et al. Shift from Conn's syndrome to Cushing's syndrome in a recurrent adrenocortical carcinoma. Eur J Endocrinol. 2005;153:629–636. [DOI] [PubMed] [Google Scholar]
- 19. Bonnet S, Gaujoux S, Launay P, et al. Wnt/β-catenin pathway activation in adrenocortical adenomas is frequently due to somatic CTNNB1-activating mutations, which are associated with larger and nonsecreting tumors: a study in cortisol-secreting and -nonsecreting tumors. J Clin Endocrinol Metab. 2011;96:E419–E426. [DOI] [PubMed] [Google Scholar]
- 20. Tauber P, Penton D, Stindl J, et al. Pharmacology and pathophysiology of mutated KCNJ5 found in adrenal aldosterone-producing adenomas. Endocrinology. 2014;155:1353–1362. [DOI] [PubMed] [Google Scholar]
- 21. Enyeart JJ. Biochemical and Ionic signaling mechanisms for ACTH-stimulated cortisol production. Vitam Horm. 2005;70:265–279. [DOI] [PubMed] [Google Scholar]