

Abstract
Atorvastatin (ATV) has an important pro-survival role in cardiomyocytes after acute myocardial infarction (AMI). The objectives of this study were to: 1) determine whether ATV could affect autophagy of cardiomyocytes via the AMP-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) pathway, and 2) investigate the balance between autophagy and apoptosis pathways. Male Wistar rats (n = 100) were randomly divided into sham, control, ATV, Compound C, and ATV+ Compound C groups. In this AMI model, drug treatments were administered for 1 week before induction of MI by surgical ligation, and measurements were taken 1 and 4 weeks after AMI induction. Transthoracic echocardiography showed that the ejection fraction in the ATV group increased by 11.7% ± 6.83% over the control group 4 weeks after AMI. The fibrosis, infarcted area, and inflammatory level were determined by pathological and histological studies; these were found to be decreased substantially with ATV treatment (P<0.05). The expression of apoptotic, autophagic, and AMPK pathway proteins was detected by immunohistochemical staining and western blotting, while expression of their corresponding genes was measured with real-time polymerase chain reaction (PCR). ATV treatment increased AMPK/mTOR activity and the expression of autophagic protein LC3 in infarcted myocardium (P<0.05). The treatment also inhibited induction of pro-apoptotic protein Bax. AMPK inhibitor Compound C reversed these beneficial effects. In conclusion, ATV improves survival of cardiomyocytes and decreases alterations in morphology and function of infarcted hearts by inducing autophagy and inhibiting apoptosis through the activation of AMPK/mTOR pathway.
Keywords: Myocardial infarction, atorvastatin, apoptosis, autophagy, AMPK, mTOR
Introduction
Acute myocardial infarction (AMI), caused by coronary artery occlusion, accounts for a large percentage of mortality all over the world. The occlusion of blood flow to the myocardium causes death of cardiomyocytes and myocardial remodeling, leading to myocardial fibrosis, reduced left ventricular contractility, and eventually, heart failure [1]. Although many therapeutic strategies focus on AMI, heart failure remains a difficult problem to resolve because cardiomyocytes cannot regenerate [2-5]. New methods are needed to prevent cardiomyocytes from dying and to reduce myocardial fibrosis after AMI, thereby maintaining cardiac function despite injury.
Autophagy, a physiologic process of “self-eating” through the lysosomal degradation pathway [6], serves either a pro-survival or pro-death role under different cellular settings and conditions [7-9]. Cell death can occur by autophagy or apoptosis, depending on the stimulus and cell environment [10]. Autophagy and apoptosis share several molecular regulation mechanisms and can work either synergistically or antagonistically to each other [7]. Autophagy can be induced by cardiac ischemic injury; however, the role of autophagy and its relationship to apoptosis during myocardial infarction remains controversial [11].
Statins, a group of HMG-CoA reductase inhibitors, have proven to be beneficial in ischemic heart disease in several ways that are independent of their lipid-lowering ability, including anti-thrombotic, anti-inflammatory, and anti-oxidant effects [12,13]. We have shown that one statin, ATV protects stem cells in infarcted heart from apoptosis and reduces myocardial ischemic and reperfusion injury [14,15]. However, the impact and underlying mechanisms of ATV therapy on AMI are not yet understood. There is no evidence to show whether ATV has an effect on the autophagy of myocardial cells or how it regulates cellular decision to take either the apoptosis or autophagy pathway after AMI.
AMPK has an important effect on mediating energy metabolism and cell survival. It could respond to the decreased ATP/AMP ratio, suppressing mTOR activation, which could stimulate autophagy [16,17]. Furthermore, previous reports showed that statins could activate AMPK to prevent apoptosis in mesenchymal stem cells (MSCs) [18]. Still, the regulation of ATV on myocardial cells requires further elucidation. Therefore, this study aimed to determine whether ATV treatment during the peri-infarct period has an effect on autophagy and apoptosis in cardiomyocytes through the AMPK/mTOR signaling pathway. We hypothesized that ATV could induce autophagy and attenuate apoptosis in cardiomyocytes after AMI by regulating AMPK pathway proteins, which could facilitate cardiomyocyte survival and improve cardiac function.
Materials and methods
Animal care and experimental design
Male Wistar rats, 2 months old and weighing 200-220 g, were assigned to five groups randomly (n = 20 for each group): (1) No intervention (group sham), (2) AMI alone (group control), (3) ATV treatment (10 mg/kg/d orally from 1 week before MI to 4 weeks thereafter; group ATV), (4) Compound C treatment (an AMP inhibitor, 20 mg/kg intraperitoneal injection per week; group CC), or (5) Combined therapy of ATV and Compound C (group CC+ATV). As previous described, the MI model was established in all rats except the sham group by ligating the left anterior descending coronary artery (LAD) [19]. Sham operations were performed by passing a suture around the LAD without ligation. All rats in our study received humane care according to the Guide for the Care and Use of Laboratory Animals established by the National Institutes of Health, USA. Experimental protocols were approved by the Care of Experimental Animals Committee of Fuwai Hospital.
Echocardiographic assessment
Transthoracic echocardiography was used to measure cardiac function and ventricular dimensions of the rat hearts (Sonos 7500; Phillips; equipped with a 12-MHz phased-array transducer, n = 8). Function and dimensions were measured 1 week after AMI to provide baseline data and 4 weeks after AMI to provide endpoint data, as previously described [20]. All rat hearts were recorded at the papillary muscle level in 2D and M-mode. Left ventricular end-systolic dimension (LVESd) and left ventricular end-diastolic dimension (LVEDd) were measured for at least three consecutive cardiac cycles. Left ventricular fractional shortening (LVFS) was calculated as [(LVEDd-LVESd)/LVEDd] × 100%, and left ventricular ejection fraction (LVEF) was calculated as [(LVEDd)3-(LVESd)3]/(LVEDd)3] × 100%. All data were collected by an independent, blinded sonographer.
Histological analysis
All rats were sacrificed after the endpoint echocardiography measurement, and then the hearts were fixed with 10% formalin. The pathological samples were cut into 4 μm paraffin sections at the mid-left ventricular (LV) level. Hematoxylin-Eosin (H&E) staining was used to evaluate the degree of inflammatory cell infiltration. Masson’s trichromatic stain was used to assess the size of the infarct and the fibrotic area. Image-Pro-Plus software was used to measure the total LV size and infarcted area on the images. The fibrotic percentage of total LV was expressed as (fibrotic area/total LV area) × 100%. At least five sections of each heart were stained.
ELISA assay
The tissue from infarcted hearts was separated and used to measure the levels of inflammatory factors, including tumor necrosis factor (TNF-α) and interleukin-6 (IL-6), by ELISA in accordance with the manufacturer’s instructions. Expression of TNF-α and IL-6 was detected with the respective ELISA kits (R&D Systems, USA). All samples were analyzed in duplicate.
Immunohistochemical analysis of TUNEL and LC3
Myocardial tissue from the peri-infarct areas of each rat heart was obtained 4 weeks after AMI. A TUNEL assay (Roche, Germany) was conducted to assess the level of apoptosis. Immunohistochemistry was conducted on paraffin-embedded sections using rabbit polyclonal primary antibodies against LC3 (polyclonal; Abcam, USA; 1:100 dilution). The secondary antibody was Peroxidase-conjugated Affinipure Goat Anti-Rabbit IgG(H+L) (Proteintech, USA; 1:300 dilution).
Western blot analysis
Tissues were extracted from infarcted and peri-infarcted regions of the myocardium. Protein concentrations were measured with a BCA assay. To detect the expression of Bcl-2, Bax, Beclin, LC3, AMPK, phospho-AMPK (p-AMPK), mTOR, phospho-mTOR (p-mTOR), p53, and phospho-p53 (p-p53) in the heart tissue, 50 mg of protein lysate was resolved by SDS-PAGE, transferred to nitrocellulose membranes (Life Technologies), and blocked with 5% non-fat dry milk. The primary antibodies (Cell Signaling Technology), were diluted 1:1000. The membranes were incubated with primary antibodies overnight at 4°C. Then they were incubated with peroxidase-conjugated secondary antibodies diluted 1:5000. After washing, the membranes were visualized with the Chemiluminescence Detection Kit (Pierce). Target protein signals were normalized to β-actin as a loading control (1:1000 dilution; Zhongshanjinqiao, China). Densitometry analysis was completed using Quantity One software.
Real-time quantitative PCR
Expression of Bax and LC3 was measured by real-time quantitative PCR. Total RNA from each sample was isolated using TRIzol reagent and reverse transcribed using Super Script III (Life Technologies). From the cDNA libraries, target genes were amplified using TaqPCRx DNA Polymerase and measured using SYBRgreen chemistry. Data were collected from the linearity phase of the exponential reaction for each gene. GAPDH was selected as an internal control to correct for sample variation.
Statistical analysis
The data were analyzed using SPSS Software (version 18.0). Continuous variables were expressed as mean ± standard deviation (SD). Student’s t-tests and ANOVA analysis of variance were used to compare between two groups and among three or more groups, respectively. Two-sided P-values of <0.05 were considered statistically significant.
Results
Of the 100 rats, 11 (11%) rats died during the AMI operation, another 10 (10%) died within the first week after the procedure. These animals were not included in the subsequent experiments or statistical analyses, and sample sizes were adjusted accordingly.
Effect of ATV on the cardiac function
Echocardiograms were taken for all rats at 1 week after AMI (baseline) and 4 weeks after AMI (endpoint). As shown in Table 1, AMI model was both successful and stable, as there were no significant differences between the various treatment groups and the sham group at the time of the baseline echocardiography analysis (Figure 1 and Table 1). However, at the endpoint, the LVEF and LVFS in the ATV group showed significant improvement over the AMI group (P<0.05). However, there were no remarkable differences between the CC group or the CC+ATV group and the AMI group (Figure 1 and Table 1). These results suggest that ATV significantly improved recovery of cardiac function and reduced cardiac ventricular remodeling. Combined treatment of ATV and Compound C markedly attenuated the protective effect of ATV.
Table 1.
Effects of ATV on left ventricle dimensions and function after induced AMI
| Group | Sham | AMI | ATV | CC | CC+ATV |
|---|---|---|---|---|---|
| LVEDd (mm) | |||||
| Baseline | 6.28 ± 0.45 | 7.43 ± 1.10 | 7.32 ± 0.49 | 7.46 ± 1.30 | 7.39 ± 0.54 |
| Endpoint | 6.70 ± 0.77 | 8.40 ± 1.31 | 8.75 ± 1.74 | 7.80 ± 1.64 | 9.05 ± 1.51 |
| Difference | 0.42 ± 0.70 | 0.98 ± 1.80 | 1.43 ± 1.71 | 0.34 ± 1.85 | 1.66 ± 1.33 |
| LVESd (mm) | |||||
| Baseline | 3.56 ± 0.36 | 5.80 ± 0.96 | 5.57 ± 0.43 | 5.87 ± 1.34 | 5.74 ± 0.71 |
| Endpoint | 3.71 ± 0.68 | 6.83 ± 0.68 | 6.47 ± 1.20 | 6.83 ± 1.56 | 7.45 ± 1.50 |
| Difference | 0.14 ± 0.54 | 1.03 ± 1.71 | 0.80 ± 1.27 | 0.51 ± 1.64 | 1.71 ± 1.22 |
| LVEF (%) | |||||
| Baseline | 79.20 ± 6.53 | 49.90 ± 6.16 | 49.06 ± 4.05 | 49.50 ± 9.45 | 49.86 ± 7.89 |
| Endpoint | 80.28 ± 5.10 | 44.41 ± 10.85 | 56.19 ± 5.31 | 43.45 ± 6.70 | 43.66 ± 6.37 |
| Difference | 1.08 ± 3.90 | -5.49 ± 9.68# | 7.13 ± 6.18* | -6.05 ± 8.69# | -6.20 ± 8.03# |
| LVFS (%) | |||||
| Baseline | 43.05 ± 6.25 | 22.06 ± 3.30 | 21.46 ± 2.26 | 22.09 ± 4.97 | 22.11 ± 4.54 |
| Endpoint | 44.96 ± 5.94 | 19.45 ± 5.78 | 25.94 ± 3.36 | 18.64 ± 3.35 | 18.94 ± 3.30 |
| Difference | 1.91 ± 4.73 | -2.61 ± 5.02# | 4.48 ± 3.85* | -3.45 ± 4.63# | -3.18 ± 4.58# |
Note: All values are expressed as mean ± SD. Baseline = 7 days after AMI; Endpoint = 28 days after AMI. LV, left ventricle; LVEDd, left ventricular end-diastolic dimension; LVESd, left ventricular end-systolic dimension; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening.
P<0.05 vs. AMI;
P<0.05 vs. ATV.
n = 8 for each group.
Figure 1.

Echocardiography assessment of cardiac function. A: M-mode echocardiograms are shown at 1 week after myocardial infarction (baseline) and 4 weeks after myocardial infarction (endpoint). B and C: Indices of cardiac function at baseline and endpoint. *P<0.05 vs. AMI; #P<0.05 vs. ATV; n = 8 for each group.
Effect of ATV on myocardial infarct size, fibrosis, and inflammation
Histological analysis was conducted 4 weeks after AMI. As shown in Figure 2A, there was more surviving myocardium, less fibrotic area, and accumulation of extracellular matrix in the peri-infarct heart in the ATV group than in the other groups. The fibrosis of LV was dramatically decreased in the ATV group (18.40 ± 2.46%) than the AMI group (29.84 ± 7.06%, P<0.05), but no significant differences were found between groups CC (30.80 ± 6.72%, P>0.05) and CC+ATV (24.50 ± 4.85%, P>0.05) (Figure 2B). H&E staining was applied to show the infiltration of inflammatory cells in the peri-infarct hearts. Fewer inflammatory cells were found in the ATV group than in the other groups (Figure 2C).
Figure 2.
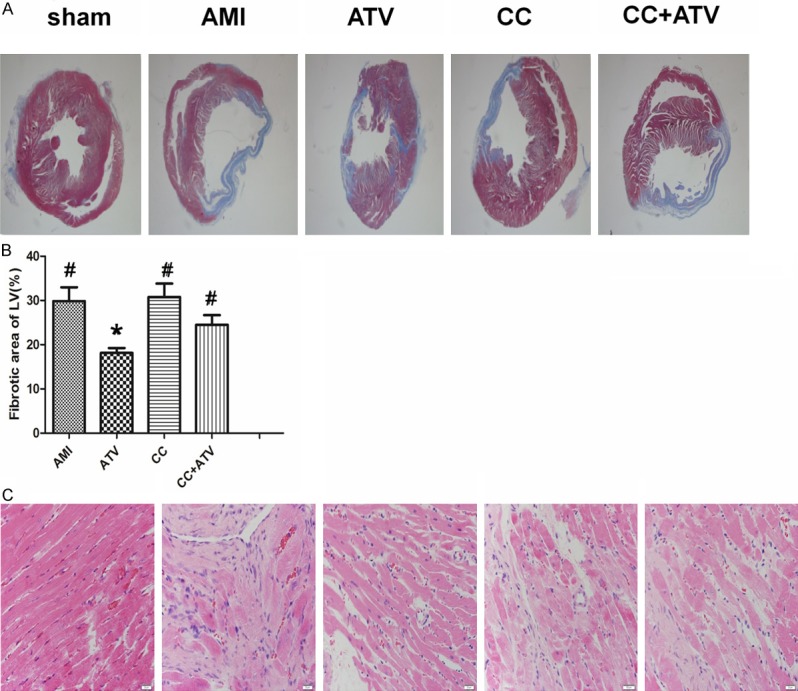
Results of hematoxylin & eosin and Masson’s trichome staining. (A) Representative Masson’s trichome staining images are shown each group. (B) Quantitative analysis of the fibrotic area. (C) Inflammatory cells infiltration in the peri-infarct area. The magnification for (A) is × 12.5, for (C) is × 400; n = 5 for each group; LV, left ventricle. *P<0.05 vs. AMI, #P<0.05 vs. ATV.
Moreover, the results from the ELISAs indicated that at 4 weeks after AMI the level of inflammatory factors including IL-6 and TNF-α was significantly increased in other four groups than in the sham group (Figure 3). ATV treatment dramatically reduced the expression of IL-6 compared with the AMI group (down to 36.98 ± 5.62 pg/mL in the ATV group from 102.85 ± 66.65 pg/mL in the AMI group). Compound C moderately elevated the level of IL-6 (124.28 ± 17.33 pg/mL) over the AMI group. In the CC+ATV group, the expression of IL-6 significantly increased (84.17 ± 11.50 pg/mL) than the ATV group. The levels of TNF-α revealed a similar pattern, but showed no significant difference between groups.
Figure 3.
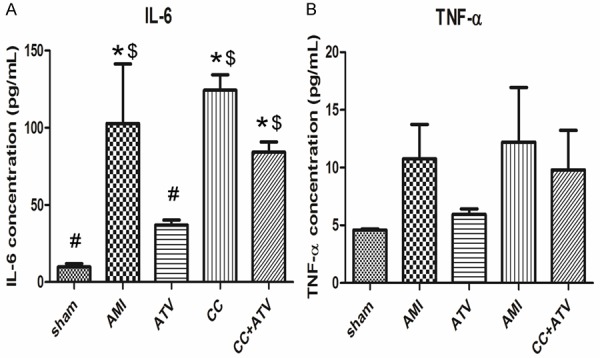
Inflammatory cytokines level in the peri-infarct myocardium. Inflammatory cytokines were analyzed by ELISA 4 weeks after AMI. n = 3 in each group. *P<0.05 vs. sham; #P<0.05 vs. AMI; $P<0.05 vs. ATV.
Effect of ATV on the apoptosis of cardiomyocytes
TUNEL assays were used to investigate whether ATV treatment had an effect on the survival of the myocardium in infarcted hearts. The number of apoptotic cells in peri-infarcted areas of hearts from the ATV group was significantly less than that in untreated rats (Figure 4A). Apoptosis of myocardial cells in Compound C-treated rats was greater than that in animals without the treatment, but this difference was not significant. The apoptosis index in the CC+ATV group was not significantly different than the CC group. Further analysis by PCR indicated that the mRNA expression of Bax, which is related to the promotion of apoptosis, decreased significantly in the ATV group compared with other groups (Figure 4B). Apoptosis-related proteins were detected by western blot, showing that expression of Bax, which is a pro-apoptotic protein, was remarkable down-regulated (P<0.05) and anti-apoptotic protein Bcl-2 was upregulated (P<0.05) in rats with ATV treatment relative to the untreated group. The effect of ATV could be abrogated by Compound C, thus the expression of these proteins were similar among AMI group, CC group, and CC+ATV group (Figure 4C-E).
Figure 4.
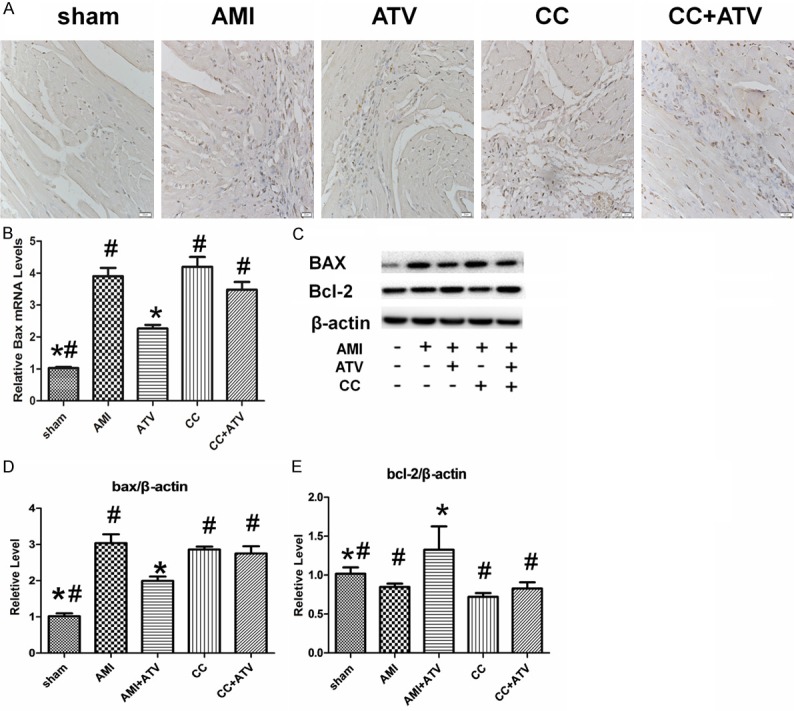
Detection of apoptosis in infarcted rat hearts. (A) Apoptotic cells detected by the TUNEL assay in infarcted hearts. (B) Quantitative analysis of Bax mRNA levels detected by real-time quantitative PCR. Expression values are reported relative to GAPDH and to the sham treatment group. (C) The expression of Bax, Bcl-2, and β-actin detected by western blot analysis. (D and E) Quantitative analysis of the expressions of Bax and Bcl-2 normalized to β-actin. Each column represents mean ± SD of three independent replicates. The magnification for (A) is × 400; *P<0.05 vs. AMI; #P<0.05 vs. ATV.
Effect of ATV on the autophagy of cardiomyocytes
Immunohistochemical staining was conducted to detect the autophagy of cardiomyocytes in infarcted hearts in each treatment group. LC3, an autophagy-specific marker, had a substantial increase in the peri-infarcted hearts from the ATV group compared with untreated rat hearts. LC3 protein levels in hearts from the CC group were slightly, but not significantly, decreased relative to those from untreated animals. No significant differences in LC3 protein levels were found between the CC and ATV+CC groups (Figure 5A). Further analysis by PCR indicated that the expression of Lc3b, which is related to the promotion of autophagy, increased significantly in the ATV group compared with other groups (Figure 5B). Additionally, the expression of LC3 was detected by western blot. It was significantly upregulated (P<0.05) in ATV group compared with other groups. However, levels of another autophagic protein, Beclin, had no significant difference between the groups (P>0.05). Furthermore, the pro-autophagic effect of ATV was abrogated by Compound C (Figure 5C-E).
Figure 5.
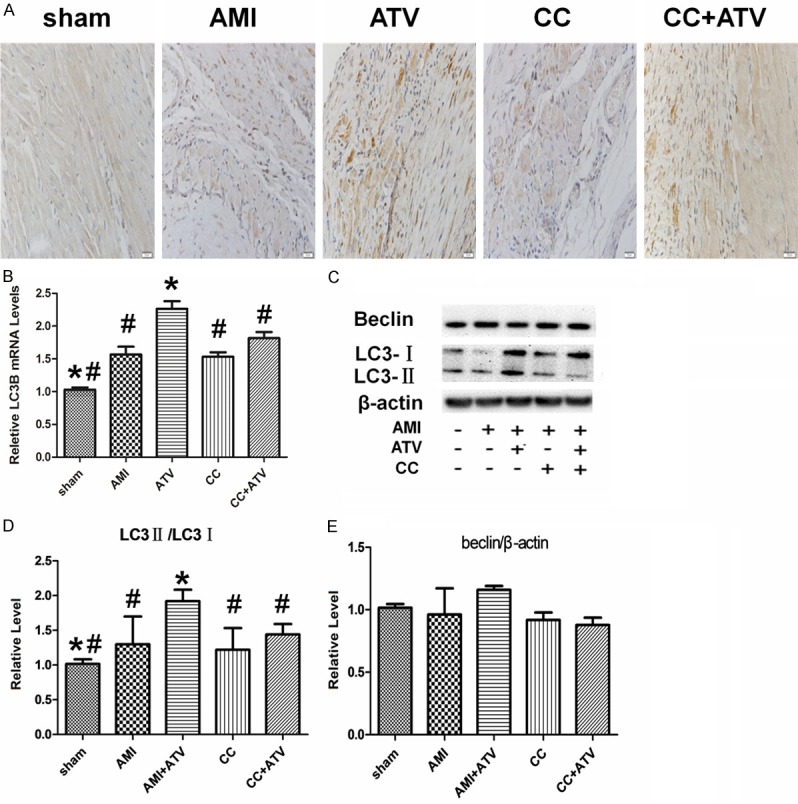
Detection of autophagy in infarcted rat hearts. (A) Autophagic cells detected by the immunohistochemistry with LC3B antibodies. (B) Quantitative analysis of LC3B mRNA levels measured by real-time quantitative PCR. Expression values were normalized to GAPDH and to sham. (C) The expression of Beclin, LC3I, LC3II, and β-actin detected by western blot analysis. (D and E) Quantitative analysis of the expression of Beclin/β-actin and LC3II/ LC3I was shown in the bar graphs. Each column represents mean ± SD of three independent experiments. The magnification for (A) is × 400; *P<0.05 vs. AMI; #P<0.05 vs. ATV.
Effect of ATV on phosphorylation of AMPK signaling proteins
To observe whether ATV could regulate the apoptosis and autophagy through the AMPK/mTOR signaling pathway, the AMPK inhibitor Compound C was used. The expression of AMPK/mTOR signaling proteins were evaluated 4 weeks after AMI (Figure 6). The phosphorylation of AMPK was upregulated (P<0.05) and the phosphorylation of p53 and mTOR was down-regulated (P<0.05) in ATV-treated rats compared to sham-treated rats. In contrast, the levels of total AMPK and mTOR were not different between the five groups. Compound C reduced the phosphorylation of AMPK dramatically and abrogated the effects of ATV on the down-regulation of p53 and mTOR phosphorylation significantly.
Figure 6.
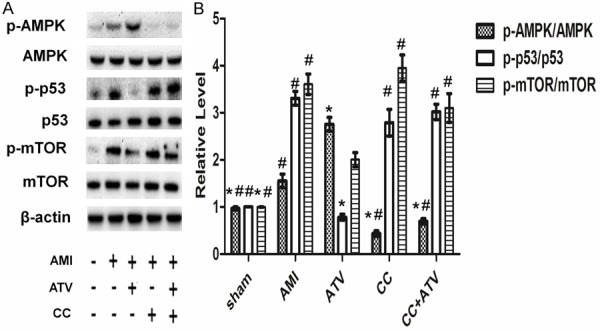
AMPK/mTOR pathway proteins detected by western blot in infarcted rat hearts. (A) AMPK/mTOR pathway proteins, including p-AMPK, AMPK, p-p53, p53, p-mTOR, and mTOR, were examined by western blot analysis. (B) Quantitative analysis of the proportion of p-AMPK/AMPK, p-p53/p53, and p-mTOR/mTOR. p-AMPK, phosphorylation of AMP-activated protein kinase; p-p53, phosphorylation of p53; p-mTOR, phosphorylation of mammalian target of rapamycin. Each column represents mean ± SD of three independent experiments. The magnification for (A) is × 400; *P<0.05 vs. AMI; #P<0.05 vs. ATV.
Discussion
This study used an in vivo model of AMI to investigate the protective effects of ATV on rat cardiomyocytes post-infarct and determine the underlying mechanisms of this effect. The major findings are: (1) ATV treatment significantly induced autophagy and decreased apoptosis of cardiomyocytes, leading to improved LV function, inhibition of myocardial inflammation, and reduction of myocardial infarcted size and fibrosis; (2) The AMPK/mTOR signaling pathway had a critical effect on the protective roles of ATV. To our knowledge, the present study is first to investigate the pro-autophagic effect of ATV after AMI in vivo and to elucidate the underlying molecular mechanism.
Statins are some of the most commonly used agents in patients with coronary diseases. In addition to its lipid-lowering effect, ATV has a number of additional benefits, including anti-inflammatory, anti-apoptotic, anti-fibrotic, angiogenesis-promoting effects [21,22]. These properties are predicted to improve the harsh microenvironment of the peri-infarcted area after AMI. Previous studies have reported that pretreatment with ATV protected heart tissue from acute ischemia/reperfusion injury, but no long-term effects were observed [23]. In this study, we found that treatment with ATV dramatically reduced cardiac function deterioration 4 weeks after AMI in rats. In contrast to the control group, ATV treatment remarkably elevated LVES and LVEF. The precise mechanism through which ATV treatment improved LV dysfunction remains undetermined; however, several of the known benefits of ATV could be contributing to the observed improvements in cardiac function observed here.
The inflammatory response has an essential role in myocardial ischemic injury [24]. Endothelial cells are damaged, activated, and interact with inflammatory corpuscle during ischemia and reperfusion injury, causing the release of inflammatory cytokines and damage to cardiomyocytes [25,26]. Our study demonstrated that ATV treatment reduced the release of inflammatory factors and inhibited the neutrophil infiltration, as shown by ELISA and H&E staining results, respectively. Previous studies have shown that protein kinase A (PKA)-related effects, including reduction of inflammatory factors expression and the suppression of NF-κB activation, were related to the anti-inflammatory effects of ATV after cardiac ischemic injury [27,28]. In addition, we found that the activation of AMPK pathway, which mediates inhibition of the inflammatory response, is another critical mechanism for ATV against ischemic injury.
Ventricular remodeling, caused by myocardial injury in response to the pressure and volume overload experienced during AMI, is harmful and ultimately results in heart failure [29]. The pathological process of cardiac ventricular remodeling mainly involves cardiomyocyte reconstruction, changes in the myocardial extracellular matrix, and fibroblast hyperplasia or fibrosis [30]. MMP-2, a member of the matrix metalloprotease (MMP) family, has a digestive function of collagen types I, II, and III, and its activity could be strongly suppressed by the tissue matrix metalloproteinase inhibitor (TIMP), TIMP-2 [31]. ATV has been shown to improve ventricular remodeling by inhibiting fibroblast proliferation and increasing the MMP/TIMP ratio, leading to reduced collagen synthesis, extracellular matrix deposition, and myocardial fibrosis during the peri-infarct period [32]. These data are consistent with our results. We found that ATV can reduce the fibrotic area of the LV dramatically 4 weeks after AMI, while addition of the AMPK inhibitor, Compound C, attenuated this effect. These data implicate the AMPK pathway with the anti-fibrotic effect of ATV during heart infarction.
Previous studies have reported that both autophagy and apoptosis contribute to cardiomyocyte death during AMI [33,34]. Because autophagy and apoptosis have opposing influences on the cell survival and cell death, the balance between these cellular decisions is complicated. In some studies, autophagy was found to promote apoptosis of cardiomyoblasts during short-term hypoxia/reoxygenation injury [35]. In other studies, autophagy was shown to have a protective role by reducing myocardial reperfusion injury [36,37]. The relationship between apoptosis and autophagy has been discussed in various contexts; however, it has not been extensively studied in cardiomyocytes in long-term infarction after AMI. Our results showed that autophagy was induced by infarcted injury 4 weeks after AMI in rat hearts, and this effect was enhanced by ATV treatment. These findings indicate that the role of autophagy in long-term infarcted injury may be beneficial, not harmful. An increase in the ratio of autophagic markers LC3II/LC3I showed that the autophagy pathway was activated effectively in injured cardiomyocytes.
Notably, the expression of Beclin-1, which participates in autophagy regulation, did not change significantly in any AMI group tested, and the modulation of autophagy by ATV did not change the expression of Beclin-1. A probable explanation may be that the level of Beclin-1 is affected by many factors, such as Bcl-2, Bcl-xL, and class III PI3K [38]. Thus, the increase in Bcl-2 observed in our study may have had an opposing influence on the expression of Beclin-1, resulting in a net change of zero in Beclin-1 levels. This hypothesis is supported by other studies. For example, two studies reported that ischemia/reperfusion injury did not affect Beclin-1 expression in MSCs or cardiac microvascular endothelial cells (CMECs) [34,35]. Apoptosis also contributes to cell death after ischemia/reperfusion injury. Our results revealed that ATV therapy dramatically decreased apoptosis in cardiomyocytes, as shown by increased expression of Bcl-2 and decreased Bax expression.
Apoptosis, which has been received extensive attention for its role in the pathogenesis of many diseases, is not the only influencer of cellular fate. Autophagy also participates as a cell survival pathway in several settings. Furthermore, it can function with apoptosis to lead to cell death or serve as a back-up program when apoptosis is defective. Although the crosstalk between these two pathways is critical and complex, little research has focused on the influence ATV has on each of them during AMI [39,40]. Our study showed that apoptosis and autophagy functioned antagonistically during ischemic injury in rat hearts, suggesting that autophagy blocked apoptotic cell death and promoted cell survival. ATV treatment induced autophagy and suppressed apoptosis in myocardial cells, thus facilitating survival and helping to preserve cardiac function. In addition, Compound C inhibited these effects, indicating that the AMPK pathway may be one essential signal-transduction pathway in the process.
AMPK, activated by the increased AMP/ATP ratio, when energy and nutrients are absent, promotes autophagy by stimulating tuberous sclerosis complex 1/2 (TSC1/TSC2) activity through a phosphorylation signal [41]. mTOR is negatively regulated by TSC1/TSC2, and thus will be inactive when AMPK is activated and autophagy is induced [42]. As the downstream molecule of many other signals, mTOR has an effect on apoptosis as well [43]. p53, a known activator of apoptosis, is capable of elevating the pro-apoptotic protein Bax, and inhibiting the anti-apoptotic protein Bcl-2 [44]. Moreover, p53 is also involved in the positive regulation of autophagy through AMPK activation [45]. Thus, this signal pathway can lead to both apoptosis and autophagy. Previous reports have found that ATV has protective roles in certain conditions through the activation of AMPK [46,47]. In consistent with these reports, we found that phosphorylation of AMPK increased and the phosphorylation of p53 and mTOR decreased significantly with ATV treatment 4 weeks after AMI, while Compound C inhibited these effects. The possible mechanism may be that the activation of AMPK by ATV reduces the phosphorylation of mTOR, resulting in the promotion of autophagy and inhibition of apoptosis.
There are some limitations in this study. First, only one time point (4 weeks post-AMI) was selected for data collection. Further study is still needed to investigate the effects at immediate and long-term consequences of ATV treatment on infarcted hearts. Additionally, only AMPK signaling proteins were measured for this study. It is possibly that other pathways may have an effect on the regulation of autophagy and apoptosis.
In conclusion, we have demonstrated that ATV could efficiently promote autophagy and reduce apoptosis by signaling through the AMPK/mTOR pathway in infarcted rat hearts after AMI. Our study helps to provide a more comprehensive understanding of the mechanism by which ATV improves cardiac function and modulates crosstalk between autophagy and apoptosis during AMI. Thus ATV has the potential to be a new target for therapeutic development through its ability to modulate autophagy, reduce apoptosis, save cardiomyocytes, and improve the function and morphology of infarcted hearts.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81200107, 81000091, and 81170129), the 863 Program of China (2013AA020101), and the 973 Program of China (2012CB518602).
Disclosure of conflict of interest
None.
References
- 1.Lucas A, Mialet-Perez J, Daviaud D, Parini A, Marber MS, Sicard P. Gadd45γ regulates cardiomyocyte death and post-myocardial infarction left ventricular remodelling. Cardiovasc Res. 2015;108:254–267. doi: 10.1093/cvr/cvv219. [DOI] [PubMed] [Google Scholar]
- 2.Nesselmann C, Ma N, Bieback K, Wagner W, Ho A, Konttinen YT, Zhang H, Hinescu ME, Steinhoff G. Mesenchymal stem cells and cardiac repair. J Cell Mol Med. 2008;12:1795–1810. doi: 10.1111/j.1582-4934.2008.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Awada HK, Hwang MP, Wang Y. Towards comprehensive cardiac repair and regeneration after myocardial infarction: aspects to consider and proteins to deliver. Biomaterials. 2016;82:94–112. doi: 10.1016/j.biomaterials.2015.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos-Gallego CG, Vahl TP, Goliasch G, Picatoste B, Arias T, Ishikawa K, Njerve IU, Sanz J, Narula J, Sengupta P, Hajjar RJ, Fuster V, Badimon JJ. The sphingosine 1-phosphate receptor agonist fingolimod increases myocardial salvage and decreases adverse post-infarction left ventricular remodeling in a porcine model of ischemia-reperfusion. Circulation. 2016;133:954–966. doi: 10.1161/CIRCULATIONAHA.115.012427. [DOI] [PubMed] [Google Scholar]
- 5.Yang B, Zhao H, X B, Wang YB, Zhang J, Cao YK, Wu Q, Cao F. Influence of interleukin-1 beta gene polymorphisms on the risk of myocardial infarction and ischemic stroke at young age in vivo and in vitro. Int J Clin Exp Pathol. 2015;8:13806–13813. [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 8.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 9.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011;21:113–119. doi: 10.1016/j.gde.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Przyklenk K, Dong Y, Undyala VV, Whittaker P. Autophagy as a therapeutic target for ischaemia/reperfusion injury? Concepts, controversies, and challenges. Cardiovasc Res. 2012;94:197–205. doi: 10.1093/cvr/cvr358. [DOI] [PubMed] [Google Scholar]
- 12.Yang YJ, Qian HY, Huang J, Li JJ, Gao RL, Dou KF, Yang GS, Willerson JT, Geng YJ. Combined therapy with simvastatin and bone marrow-derived mesenchymal stem cells increases benefits in infarcted swine hearts. Arterioscler Thromb Vasc Biol. 2009;29:2076–2082. doi: 10.1161/ATVBAHA.109.189662. [DOI] [PubMed] [Google Scholar]
- 13.Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, Linz W, Bohm M, Nickenig G. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2002;22:300–305. doi: 10.1161/hq0202.104081. [DOI] [PubMed] [Google Scholar]
- 14.Li N, Zhang Q, Qian H, Jin C, Yang Y, Gao R. Atorvastatin induces autophagy of mesenchymal stem cells under hypoxia and serum deprivation conditions by activating the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway. Chin Med J. 2014;6:1046–1051. [PubMed] [Google Scholar]
- 15.Zhang Q, Yang YJ, Wang H, Dong QT, Wang TJ, Qian HY, Xu H. Autophagy activation: a novel mechanism of atorvastatin to protect mesenchymal stem cells from hypoxia and serum deprivation via AMP-activated protein kinase/mammalian target of rapamycin pathway. Stem Cells Dev. 2012;21:1321–1332. doi: 10.1089/scd.2011.0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 18.Dong Q, Yang Y, Song L, Qian H, Xu Z. Atorvastatin prevents mesenchymal stem cells from hypoxia and serum-free injury through activating AMP-activated protein kinase. Int J Cardiol. 2011;153:311–316. doi: 10.1016/j.ijcard.2010.08.047. [DOI] [PubMed] [Google Scholar]
- 19.Muller-Ehmsen J. Rebuilding a damaged heart: long-term survival of transplanted neonatal rat cardiomyocytes after myocardial infarction and effect on cardiac function. Circulation. 2002;105:1720–1726. doi: 10.1161/01.cir.0000013782.76324.92. [DOI] [PubMed] [Google Scholar]
- 20.Xu H, Yang YJ, Qian HY, Tang YD, Wang H, Zhang Q. Rosuvastatin treatment activates JAK-STAT pathway and increases efficacy of allogeneic mesenchymal stem cell transplantation in infarcted hearts. Circ J. 2011;75:1476–1485. doi: 10.1253/circj.cj-10-1275. [DOI] [PubMed] [Google Scholar]
- 21.Lipinski MJ, Cauthen CA, Biondi-Zoccai GG, Abbate A, Vrtovec B, Khan BV, Vetrovec GW. Meta-analysis of randomized controlled trials of statins versus placebo in patients with heart failure. Am J Cardiol. 2009;104:1708–1716. doi: 10.1016/j.amjcard.2009.07.055. [DOI] [PubMed] [Google Scholar]
- 22.McKenney JM. Combination treatment with atorvastatin plus niacin provides effective control of complex dyslipidemias: a literature review. Postgrad Med. 2012;124:7–20. doi: 10.3810/pgm.2012.01.2513. [DOI] [PubMed] [Google Scholar]
- 23.Ye Y, Lin Y, Perez-Polo R, Huang MH, Hughes MG, McAdoo DJ, Manickavasagam S, Uretsky BF, Birnbaum Y. Enhanced cardioprotection against ischemia-reperfusion injury with a dipyridamole and low-dose atorvastatin combination. Am J Physiol Heart Circ Physiol. 2007;293:H813–H818. doi: 10.1152/ajpheart.00210.2007. [DOI] [PubMed] [Google Scholar]
- 24.Bekkers SC, Yazdani SK, Virmani R, Waltenberger J. Microvascular obstruction: underlying pathophysiology and clinical diagnosis. J Am Coll Cardiol. 2010;55:1649–1660. doi: 10.1016/j.jacc.2009.12.037. [DOI] [PubMed] [Google Scholar]
- 25.Sumagin R, Lomakina E, Sarelius IH. Leukocyte-endothelial cell interactions are linked to vascular permeability via ICAM-1-mediated signaling. Am J Physiol Heart Circ Physiol. 2008;295:H969–H977. doi: 10.1152/ajpheart.00400.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehnen TE, Lehnen AM, Tavares AM, Bello-Klein A, Markoski MM, Machado UF, Schaan B. Atorvastatin administered before myocardial infarction in rats improves contractility irrespective of metabolic changes. Clin Exp Pharmacol Physiol. 2014;41:986–994. doi: 10.1111/1440-1681.12313. [DOI] [PubMed] [Google Scholar]
- 27.Zhong C, Zhou Y, Liu H. Nuclear factor κB and anesthetic preconditioning during myocardial ischemia-reperfusion. Anesthesiology. 2004;100:540–546. doi: 10.1097/00000542-200403000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Yuan X, Deng Y, Guo X, Shang J, Zhu D, Liu H. Atorvastatin attenuates myocardial remodeling induced by chronic intermittent hypoxia in rats: partly involvement of TLR-4/MYD88 pathway. Biochem Biophys Res Commun. 2014;446:292–297. doi: 10.1016/j.bbrc.2014.02.091. [DOI] [PubMed] [Google Scholar]
- 29.Konstam MA, Kramer DG, Patel AR, Maron MS, Udelson JE. Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc Imaging. 2011;4:98–108. doi: 10.1016/j.jcmg.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 30.Tanai E, Frantz S. Pathophysiology of heart failure. Compr Physiol. 2015;6:187–214. doi: 10.1002/cphy.c140055. [DOI] [PubMed] [Google Scholar]
- 31.Kandalam V, Basu R, Moore L, Fan D, Wang X, Jaworski DM, Oudit GY, Kassiri Z. Lack of tissue inhibitor of metalloproteinases 2 leads to exacerbated left ventricular dysfunction and adverse extracellular matrix remodeling in response to biomechanical stress. Circulation. 2011;124:2094–2105. doi: 10.1161/CIRCULATIONAHA.111.030338. [DOI] [PubMed] [Google Scholar]
- 32.An Z, Yang G, He YQ, Dong N, Ge LL, Li SM, Zhang WQ. Atorvastatin reduces myocardial fibrosis in a rat model with post-myocardial infarction heart failure by increasing the matrix metalloproteinase-2/tissue matrix metalloproteinase inhibitor-2 ratio. Chin Med J (Engl) 2013;126:2149–2156. [PubMed] [Google Scholar]
- 33.Li XD, Yang YJ, Cheng YT, Dou KF, Tian Y, Meng XM. Protein kinase A-mediated cardioprotection of Tongxinluo relates to the inhibition of myocardial inflammation, apoptosis, and edema in reperfused swine hearts. Chin Med J (Engl) 2013;126:1469–1479. [PubMed] [Google Scholar]
- 34.Cui H, Li X, Li N, Qi K, Li Q, Jin C, Zhang Q, Jiang L, Yang Y. Induction of autophagy by tongxinluo through the MEK/ERK pathway protects human cardiac microvascular endothelial cells from hypoxia/reoxygenation injury. J Cardiovasc Pharm. 2014;64:180–190. doi: 10.1097/FJC.0000000000000104. [DOI] [PubMed] [Google Scholar]
- 35.Zhang ZL, Fan Y, Liu ML. Ginsenoside Rg1 inhibits autophagy in H9c2 cardiomyocytes exposed to hypoxia/reoxygenation. Mol Cell Biochem. 2012;365:243–250. doi: 10.1007/s11010-012-1265-3. [DOI] [PubMed] [Google Scholar]
- 36.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32:329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 37.Loos B, Genade S, Ellis B, Lochner A, Engelbrecht AM. At the core of survival: autophagy delays the onset of both apoptotic and necrotic cell death in a model of ischemic cell injury. Exp Cell Res. 2011;317:1437–1453. doi: 10.1016/j.yexcr.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 38.Huang J, Nakamura K, Ito Y, Uzuka T, Morikawa M, Hirai S, Tomihara K, Tanaka T, Masuta Y, Ishii K, Kato K, Hamada H. Bcl-xL gene transfer inhibits Bax translocation and prolongs cardiac cold preservation time in rats. Circulation. 2005;112:76–83. doi: 10.1161/CIRCULATIONAHA.105.535740. [DOI] [PubMed] [Google Scholar]
- 39.Shao BZ, Han BZ, Zeng YX, Su DF, Liu C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol Sin. 2016;37:150–156. doi: 10.1038/aps.2015.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oral O, Akkoc Y, Bayraktar O, Gozuacik D. Physiological and pathological significance of the molecular cross-talk between autophagy and apoptosis. Histol Histopathol. 2016;31:479–98. doi: 10.14670/HH-11-714. [DOI] [PubMed] [Google Scholar]
- 41.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 42.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 43.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation, and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamada R, Toguchi Y, Nomura T, Imagawa T, Sakaguchi K. Tetramer formation of tumor suppressor protein p53: structure, function, and applications. Biopolymers. 2016;106:598–612. doi: 10.1002/bip.22772. [DOI] [PubMed] [Google Scholar]
- 45.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia F, Wu C, Chen Z, Lu G. Atorvastatin inhibits homocysteine-induced endoplasmic reticulum stress through activation of AMP-activated protein kinase. Cardiovasc Ther. 2012;30:317–325. doi: 10.1111/j.1755-5922.2011.00287.x. [DOI] [PubMed] [Google Scholar]
- 47.Song L, Yang YJ, Dong QT, Qian HY, Xu H, Meng XM, Tang Y. Atorvastatin protects swine bone marrow mesenchymal stem cells from apoptosis through AMPK, but not PI3K/Akt pathway. Zhonghua Xin Xue Guan Bing Za Zhi. 2011;39:1033–1038. [PubMed] [Google Scholar]