

Abstract
Cancer cell epithelial-mesenchymal transition (EMT) is the crucial event for cancer progression and plays a vital role in the metastasis of cancer cells. Activation of Polo-like kinase 1 (PLK1) signaling has been implicated as the critical event in several tumor metastasis and EMT, however, whether PLK1 participates in gastric carcinoma metastasis and EMT still remains unclear. For this study, we elucidated the potential physiological function of PLK1 in the metastasis of gastric tumors, as well its distinct role in cells EMT and subsequently determined the mechanism involved in PLK1 regulated. Immunoblotting assay and Oncomine data mining analysis indicated that PLK1 expression was highly up-regulated in gastric carcinoma. Kaplan-Meier survival analysis for the relationship between survival outcomes and PLK1 expression in gastric carcinoma was performed with an online Kaplan-Meier plotter (http://kmplot.com/analysis/). Over-expression of PLK1 in gastric cancer cells SGC-7901 and MKN-28 significantly promoted cells profound morphological changes and enhanced metastatic ability of tumor cells. On the contrary, silencing of PLK1 induced mesenchymal epithelial transition (MET)-like morphological and inhibited the metastatic process. Furthermore, we found that the metastatic characters promoting effects of PLK1 in gastric carcinoma was related to the activation of protein kinase B (AKT). Our mechanistic investigations revealed that AKT inhibition reversed PLK1-induced EMT, blocked gastric carcinoma cells invasiveness and metastasis. Additionally, over-expression of AKT promoted the migratory and invasion ability of the two cell lines, which was disrupted by PLK1 down-regulation. To conclude, our findings demonstrate that PLK1 accelerates the metastasis and epithelial-mesenchyme transition of gastric cancer cells through regulating the AKT pathway.
Keywords: PLK1, EMT, metastasis, gastric carcinoma
Introduction
Gastric carcinoma, also known as stomach cancer, is the typical metastasis form of cancer and remains one of the lethal malignancies with poor prognosis. Advanced gastric carcinoma is associated with substantial mortality because it metastasizes to lymph nodes, peritoneum, lung and liver [1]. Tumor cell metastasis is a complex cell biology and molecular process, among which epithelial-mesenchymal transition (EMT) is an initial step in the metastasis process, during which non-motile, epithelial tumor cells progressively alter their polarity complexes and lost their cell-cell adhesion to acquire motile and plastic characteristics of mesenchymal cells [2]. An extensive body of evidences indicates that EMT plays a vital role in a plethora of cancer-related progression, including cancer cells migration, invasion, angiogenesis, and chemotherapy-resistant. EMT program is functionally triggered and controlled by multiple signaling pathways, including cytokine receptors, transforming growth factor-β (TGF-β), glycogen synthesis kinase-3β (GSK-3β), Wingless and INT-1 (Wnt) signaling and down-stream transcriptional regulators, such as Twist and Snail [3]. Once activated, these pathways transcriptional regulate a range of genes that are crucial for the maintenance of tumor cell epithelial characteristics and lead to the functional loss of E-cadherin, which is one of the typical hallmarks of EMT program [4]. Accompany with the decreasing expression of E-cadherin, catenin beta-1 also known as β-catenin, which in complexes with cadherin cell adhesion molecules of tumor cells, release into cytosol and subsequent nuclear translocation, where β-catenin becomes a co-activator for TCF and LEF to activate the transcription factors including, Snail, Slug and Twist [5]. These molecular alterations cause dysfunctional cell-cell adhesion and cell-cell junctions break off, thereby allowing dissemination of gastric carcinoma cells from the primary situ and spread to distal tissue sites.
Serine/threonine-protein kinase Polo-like kinase 1 (PLK1) is an enzyme, which functions as a key regulator of the mitotic phase, including centrosome maturation, chromosome segregation, bipolar spindle formation, mitotic entry, and cytokinesis execution [6]. PLK1 consists of 603 amino acids and contains a conserved N-terminal kinase domain and two conserved polo-box regions of 30 amino acids at the C-terminus. Previous studies shown that the loss or mutation of PLK1 in tumor cells directly trigger pro-apoptotic pathways, blocks anti-apoptotic pathways and inhibits the growth of cancerous tumors [7]. In addition, recent studies suggest that PLK1 may have other important functions such as regulation of DNA synthesis, p53 transactivation, recovery from the G2 DNA damage checkpoint and drives tumor cell metastasis [8]. An extensive body of research has demonstrated that the proto-oncogene PLK1 is over-expressed and highly activated in a wide variety of human tumors, including melanoma, breast, non-small cell lung cancer, colorectal, prostate, ovarian, and head and neck cancers, pancreatic carcinoma, as well as non-Hodgkin’s lymphomas and acute myeloid leukemia (AML) [9]. Its expression level frequently positively correlates with increased indefinite proliferation of tumor cells, enhanced metastatic potentiality and survival outcomes in patients with several different cancers. For example, PLK1 overexpression is commonly observed in prostate cancer, and over-expression of PLK1 is linked to higher tumor grade and correlates with a poor prognosis, suggesting that PLK1 plays a central role in prostate cancer etiology [10]. Impressively, increased PLK1 expression levels positively correlate with the invasiveness of colorectal carcinoma, breast cancer, and thyroid tumor cells and highly correlated with increased metastasis activity. PLK1 is certified to be critical for the viability of cancer cells harboring activated RAS oncogene or inactivated p53 transcription factor, particularly in the presence of genotoxic damage [11]. Some published results have shown RNA interference (RNAi) entails the potential for novel therapeutic strategies through the silencing of cancer-causing genes. In orthotopic breast cancer models, PLK1 siRNAs significantly impair human breast cancer growth and induce apoptosis in breast cancer cell lines. The most impressive thing is intravenously injected PLK1-siRNA into breast cancer xenograft suppressing PLK1 expression and reducing breast cancer cells metastasis [12]. Finally, the PLK1 inhibitor causes a mitotic arrest and induces apoptosis in human cancer cell lines of diverse tissue origin and oncogene signature, suggesting that the potential druggability of PLK1 and provide the molecular details of targeting PLK1 may be considered as a cancer therapeutic option [13]. All these data imply a possible role for PLK1 in tumor progression including proliferation, invasion and metastasis; however, direct evidence supporting this hypothesis and mechanisms of the pro-metastasis activity of PLK1 in gastric carcinoma are lacking.
In the present study, we evaluated the expression of PLK1 by molecular-biological analysis as well as bioinformatics, and explored the roles of PLK1 in control the growth and motility of gastric carcinoma. Our data highlight PLK1 as a crucial positive regulator of gastric carcinoma cell migration, invasion and EMT. The present study suggests that pro-metastasis activity of PLK is mediated by induction of the metastasis and EMT via activation of protein kinase B (AKT) signaling pathway, thereby may be a promising therapeutic target of gastric carcinoma metastasis.
Materials and methods
Cell culture and reagents
Human gastric mucosal epithelial cell line GES-1, gastric carcinoma cells BGC-823, SGC-7901, MKN-45, MKN-28 and MGC-803 were purchased from American Type Culture Collection (ATCC). Cells were cultured in DMEM or RPMI-1640 with 10% fetal bovine serum (FBS), and 1% penicillin-streptomycin solution (Gibco, Invitrogen, USA), and cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. AKT inhibitor (GSK690693) was obtained from Selleck (USA). Lipofectamine 2000 was purchased from Invitrogen company.
Gene transfection
SGC-7901 and MKN-28 cells were transiently transfected with plasmids (2 μg/well for 6 well culture plates) containing the mammalian expression vector pcDNA3 (plasmid 14751) encoding HA-tagged constitutively active Akt (T308D; S473D) mutant, which was obtained from Addgene (Cambridge, MA, USA) and pcDNA3.0 empty vector (Invitrogen, Carlsbad CA, USA) as control using the opti-MEM medium (Gibco-BRL/Invitrogen, Carlsbad, CA) plus Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s recommendations.
Plasmids and transfection
A PLK1 expression construct was generated by sub-cloning PCR-amplified full-length human PLK1 cDNA into the pIRES2-EGFP vector (Clontech, USA). Transfection was performed with lipofectamine 2000 according to the manufacture’s manual. Retroviral production and infection were performed as we previously described, and stable cell lines were selected by treatment with 0.5 μg/ml puromycin for 10 days, beginning at 48 h after infection. After 10 days post-transfection, the cells were treated with indicated agents for western blot analyses and biological behavior assay [14].
RNA interference
Oligonucleotides for human PLK1 siRNA kit were purchased from OriGene (Rockville, MD, USA). The kit contains three predesigned duplexes targeting a specific gene of interest, and we used a pool of three target siRNAs to ensure work efficiency. Cells were transfected with PLK1 siRNA or non-specific siRNA (2 μg/well for 6 well culture plates) using the opti-MEM plus X-tremeGENE siRNA transfection reagent (Roche, Mannheim, Germany) according to the instruction manual. After 48 h post-transfection, the cells were further tested for western blot analyses, migration and invasion assay [15].
Analysis of oncomine data
To determine the expression pattern of PLK1 in gastric carcinoma, two datasets, including Badea Pancreas and Segara Pancreas in Oncomine database (www.oncomine.org) were used. The gene expression of PLK1 was compared between gastric carcinoma tissues with normal according to the standard procedures as previously described [16].
Wound healing assay
Briefly, SGC-7901 and MKN-28 cells were seeded in six-well culture dishes and cultured at 37°C to form a confluent monolayer. After starvation in serum-free medium for 24 h, a wound was made by scratching the monolayer with a 100 µl pipette tip. Then, the wounded monolayer was washed to remove cell debris with PBS and incubated with fresh normal medium. After scratching, the area of cell-free scratch was photographed at 0 h and 24 h. The wound healing effect was assayed by measuring the percentage of the remaining cell-free area compared with the area of the initial wound [17].
Cells invasion assay
Invasion of gastric carcinoma cells was determined by BD BioCoat™ Matrigel™ Invasion Chamber (BD Biosciences, USA) assay in vitro according to the manufacturer’s instructions. In brief, 1 × 105 cells with 500 µl in serum-free medium were added into the upper chamber and 600 µl of medium conditioning with 10% FBS was added into the lower chamber, function as chemo-attractant to induce cells invasion. After incubation in culture incubator, 37°C, 5% CO2 atmosphere for 6 h, the noninvasive cells in the upper surface of the membrane were removed and the invasive gastric carcinoma cells migrating to the lower surface of the membrane were fixed and stained with 0.1% crystal violet for 15 min. Finally, cell counting was carried out by photographing the membrane through the microscope [18].
Western blotting
After washing with PBS (0.5 mM KH2PO4, 3.2 mM Na2HPO4, 140 mM NaCl, and 1.3 mM KCl), SGC-7901 and MKN-28 cells were extracted with cold lysis buffer (100 mM NaCl, 20 mM Tris, 5 mM EDTA, 5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM b-glycerolphosphate, 1 mM Na3VO4, 1 mM PMSF, and protease inhibitors) and centrifuged at 15,000 g for 15 min at 4°C. Protein concentration of the supernatants was determined with Bradford assay (Biorad, USA). 30 µg of samples was separated by electrophoresis on 8% SDS-PAGE and transferred to Polyvinylidene fluoride membrane (PVDF) (Millipore, USA). After blocking with 5% skimmed milk, PVDF membranes were incubated with different specific primary antibodies (anti-GSK-3β, phospho-GSK-3β, NF-κB, phospho-NF-κB, FAK, phospho-FAK, AKT, phospho-AKT, Wnt, phospho-Wnt, E-cadherin, N-cadherin, Slug Twist and GAPDH from Cell Signaling Technology). After washing with PBST for 30 min, the membranes were further incubated with corresponding HRP-conjugated secondary antibodies and developed with Pierce’s West Pico chemiluminescence substrate (Millipore, USA). GAPDH was used as a control to verify equal protein loading.
Quantitative realtime PCR
Total RNA was extracted from cell lines using TRIzol reagent (Invitrogen) and we synthesized cDNA was using SuperScript II Reverse Transcriptase (Invitrogen). Quantitative PCR was performed using an ABI Prism 7900HT Sequence detection system (Applied Biosystems, Foster City, CA, USA). PLK1 amplification was performed using the following primers: 5’-ACCAGCACGTCGTAGGATTC-3’; and reverse 5’-ATAACTCGGTTTCGGTGCAG-3’. Primers used for the GAPDH gene were 5’-AACGGATTTGGTCGTATTGG-3’ (forward) and 5’-GGATCTCGCTCCTGGAAGAT-3’ (reverse) Samples were run in triplicates, and the samples were normalized to the internal control gene (GAPDH) using the comparative Ct method (ΔΔCt).
Statistical analysis
Statistical analysis was performed using SPSS 13.0 software package (SPSS Inc, Chicago, IL, USA). Data were expressed as mean ± standard deviation from three independent experiments. Statistical differences between the groups were compared using Student’s t test. A P value of less than 0.05 was considered statistically significant.
Results
Elevated expression of PLK1 in gastric cancer
To determine the potential role of PLK1 in gastric cancer, we examined its expression in several gastric cancer cell lines and the human gastric mucosal epithelial cell line GES-1 by western blotting analysis. Quantitative analysis of the western blotting results was performed by normalizing the gray intensity of PLK1 band to that of GAPDH. As shown in Figure 1A, the expression of PLK1 was increased in BGC-823, SGC-7901, MKN-45, MKN-28 and MGC-803, as compared with the expression of PLK1 in the control cell line GES-1, which suggesting that the expression of PLK1 elevated in gastric cancer cell lines. To further assess the expression level of PLK1 in gastric cancer, we performed quantitative real-time PCR to assess the mRNA level of PLK1 in gastric carcinoma cells. In agreement with the western blotting data, increase in PLK1 mRNA levels was observed in most gastric cancer cell lines as compared with that in GES-1 (Figure 1B). Next, PLK1 in human gastric cancer tissues was investigated using three datasets from the publicly available Oncomine database. As shown in Table 1 and Figure 2A, Oncomine analysis of neoplastic vs. normal tissue showed that PLK1 was significantly overexpressed in gastric cancer in different datasets (P < 0.05). To further extend our observations to the relevant of PLK1 and gastric cancer patient survival outcomes, we performed Kaplan-Meier survival analysis of PLK1 with an online tool (http://kmplot.com/analysis/). The results showed that higher PLK1 expression was associated with a worse overall survival for patients with gastric cancer (Figure 2B).
Figure 1.

PLK1 is highly expressed in gastric cancer cells. A. PLK1 protein expression in gastric cancer lines (BGC-823, SGC-7901, MKN-45, MKN-28, and MGC-803) and gastric mucosal epithelial cell line (GES-1) was detected by western blotting analysis. GAPDH was used as an internal standard. B. Total RNAs were extracted from gastric cancer cells. PLK1 mRNA level was determined by means of quantitative real-time PCR and normalized to the level of GAPDH mRNA. The fold changes of mRNA expression of PLK1 gene were compared as a ratio to the GES-1 cells. The data are shown as mean ± SD of triplicates experiments. *P < 0.05, **P < 0.01 compared with the GES-1 cells.
Table 1.
Changes in PLK1 gene expression in gastric carcinoma
Figure 2.
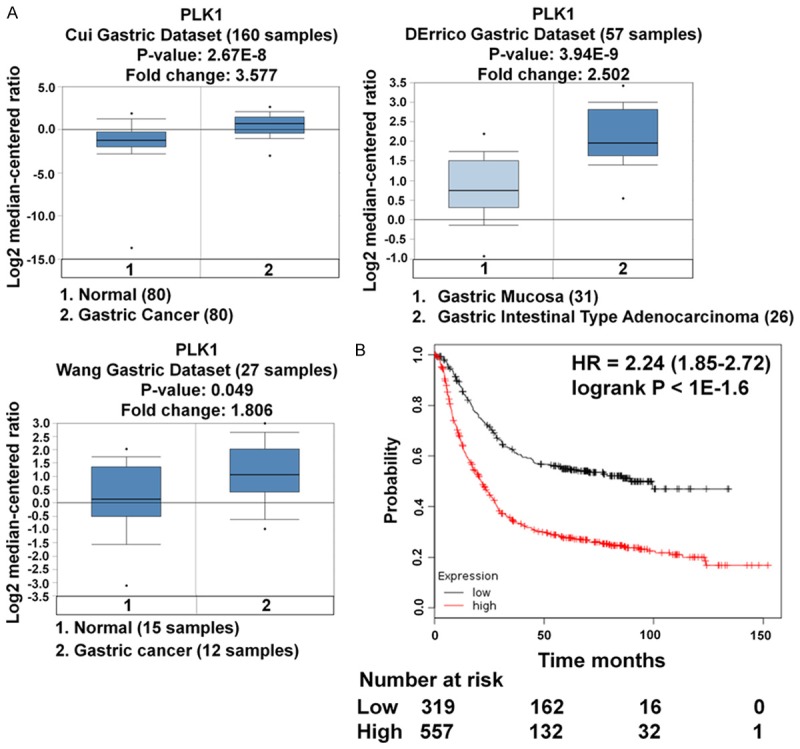
PLK1 expression is over-expressed in gastric cancer and correlates with survival time. A. Box plots derived from gene expression data in Oncomine comparing expression of PLK1 gene in normal tissue (left plot) and gastric cancer tissue (right plot). Oncomine box plots were retrieved from serous gastric carcinoma and normal tissue. B. Kaplan-Meier survival analysis for the relationship between survival time and PLK1 signature in gastric cancer was performed by using the online tool (http://kmplot.com/analysis/).
Crucial function of PLK1 signaling on invasiveness and metastatic potential of gastric cancer cells
To investigate the role of PLK1 signaling as it relates to invasive phenotypes and the motility of gastric cancer cells, SGC-7901 and MKN-28 cell lines that stably expressed PLK1 were established. The transfection efficiency was confirmed by evaluating the expression of green fluorescence protein (GFP) (Figure 3A) and immunoblotting with PLK1 antibody (Figure 3B). To further confirm the role of PLK1 in cell motility, we tested the effects of PLK1 on the motility of SGC-7901 and MKN-28 cells in a wound healing assay. As shown in Figure 3C, a significant increase in the percentage of total area that was covered by cells that ectopic expression of PLK1. Next, the effect of PLK1 signaling on the invasion of gastric cancer cells was measured using a Matrigel invasion assay. The ability of gastric cancer cells to invade Matrigel was markedly enhanced by over-expression PLK1 (Figure 3D). These results provided evidence for the role of PLK1 in promotion of metastasis capability in gastric carcinoma.
Figure 3.

PLK1 overexpression promotes gastric cancer cell mobility. A. PLK1 was cloned into pIRES2-EGFP vector and then transfected into SGC-7901 and MKN-28 cells. Cells transfected with the empty vector were used as control. The transfection efficiency was evaluated by assayed the green fluorescence protein expression. B. After 24 h post transfections, protein samples were subjected to western blot for measuring PLK1. C. Confluent SGC-7901 and MKN-28 cell monolayers were wounded with 100 µl pipette tip. Cell migration to the wound area was monitored by microscope for 24 h post-wound. The percentage of wound area covered by cells was assessed using the ImageJ program. Bars represent the standard deviation of three independent experiments conducted in triplicate. **P < 0.01 compared with untreated control groups. D. SGC7901 and MKN-28 cells (1 × 105 cells/well) were seeded in the upper chamber, and medium containing 10% FBS was added to the lower chamber. After incubation for 6 h, the cells that invaded the lower membrane of the insert were stained with 0.1% crystal violet and counted by microscopy. The data are expressed as the means of three independent experiments ± standard deviation. **P < 0.01 compared with untreated control groups.
Confirmation of the role of PLK1 in gastric cancer cells metastasis by gene silencing
To ascertain PLK1 was indeed responsible for metastasis in both SGC-7901 and MKN-28 cells, we investigated whether reduced metastasis of SGC-7901 and MKN-28 cells could be reproduced by gene silencing with siRNA. To test this experiment, we silenced the expression of PLK1 in SGC-7901 and MKN-28 cells with a siRNA-incorporated plasmid targeting a specific site of PLK1 mRNA. As shown in Figure 4A, the expression of PLK1 in cells transfected with siRNA plasmid (siPLK1) was significantly decreased compared with the cells transfected with scrambled siRNA (siCTL). The infection efficiency was also confirmed by evaluating mRNA expression levels (Figure 4B). Then, the transfected cells were subjected to both wound healing and Transwell assay to evaluate cells metastasis potential. Both SGC-7901 and MKN-28 cells transfected with control siRNA were able to close the artificial wound by 24 h. Nevertheless, the wound inflicted on SGC-7901 and MKN-28 cells transfected with PLK1 siRNA had not yet closed up at this time (Figure 4C). Consistently, results from invasion assay also showed that the cell number of siPLK1 cells migrated across the membrane was much fewer than the control cells (Figure 4D). Taken together, these results indicated that silencing of PLK1 drastically reduced the cell motility of gastric cancer cells.
Figure 4.
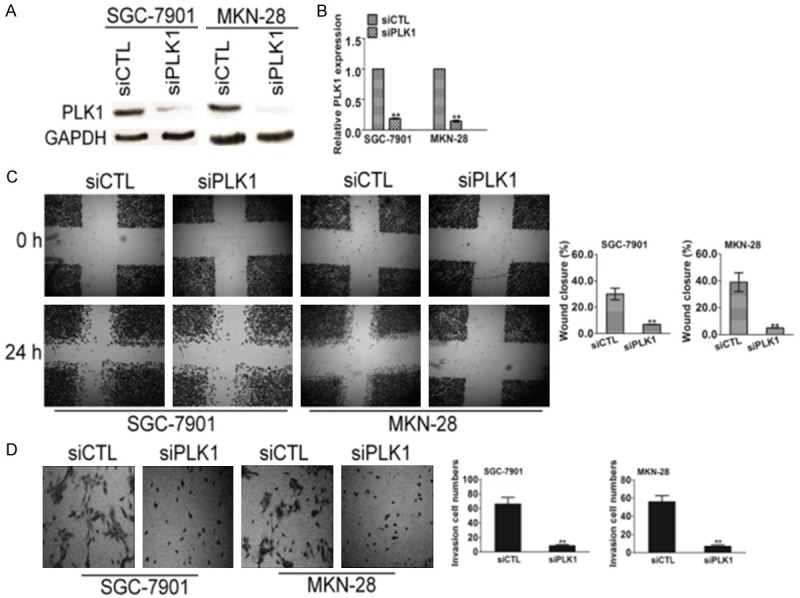
PLK1 silencing decreases the migration and invasion of SGC-7901 and MKN-28 cells. A. Cells transfected with the empty vector were used as control cells. The expression of PLK1 was determined western blot with PLK1 antibody. B. RT-PCR analyses showed effective down-regulation of PLK1 following siRNA transfection. C. PLK1- and control-siRNA transfected SGC-7901 and MKN-28 cells were wounded with pipette and wound closure percentage was quantified 24 h after scratch relative to that at 0 h. *P < 0.05, **P < 0.01 compared to the control cells. D. SGC-7901 and MKN-28 cells transfected with PLK1-siRNA or control siRNA were seeded in the upper chamber. After 6 h, cells invaded through the membrane were stained and counted in five random microscopic fields.
PLK1 induces EMT of gastric cancer cell line
EMT has been considered to be the critical step involved in cancer metastasis. Core elements related to EMT include reduction of cell-cell adherence via the delocalization of cadherins and transcriptional repression, and functional loss of E-cadherin, which is a well-known hallmark of EMT. Expression of epithelial intermediate filaments is typically reduced and the equivalent non-epithelial intermediate filaments (vimentin) increased. To investigate what role of PLK1 might play in the gastric cancer cell EMT, the morphological alterations of SGC7901 and MKN-28 cells, and epithelial or mesenchymal marker changes in PLK1-expressing cells were assessed by microscopy and western blotting, respectively. As shown in Figure 5A, while control cells maintained organized cell-cell adhesion, overexpression of PLK1 in SGC7901 and MKN-28 cells by lentiviruses infection result in loss of cell-cell contacts; and these cells became scattering and gain a variable cell shape. Consistently, overexpression of PLK1 in SGC7901 and MKN-28 cells led to reduction of epithelial markers E-cadherin, and induction of mesenchymal markers, including N-cadherin, Slug and Twist (Figure 5B). TGF-β elicits tumor promoting effects through its ability to induce EMT which enhances invasiveness and metastasis. Whether depletion of PLK1 could induce mesenchymal-epithelial transition (MET) was also investigated in SGC7901 and MKN-28 cell lines. As shown in Figure 5C, while control cells appeared to had a typical spindle-like fibroblastic morphology, PLK1 siRNA cells exhibited a cobble stone like appearance. Consistently, showed a significantly reduction in N-caderhein, Slug, Twist, and increasing of E-caderhein (Figure 5D). These results indicate that overexpression of PLK1 promotes EMT in gastric cancer cells. Taken together, these results indicate that overexpression of PLK1 induce mesenchymal phenotypes, which revealing that PLK1 play an important role in EMT program.
Figure 5.
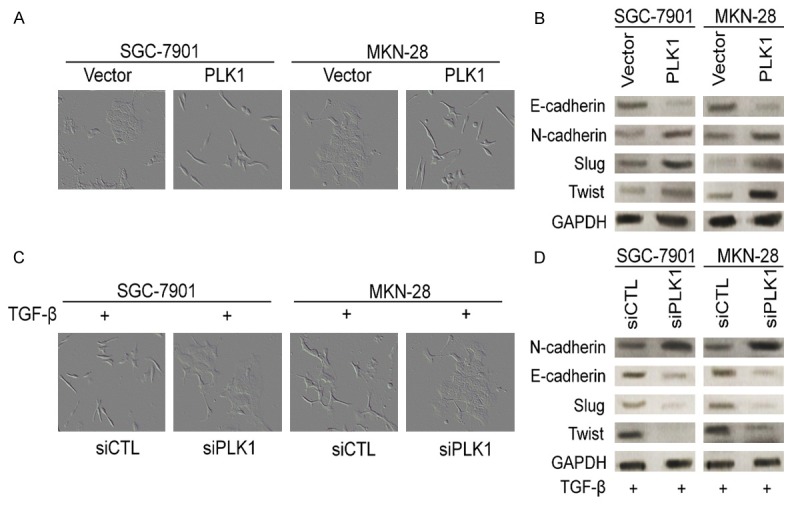
PLK1 regulates EMT in gastric cancer cells. A. Overexpression of PLK1 induced EMT in SGC7901 and MKN-28 cells. SGC7901 and MKN-28 cells were infected with empty vector or PLK1, the morphological change was examined after 10 days. B. SGC7901 and MKN-28 cells were infected with empty vector or PLK1 and the expression of the indicated EMT markers was detected by western blotting. GAPDH was used as an internal standard. C. Control siRNA or siRNA against PLK1 were transfected into SGC7901 and MKN-28 cells. After 48 h post transfections, morphology of the transfected cells were examined. D. SGC7901 and MKN-28 cells were infected with control siRNA or siRNA against PLK1 and the expression of the indicated MET markers was detected by western blotting. GAPDH was used as an internal standard.
AKT pathway is involved in PLK1-mediated gastric cancer cell migration and invasion
Next, we investigated the signaling pathway which is responsible for the induction of migration and invasion in PLK1-overexpressing gastric cancer cells. To determine the signaling pathway which was involved in PLK1-mediated gastric cancer cell metastasis, multiple potential signaling pathways related to metastasis of tumor cells were measured. As shown in Figure 6A, only the basal level of AKT phosphorylation was found to be significantly up-regulated in cells overexpressing PLK1. In contrast, we observed no obvious difference in many other signaling pathways, such as focal adhesion kinase (FAK) and Wnt activity. Aberrant activation of AKT pathway has been shown to contribute to tumor metastasis and progression, and has been linked to the tumor EMT process. Consistently, when PLK1 was silenced by siRNA in SGC7901 and MKN-28 cells, AKT activation was also down regulated (Figure 6B). To confirm the role of AKT in PLK1-regulated cell metastasis, constitutively active form of AKT (D2AKT) was transfected into cells after PLK1 silenced and the expression of D2AKT was confirmed by western blot with anti-HA-tag as well as specific AKT antibody (Figure 6C). As expected, active AKT obviously restored the impaired invasion in PLK1-silenced cells (Figure 6D). In addition, GSK690693, which is an AKT specific inhibitor, was also employed to confirm the role of AKT in SGC7901 and MKN-28 cells invasion. As shown in Figure 6E, PLK1-mediated AKT phosphorylation was completely inhibited by GSK690693. As a result, PLK1-promoted invasion in SGC-7901 and MKN-28 cells were also blocked by GSK690693, as shown by the Transwell invasion assay (Figure 6F). To conclude, these data indicated that AKT was involved in PLK1 promoted metastasis of gastric cancer cells.
Figure 6.

PLK1 facilities invasion of gastric cancer cells through AKT. A. Western blot result shown that the phosphorylation of AKT at Ser-473 was elevated in cells transfected with pIRES2-EGFP-PLK1. Total AKT expression was used as a loading control. B. The phosphorylation of AKT at Ser-473 was perturbed in cells transfected with PLK1 siRNA plasmid. Total AKT expression was used as a loading control. C. Expression of D2AKT was confirmed by western blot with antibody against AKT, and GAPDH was used as loading control. D. Transwell assay was performed to determine the invasion of cells co-transfected with PLK1 silencing plasmid and D2AKT plasmid. E. In the presence of GSK690693, cells were incubated for 6 h, protein extracts were analyzed by western blot with antibodies against phosphorylated AKT (S473) or AKT. F. In the presence of GSK690693 (2 μM), transwell invasion assay was conducted to evaluate the cell invasiveness after transfection. Representative pictures were taken after staining with crystal violet.
Discussion
Abundant clinical and pathologic evidence indicates that PLK1 signaling is involved in tumorigenesis and might be a novel biomarker for tumor progression [19]. EMT is a pathological progress that epithelial cells lose their characteristic of cell polarity and adhesion, which acquire a mesenchymal phenotype [20]. EMT process results in enhanced ability of mobility and invasiveness for carcinoma cells, which is considered as a crucial early step in cancer metastasis. Although some studies have suggested that PLK1 expression in tumors is correlated with aggressiveness of cancer, none of these studies clearly indicate the exact role of PLK1 in the metastatic progression of gastric cancer and the mechanisms underlying PLK1 induced EMT in have not been completely determined. Here, we have found that PLK1 is over-expressed in metastatic breast cell lines. The analysis of oncomine reveals that PLK1 protein expression is significantly higher in primary cancers than that of the matched noncancerous tissue. We also investigated the functional role of PLK1 signaling in gastric cancer metastasis and cell EMT using multiple experiments. We found that PLK1 promotes gastric cancer cells metastasis in vitro through the AKT signaling pathway. Our results provided a new target for intervention in the gastric cancer metastasis treatment and might improve the future gastric carcinoma prognosis.
PLK1 is a versatile and critical mitotic kinase that regulates tumor cell cycle progression from G2 into mitosis, progression through mitosis, and the completion of cytokinesis, thus plays an important role in the maintenance of genomic stability [21]. PLK1 levels are tightly regulated, peaking at the onset of mitosis and declining upon mitotic exit. Over-expression of PLK1 has been shown to stimulate cell proliferation and oncogenic transformation in NIH 3T3 while PLK1 depletion leads to mitotic arrest and apoptosis in various human cancer cell lines [22]. A recent study showed that PLK1 controls prostate epithelial cell motility by mechanisms requiring transcriptional reprogramming and dramatic alterations of cell phenotype. Indeed, over-expression of PLK1 induced EMT-like alterations in prostate epithelial cells, whereas PLK1 knockdown restored epithelial features of invasive prostate cancer cell lines [23]. Importantly, previous studies proved that increased PLK1 expression levels positively correlate with the invasiveness of colorectal, breast, and thyroid tumors. These data imply possible role for PLK1 in tumor metastasis; however, the definite mechanism underlying PLK1 regulating gastric cancer cells metastasis is still not clear [24]. Our experiments demonstrated that knockdown of PLK1 by siRNA significantly attenuated the migratory and invasive potency of gastric cancer cells. Moreover, down-regulation PLK1 induced epithelial like phenotypes and caused a shift from mesenchymal markers to epithelial markers, which was up-regulation of E-cadherin, and down-regulation of N-cadherin. The expression of Snail, Slug and Twist, three well-known EMT-promoting transcriptional factors, were also found to be positively regulated by PLK1. In contrast, the overexpression of PLK1 in cancer cells enhanced the migration of and led to the decrease of N-cadherin, a hallmark of EMT. Our findings identified PLK1 as a novel promoter of tumor EMT and a potential enhancer of the metastatic potential in cancer.
The molecular mechanism for PLK1-meditated gastric cancer cell metastasis is identified to be related to AKT signaling pathway. Previous studies demonstrated that AKT signaling plays a crucial role in regulation of EMT as well as metastasis, including migration and invasion process in several cancers [25]. The pro-metastatic potential of AKT pathway can be supported by the involvement of up-regulation of phosphorylated AKT in severely dysplastic nevi and metastatic melanomas compared with normal or mildly dysplastic nevi. Our results indicated that overexpression of PLK1 in SGC-7901 and MKN-28 gastric cancer cells induced up-regulation of AKT phosphorylation and PLK1 silencing led to reducing AKT phosphorylation. Furthermore, restoring AKT by an active form AKT plasmid rescued the impaired metastasis of cells induced by PLK1 silencing. In the presence of a specific inhibitor of AKT signaling pathway (GSK690693) PLK1 overexpression-promoted migration and invasion were significantly inhibited as revealed by the invasion assay. Consistently, reported that PLK1 siRNA inhibits AKT pathway and reduces pancreatic cancer cell proliferation. This may add an additional layer of PLK1 targeting AKT signaling to future regulate tumor growth and metastasis.
In conclusion, our observations demonstrate for the first time that PLK1 drives cell metastasis of cancer cells by mechanisms induction EMT and improve our understanding of the molecular mechanism by which PLK1 signaling activation occurs as it relates to the metastatic behavior of gastric cancer cells. Due to the important roles of PLK1 in the EMT and metastasis of gastric carcinoma cells, it may serve as an attractive target for molecular targeting cancer therapy.
Disclosure of conflict of interest
None.
References
- 1.Zhang Z, Zhang G, Kong C. FOXM1 participates in PLK1-regulated cell cycle progression in renal cell cancer cells. Oncol Lett. 2016;11:2685–2691. doi: 10.3892/ol.2016.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kadletz L, Bigenzahn J, Thurnher D, Stanisz I, Erovic BM, Schneider S, Schmid R, Seemann R, Birner P, Heiduschka G. Evaluation of Polo-like kinase 1 as a potential therapeutic target in Merkel cell carcinoma. Head Neck. 2016;38(Suppl 1):E1918–1925. doi: 10.1002/hed.24349. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez-Acenero MJ, Cortes D, Gomez Del Pulgar T, Cebrian A, Estrada L, Martinez-Useros J, Celdran A, Garcia-Foncillas J, Pastor C. PLK-1 Expression is Associated with Histopathological Response to Neoadjuvant Therapy of Hepatic Metastasis of Colorectal Carcinoma. Pathol Oncol Res. 2016;22:377–383. doi: 10.1007/s12253-015-0015-8. [DOI] [PubMed] [Google Scholar]
- 4.Zhao CL, Ju JY, Gao W, Yu WJ, Gao ZQ, Li WT. Downregulation of PLK1 by RNAi attenuates the tumorigenicity of esophageal squamous cell carcinoma cells via promoting apoptosis and inhibiting angiogenesis. Neoplasma. 2015;62:748–755. doi: 10.4149/neo_2015_089. [DOI] [PubMed] [Google Scholar]
- 5.Jin HY, Qiu XG, Yang B. The MicroRNA3686 Inhibits the Proliferation of Pancreas Carcinoma Cell Line by Targeting the Polo-Like Kinase 1. Biomed Res Int. 2015;2015:954870. doi: 10.1155/2015/954870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun W, Su Q, Cao X, Shang B, Chen A, Yin H, Liu B. High expression of polo-like kinase 1 is associated with early development of hepatocellular carcinoma. Int J Genomics. 2014;2014:312130. doi: 10.1155/2014/312130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang G, Zhang Z, Liu Z. Polo-like kinase 1 is overexpressed in renal cancer and participates in the proliferation and invasion of renal cancer cells. Tumour Biol. 2013;34:1887–1894. doi: 10.1007/s13277-013-0732-0. [DOI] [PubMed] [Google Scholar]
- 8.Song B, Liu XS, Rice SJ, Kuang S, Elzey BD, Konieczny SF, Ratliff TL, Hazbun T, Chiorean EG, Liu X. Plk1 phosphorylation of orc2 and hbo1 contributes to gemcitabine resistance in pancreatic cancer. Mol Cancer Ther. 2013;12:58–68. doi: 10.1158/1535-7163.MCT-12-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo MA, Kang KS, Di Cristofano A. The PLK1 inhibitor GSK461364A is effective in poorly differentiated and anaplastic thyroid carcinoma cells, independent of the nature of their driver mutations. Thyroid. 2013;23:1284–1293. doi: 10.1089/thy.2013.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Du XL, Wang CJ, Lin DC, Ruan X, Feng YB, Huo YQ, Peng H, Cui JL, Zhang TT, Wang YQ, Zhang H, Zhan QM, Wang MR. Reciprocal activation between PLK1 and Stat3 contributes to survival and proliferation of esophageal cancer cells. Gastroenterology. 2012;142:521–530. e523. doi: 10.1053/j.gastro.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 11.Zhang XG, Lu XF, Jiao XM, Chen B, Wu JX. PLK1 gene suppresses cell invasion of undifferentiated thyroid carcinoma through the inhibition of CD44v6, MMP-2 and MMP-9. Exp Ther Med. 2012;4:1005–1009. doi: 10.3892/etm.2012.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mok WC, Wasser S, Tan T, Lim SG. Polo-like kinase 1, a new therapeutic target in hepatocellular carcinoma. World J Gastroenterol. 2012;18:3527–3536. doi: 10.3748/wjg.v18.i27.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao C, Gong L, Li W, Chen L. Overexpression of Plk1 promotes malignant progress in human esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2010;136:9–16. doi: 10.1007/s00432-009-0630-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lan H, Zhu J, Ai Q, Yang Z, Ji Y, Hong S, Song F, Bu Y. Rapid functional screening of effective siRNAs against Plk1 and its growth inhibitory effects in laryngeal carcinoma cells. BMB Rep. 2010;43:818–823. doi: 10.5483/BMBRep.2010.43.12.818. [DOI] [PubMed] [Google Scholar]
- 15.Gerster K, Shi W, Ng B, Yue S, Ito E, Waldron J, Gilbert R, Liu FF. Targeting polo-like kinase 1 enhances radiation efficacy for head-and-neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2010;77:253–260. doi: 10.1016/j.ijrobp.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 16.Ortega CE, Seidner Y, Dominguez I. Mining CK2 in cancer. PLoS One. 2014;9:e115609. doi: 10.1371/journal.pone.0115609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Zhu L, Fang J, Ge Z, Li X. LRG1 modulates epithelial-mesenchymal transition and angiogenesis in colorectal cancer via HIF-1alpha activation. J Exp Clin Cancer Res. 2016;35:29. doi: 10.1186/s13046-016-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuang J, Li L, Guo L, Su Y, Wang Y, Xu Y, Wang X, Meng S, Lei L, Xu L, Shao G. RNF8 promotes epithelial-mesenchymal transition of breast cancer cells. J Exp Clin Cancer Res. 2016;35:88. doi: 10.1186/s13046-016-0363-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng YB, Lin DC, Shi ZZ, Wang XC, Shen XM, Zhang Y, Du XL, Luo ML, Xu X, Han YL, Cai Y, Zhang ZQ, Zhan QM, Wang MR. Overexpression of PLK1 is associated with poor survival by inhibiting apoptosis via enhancement of survivin level in esophageal squamous cell carcinoma. Int J Cancer. 2009;124:578–588. doi: 10.1002/ijc.23990. [DOI] [PubMed] [Google Scholar]
- 20.Gao H, Zhong F, Xie J, Peng J, Han Z. PTTG promotes invasion in human breast cancer cell line by upregulating EMMPRIN via FAK/Akt/mTOR signaling. Am J Cancer Res. 2016;6:425–439. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Nappi TC, Salerno P, Zitzelsberger H, Carlomagno F, Salvatore G, Santoro M. Identification of Polo-like kinase 1 as a potential therapeutic target in anaplastic thyroid carcinoma. Cancer Res. 2009;69:1916–1923. doi: 10.1158/0008-5472.CAN-08-1693. [DOI] [PubMed] [Google Scholar]
- 22.Kanaji S, Saito H, Tsujitani S, Matsumoto S, Tatebe S, Kondo A, Ozaki M, Ito H, Ikeguchi M. Expression of polo-like kinase 1 (PLK1) protein predicts the survival of patients with gastric carcinoma. Oncology. 2006;70:126–133. doi: 10.1159/000093003. [DOI] [PubMed] [Google Scholar]
- 23.Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, Loening S, Dietel M, Kristiansen G. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate. 2004;60:240–245. doi: 10.1002/pros.20050. [DOI] [PubMed] [Google Scholar]
- 24.Weichert W, Denkert C, Schmidt M, Gekeler V, Wolf G, Kobel M, Dietel M, Hauptmann S. Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br J Cancer. 2004;90:815–821. doi: 10.1038/sj.bjc.6601610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ree AH, Bratland A, Solberg Landsverk K, Fodstad O. Ionizing radiation inhibits the PLK cell cycle gene in a G2 checkpoint-dependent manner. Anticancer Res. 2004;24:555–562. [PubMed] [Google Scholar]
- 26.Cui J, Chen Y, Chou WC, Sun L, Chen L, Suo J, Ni Z, Zhang M, Kong X, Hoffman LL, Kang J, Su Y, Olman V, Johnson D, Tench DW, Amster IJ, Orlando R, Puett D, Li F, Xu Y. An integrated transcriptomic and computational analysis for biomarker identification in gastric cancer. Nucleic Acids Res. 2011;39:1197–1207. doi: 10.1093/nar/gkq960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Errico M, de Rinaldis E, Blasi MF, Viti V, Falchetti M, Calcagnile A, Sera F, Saieva C, Ottini L, Palli D, Palombo F, Giuliani A, Dogliotti E. Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur J Cancer. 2009;45:461–469. doi: 10.1016/j.ejca.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Wen YG, Li DP, Xia J, Zhou CZ, Yan DW, Tang HM, Peng ZH. Upregulated INHBA expression is associated with poor survival in gastric cancer. Med Oncol. 2012;29:77–83. doi: 10.1007/s12032-010-9766-y. [DOI] [PubMed] [Google Scholar]