

Abstract
Ferulic acid (FA), a phenolic acid which is abundant in vegetables and fruits, has been reported to exert anti-oxidative and anti-inflammatory activities. In the present study, the pharmacological effects and the underlying mechanisms of FA in mice with acetaminophen-induced hepatotoxicity were investigated. Our results revealed that FA pretreatment inhibited the augments of serum aminotransferases in a dose-dependent manner and attenuated the hepatic histopathological abnormalities and hepatocellular apoptosis in acetaminophen (APAP) exposed mice. Moreover, FA inhibited the expression of cytochrome P450 2E1 (CYP2E1), enhanced the activities of superoxide dismutase (SOD) and catalase (CAT) as well as the contents of glutathione (GSH). Furthermore, FA markedly attenuated acetaminophen-induced serum tumor necrosis factor (TNF)-α and interleukin (IL)-1β production, suppressed Toll-like receptor (TLR) 4 expression and dampened p38 mitogen-activated (MAPK) and nuclear factor kappa (NF-κB) activation. These data suggested that FA could effectively protect against APAP-induced liver injury by down-regulated expression of CYP 2E1 and the suppression of TLR4-mediated inflammatory responses.
Keywords: Ferulic acid, acetaminophen, hepatotoxicity, cytochrome P450 2E1, toll-like receptor (TLR) 4
Introduction
Acetaminophen (N-acetyl-p-aminophenol, paracetamol, APAP), a widely used analgesic and antipyretic drug, leads to acute liver injury at an overdose which has become the most frequent cause of drug-induced acute liver failure in the United States (USA) and the United Kingdom (UK) [1,2]. At therapeutic doses, most APAP is rapidly metabolized by UDP-glucuronosyltransferase (UGT) and sulfotransferase (SULT) to phenolic glucuronide and sulfate inactive conjugations and then they are excreted into the urine and bile; a small percentage of APAP is oxidized by cytochrome P450 (CYP450) enzymes to N-acetyl-p-benzoquinone imine (NAPQI), a highly reactive intermediate, which is detoxified by covalent binding with GSH [3]. APAP poisoning generates excess NAPQI which evokes the depletion of GSH and then binds to macromolecules triggering mitochondrial dysfunction, oxidative stress and ultimately resulting in the hepatocellular death [3-5]. These changes are initial events in APAP induced hepatotoxicity. However, secondary activation of innate immune system via identification of damage-associated molecular patterns (DMAPs) by pattern recognition receptors (PRRs) on immune cells, mainly involving the Toll like receptors (TLRs), exerts an essential role in determining the progression and severity of APAP induced liver injury [6-8]. Among the TLRs, TLR4 is a crucial mediator in the activation of innate immune response following increased production of pro-inflammatory cytokines and chemokines and recruitment of immune cells, thus triggers inflammatory responses exacerbating the liver injury [9]. TLR4 signaling deficiency protected against APAP-induced hepatotoxicity supported by studies that TLR4 signaling was related to APAP hepatotoxicity in TLR4 mutant mice [10] and TLR4 deficiency by knocking out or using antagonist alleviated the injury with APAP exposure [11], indicating that TLR4 signaling pathway plays an important role in the model of APAP hepatotoxicity.
N-acetyl-cysteine (NAC), as GSH precursor and antioxidant [12], is remained the cornerstone antidote in APAP poisoning. However, NAC administration is more effective at early stage, later period protection mechanisms have still been elucidated [13]. In additional, lack of formal comparative clinical trials in regiment, insufficient evidence supporting the precise dose refinement, and the adverse effects also restrict NAC use [13,14]. If NAC treatment is invalid, liver transplantation is the final strategy [2]. Therefore, it is indispensable and challenging to explore novel therapeutic drugs.
Ferulic acid (FA, 4-hydroxy-3-methoxycinnamic acid, Figure 1A), an phenolic compound found in vegetables, fruits and traditional Chinese medicines, has been known to possess numerous pharmacologic activities against many disorganized diseases related to oxidative stress and inflammation including diabetes [15,16], Alzheimer’s disease [17], cancer [18,19], cardiovascular diseases [20], and metabolic syndrome [21]. Recently, several studies have demonstrated that FA exerts hepatoprotective effects in ischemia-reperfusion induced hepatocellular apoptosis [22] and carbon tetrachloride induced liver injury [23]. Moreover, several reports have shown that FA ameliorates the inflammation via suppressing the phosphorylation of IκB, and the production of pro-inflammatory cytokines such as TNF-α and IL-6 [24,25]. These findings indicate that FA might be a promising candidate for APAP hepatotoxicity.
Figure 1.
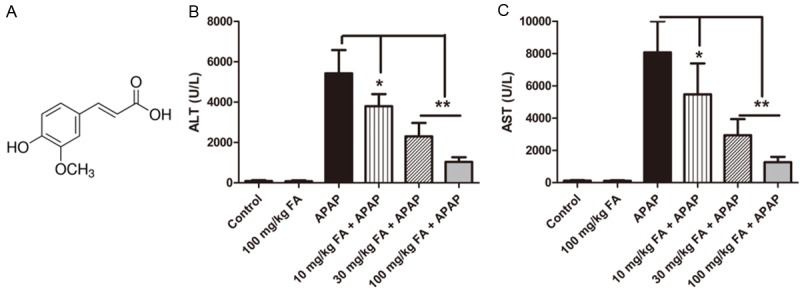
Pretreatment with FA dose-dependently supressed the elevated levels of serum aminotransferases induced by APAP. Mice were given oral gavage of vehicle or different concentration of FA (10, 30, 100 mg/kg, respectively) every 8 h one time for three times within 24 h before APAP (350 mg/kg) exposure. (A) Chemical structure of FA. The activities of serum ALT (B) and AST (C) were determined at 18 h after APAP administration. Data were expressed as mean ± SD, n=6, *P<0.05, **P<0.01, as compared with APAP group.
Therefore, in the present study, the aim was to explore whether FA attenuates APAP-induced hepatotoxicity in vivo and further search the underlying mechanisms. Our study revealed that FA alleviated APAP-induced liver injury and the protective effect was related to the suppression of CYP 2E1 and the inhibition the inflammation by TLR4-mediated MAPK and NF-κB activation.
Materials and methods
Reagents
FA (C10H10O4, MW 194.18, purity 99%) was obtained from Aladdin Industrial Corporation (Shanghai, China). APAP was supplied by Sangon Biotech (Shanghai, China). Caspase 3 colorimetric assay kit was purchased from Beyotime Institute of Biotechnology (Jiangsu, China). The kits for detection of alanine aminotransferase (ALT), asparate aminotransferase (AST), SOD, CAT and GSH were supplied by Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The enzyme-linked immunosorbent assay (ELISA) kits for measuring TNF-α and IL-1β were purchased from Bender Med Systems (Vienna, Austria). Terminal deoxynucleotidyl transferase d-uridine triphosphate nick end labeling (TUNEL) in situ cell death detection kit was supplied by Roche Diagnostics (Basel, Switzerland). Rabbit anti-mouse TLR4 and CYP 2E1 antibodies were purchased from Abcam (Cambridge, UK). Rabbit anti-mouse phospho-IRAK1, phospho-p38, and phospho-IκB were purchased from Cell Signaling Technology (Boston, MA, USA). Mouse anti-GAPDH was supplied by Santa Cruz Biotechnology (Santa Cruz, CA, USA). Bicinchoninic acid (BCA) protein assay kit, horseradish peroxidase-conjugated goat anti-rabbit antibody and enhanced chemiluminescent (ECL) reagents were obtained from Pierce Biotechnology (Rockford, IL, USA).
Animals and experimental protocols
The 6-8 week old BALB/c mice weighting 18-22 g were supplied by the Experimental Animal Center of Chongqing Medical University (Chongqing, PR China). Mice were housed in a controlled environment at temperature of 20-25°C, 55% humidity under 12 h light/dark cycles. All experimental procedures involving animals were approved by the Animal Care and Use Committee of Chongqing Medical University.
APAP solution was prepared by dissolving the compound in the warm saline before every experiment. Mice were fasted overnight with free access to water and then administrated APAP (350 mg/kg body weight) or vehicle by intraperitoneal (i.p.) injection to induce acute liver injury. Various doses of FA (10, 30, or 100 mg/kg, dissolved in 0.5% carboxylmethylcellulose sodium salt in 0.9% saline) or saline was given orally every 8 h one time for three times within 24 h prior to APAP exposure (n=6 per group). Mice were sacrificed at 18 h after APAP treatment and blood was centrifuged at 3000 rpm for 5 min and liver samples were fixed in 4% paraformaldehyde or preserved at -80°C for further analysis.
Serum aminotransferase activity
ALT and AST activities in serum were measured with detection kits as index of hepatocellular injury in accordance with the manufacturer’s directions.
Histological analysis
Liver tissues were maintained in 4% paraformaldehyde, embedded in paraffin and liver sections were sliced for hematoxylin and eosin (HE) staining and assessed using light microscopy (Nikon, Tokyo, Japan).
TUNEL and caspase 3 activity assay
Hepatocellular apoptosis was evaluated by the situ cell death detection and caspase 3 colorimetric assay kits according to the manufacturer’s instructions, respectively. Briefly, the tissue sections were dewaxed and rehydrated, and incubated with proteinase K working solution for 30 min at 37°C. Then TUNEL reaction mixture was added on samples and incubated for 1 h at 37°C in a humidified box in the dark. The TUNEL-positive cells were imaged with a light microscopy (Nikon, Tokyo, Japan). For caspase 3 activity assay, liver tissue was homogenized in cell lysis buffer, the supernatant (100 g protein) was collected from homogenates centrifuged for 1 min at 10 000 g and incubated with Ac-DEVD-pNA substrate and reaction buffer for 90 min at 37°C. Then active caspase 3 was determined at 405 nm and standardized by the total protein concentration of the same sample.
Measurement SOD activity, CAT activity and reduced glutathione content
The activities of SOD and CAT and the content of reduced GSH in liver tissues were detected to assess the anti-oxidative capacities using kits following manufacturer’s directions, respectively. All values were normalized by the total protein concentration of each sample.
ELISA for cytokines
The level of TNF-α and IL-1β in serum were determined using ELISA kits according to the instructions recommended by the manufacturer.
Western blot analysis
Total proteins were isolated from frozen liver samples using the protein extract kit (Piece Biotechnology, Rockford, USA) and then total protein concentration was measured by BCA protein assay kit. Protein extracts were fractionated on 12% polyacrylamide-sodium sulfate (SDS) gel and then transferred to polyvinylidene fluoride (PVDF) membrane. The membranes were blocked by incubating in 5% fat-free milk dissolved in Tris-buffered saline (TBS) containing 0.05% tween-20 (TBST) for 1 h at room temperature and then were incubated with primary antibody at 4°C overnight followed by incubation with HRP-conjugated secondary antibody for 1 h at room temperature. Antibody binding was visualized with an ECL chemiluminescent system and detected by Image Lab software (Bio-rad, USA).
RNA isolation and quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from liver samples with a total RNA extraction reagent (Takara, Japan) according to manufacturer’s protocol. Then the complementary DNA (cDNA) samples of CYP 2E1 were synthesized and used for Real-Time PCR reaction. The sequences of CYP 2E1 primers were 5’-CGT TGC CTT GCT TGT CTG GA-3’ (sense) and 5’-AAG AAA GGA ATT GGG AAA GGT CC-3’ (antisense) and GAPDH primers were 5’-AGG TCG GTG TGA ACG GAT TTG-3’ (sense) and 5’-TGT AGA CCA TGT AGT TGA GGT CA-3’ (antisense) which was used as an internal standard.
Statistical analysis
All data were expressed as means ± standard deviations (SD). The results were assessed by one-way analysis of variance (ANOVA) and student’s test. A p-value less than 0.05 was considered statistically significant.
Results
FA pretreatment attenuated APAP-induced hepatotoxicity
To determine the effects of FA on APAP poisoning, the levels of ALT and AST were measured as biochemical makers to evaluate the extent of liver injury. As shown in Figure 1B and 1C, after 18 h of APAP administration, the serum ALT and AST activities were significantly elevated. However, these increases were alleviated by FA (10, 30, or 100 mg/kg) in a dose-dependent manner. Therefore, FA dose at 100 mg/kg was selected for further evaluation of morphology and mechanism.
FA alleviated APAP-induced hepatic pathological damages and apoptosis
The histological analysis in Figure 2A showed that livers in control group have normal lobular architecture and cell structure. However, APAP treatment caused severe hepatocellular necrosis and neutrophil infiltration. FA at a dose of 100 mg/kg attenuated these pathological changes. Liver apoptosis was evaluated by using TUNEL staining and caspase 3 activity assay (Figure 2B and 2C). Less apoptotic cells were seen in FA pretreated group in APAP exposed mice when compared to APAP group. Meanwhile, the activity of caspase 3, a key downstream effector in apoptosis cascades, was also significantly inhibited by FA.
Figure 2.
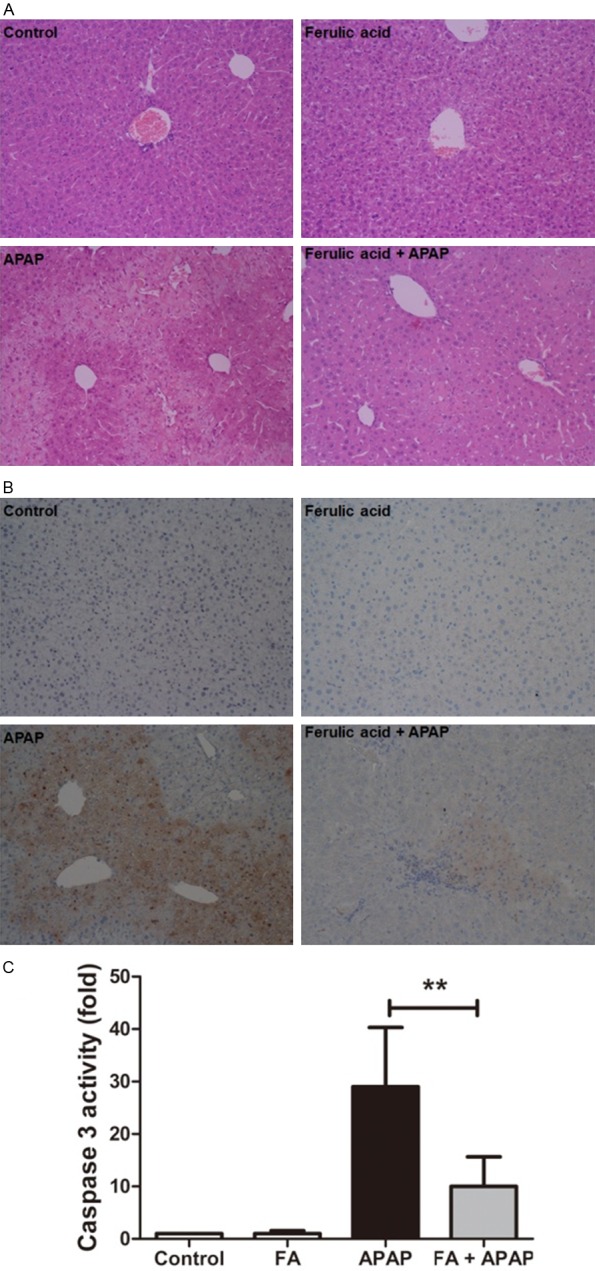
Pretreatment with FA alleviated liver histological abnormalities and apoptosis in mice challenged by APAP. Mice were given oral gavage of vehicle or FA (100 mg/kg) every 8 h one time for three times within 24 h before APAP (350 mg/kg) exposure and liver samples were harvested at 18 h after APAP administration. A: The liver sections were stained with hematoxylin-eosin for the evaluation of liver pathological changes (200× magnifications). B: Hepatocellular apoptosis was measured by TUNEL (200× magnifications). C: The caspase 3 activity was determined. Data were expressed as mean ± SD, n=6, **P<0.01, compared with APAP group.
FA abrogated APAP-induced hepatic expression of CYP 2E1
Among the CYP450 superfamily, CYP 2E1 is one of the most active members that metabolizes APAP to NAPQI [26]. Thus, the express of CYP 2E1 was measured by qRT-PCR and western blot. CYP 2E1 mRNA and protein was markedly promoted by APAP overdose. However, this upregulation was markedly inhibited by FA pretreatment (Figure 3A and 3B).
Figure 3.
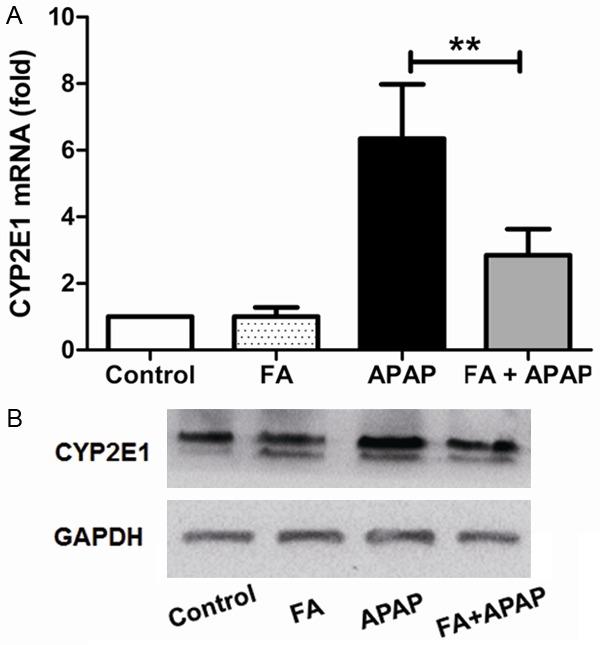
Pretreatment with FA suppressed APAP-induced CYP 2E1 expression in mice. Mice were given oral gavage of vehicle or FA (100 mg/kg) every 8 h one time for three times within 24 h before APAP (350 mg/kg) exposure and liver samples were harvested at 18 h after APAP administration. The expression of CYP 2E1 in the liver were measured by qRT-PCR (A) and western blot (B). Data were expressed as mean ± SD, n=6, **P<0.01, as compared with APAP group.
FA inhibited APAP-induced oxidative stress
Reactive oxygen species (ROS) is essential for APAP-induced acute liver failure. Normally, ROS is cleared with the antioxidants, for example, glutathione, vitamins C and E, and antioxidant enzymes such as SOD and CAT [27,28]. Overproduction of ROS causes the imbalance between pro-oxidant and anti-oxidant systems and then following oxidative stress and inflammatory response. Next, the content of GSH and the activities of SOD and CAT were determined. From Figure 4A, we could observe that GSH was depleted after APAP administration while it was reproduced with FA treatment. Moreover, the down-regulated activities of SOD and CAT after APAP poisoning were also enhanced by FA (Figure 4B and 4C), respectively. These findings indicted the restoration of antioxidant system may be a mechanism related to protection of FA against APAP-induced hepatotoxicity.
Figure 4.
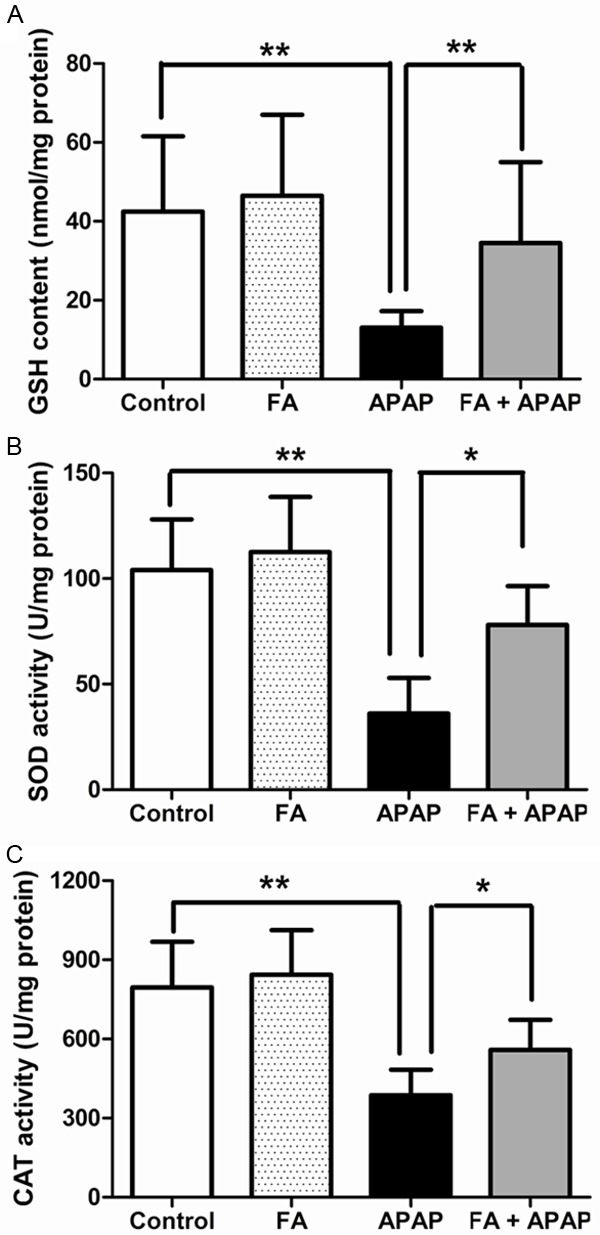
Pretreatment with FA increased GSH contents, SOD and CAT activities in the liver of APAP-challenged mice. Hepatic GSH contents (A), SOD activities (B) and CAT activities (C) were determined at 18 h after APAP administration. Data were expressed as mean ± SD, n=6, *P<0.05, **P<0.01, as compared with APAP group.
FA alleviated the pro-inflammatory cytokines in APAP exposed mice
In the model of APAP poisoning, innate immune activation triggers severe inflammatory responses which lead to a secondary hit to hepatocytes, thus amplify the liver injury. This process is complex and involves the release of pro-inflammatory cytokines and chemokines and the recruitment of immune cells. Previous studies revealed that TNF-α and IL-1β were the vital pro-inflammatory cytokines in the progress of APAP-induced liver injury [6,29]. Therefore, the level of TNF-α and IL-1β in the serum was analyzed by the ELISA kits. As shown in Figure 5A and 5B, TNF-α and IL-1β level in APAP group were markedly elevated compared to normal group. However, both of them were reduced after pretreatment with FA at a dose of 100 mg/kg.
Figure 5.
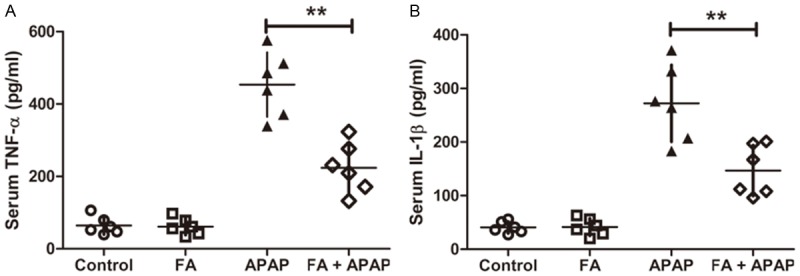
Pretreatment with FA alleviated the levels of serum TNF-α and 1L-1β in APAP-challenged mice. Mice were given oral gavage of vehicle or FA (100 mg/kg) every 8 h one time for three times within 24 h before APAP (350 mg/kg) exposure and serum were collected at 18 h after APAP administration. The serum TNF-α (A) and 1L-1β (B) were detected by ELISA. Data were expressed as mean ± SD, n=6, **P<0.01, as compared with APAP group.
FA suppressed APAP-activated TLR4 signaling pathway
To further investigate the hypothesis that the protective effect of FA on APAP-induced hepatotoxicity was related to down-regulated inflammatory responses mediated by TLR4 signaling pathway, the protein level of TLR4 and the phosphorylation of downstream mediator molecules including IRAK1, IκB, p38 were measured. In compared with the normal group, APAP administration significantly enhanced the expression of TLR4 and this increase was limited by FA pretreatment. Meanwhile, western blot analysis also shown phosphorylation of IRAK1, p38, and IκB were promoted after APAP administration. However, these elevations were abrogated with FA treatment (Figure 6). Taken together, these data suggested that FA alleviated APAP hepatotoxicity by decreasing inflammatory responses, at least in part, in a TLR4-dependent mechanism.
Figure 6.
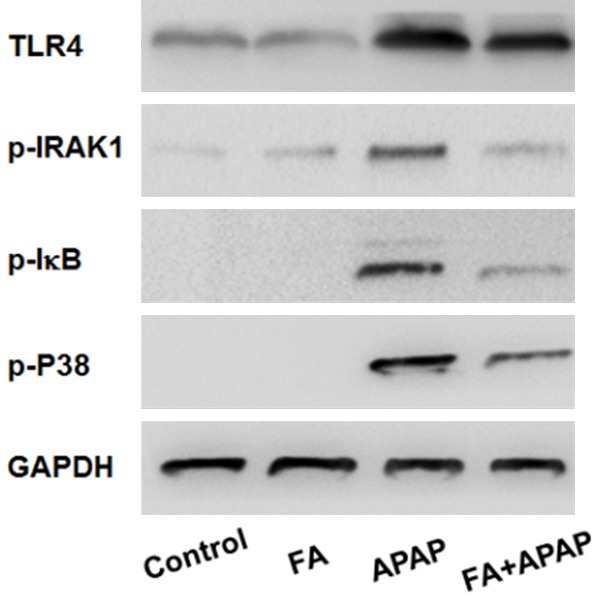
Pretreatment with FA suppressed the activation of TLR4 signaling in mice induced by APAP. Mice were given oral gavage of vehicle or FA (100 mg/kg) every 8 h one time for three times within 24 h before APAP (350 mg/kg) exposure and liver samples were harvested at 18 h after APAP administration. The protein levels of TLR4, p-IRAK1, p-IκB, p-p38 in liver tissue were determined by western blot analysis.
Discussion
APAP overdose, a leading cause of drug-induced acute liver failure, is remained a challenging issue because of limited therapeutic strategies besides liver transplantation and a lack of biomarkers indicating liver failure as early as possible [30]. At present, the explorations of natural produces increasingly establish a significant position for treating APAP-induced hepatotoxicity. Therefore, in the current study, we explored the effects of FA on APAP-induced acute liver injury. The data suggested that FA markedly attenuated liver damage in mice with APAP administration evidenced by decreased elevations of serum aminotransferase activities and improved pathologic changes.
For many years, the mechanism of hepatocellular death in APAP poisoning had focused heavily on necrosis. The release of cellular components from necrotic cells, for instance, HMGB1 and mitochondrial DNA, is responsible for evoking sterile inflammation secondary to the initiation of innate immune system [30-32]. Our results revealed severe cellular necrosis with APAP exposure by histological analysis was attenuated by FA treatment. In addition, apoptosis, associated with the release of caspase-cleaved KI8, is also a mechanism which only occupies a small part of cell death in APAP-induced hepatotoxicity [30,33]. The activation of caspase, the central downstream signaling of apoptosis, is still controversial in APAP poisoning. Several literatures revealed that there was no relevant caspase activation with or without caspase inhibitors in APAP model [34,35]. Further supporting evidences from the research that demonstrated no caspase 3 activation in patients at an overdose of APAP by analyzing the activity of caspase 3 or the protein level of the active caspase 3 [32]. In contrast, a research showed caspase 3 activity was measurable in plasma in galactosamine-induced apoptosis in rat liver [36]. In the present study, enhanced apoptotic cells and caspase 3 activity with APAP administration by TUNEL staining and the determination of caspase 3 activity were shown, which might be relevant to that the mice were fasted before APAP treatment. Starvation makes mice more susceptible to APAP-induced apoptosis owing to lower ATP content [37]. Furthermore, FA pretreatment suppressed APAP-induced apoptosis.
CYP 2E1 is the primary member of CYP450 that oxidizes APAP to NAPQI [26]. Limited CYP 2E1 is essential for metabolism and biosynthetic pathways. However, the activity and the content of CYP 2E1 are variable with stimulation such as alcohol and drugs. Previous researches demonstrated APAP-induced acute liver injury was dependent on the induction of CYP 2E1 and the damage was further aggravated by the increased CYP 2E1 with alcohol stimulation [26,38]. However, tea polyphenols alleviated APAP-induced hepatotoxicity via the suppression of CYP 2E1 [39]. Overdosed APAP elevates the expression of CYP 2E1 and promotes the percentage of CYP 2E1 metabolic pathway and then leads to overproduction of APAP protein adducts. The results from western blot and qRT-PCR in this manuscript revealed FA reversed the increased of CYP 2E1 after APAP administration. These findings demonstrated an underlying mechanism of FA protective effect on APAP hepatotoxicity is a direct influence on APAP metabolism by inhibiting CYP 2E1. Interestingly, CCl4 induced liver injury is also dependent on CYP 2E1, therefore, the suppression of CYP 2E1 after FA treatment may be responsible for the decreased liver damage induced by CCl4.
The oxidative stress triggered by the formation of covalent binding after NAPQI overload plays a central role in APAP liver injury and ROS is a key mediator for this progress [28]. In general, ROS is scavenged by endogenous antioxidants, anti-oxidative enzymes and the antioxidant defense system mainly dependent on Nrf2 signaling [27,40]. Accumulation of ROS not only results in mitochondrial dysfunction and DNA damage, but also actives JNK and leads to subsequent translocation of JNK, which in turn further induces generation of ROS and ultimately evokes a large amount of cellular death and inflammation [41]. GSH is a critical antioxidant for NAPQI, ROS and peroxynitrite scavenging in APAP hepatotoxicity. NAC could alleviate APAP-induced liver injury is dependent on resynthesis of GSH. SOD and CAT are pivotal anti-oxidative enzymes for the detoxification of ROS. SOD is responsible for turning the superoxide anion into hydrogen peroxide which is further transformed by CAT [27]. Previous researches revealed FA could restore SOD and CAT activities in diabetic rats and in hypertensive rats [20]. In this study, the activities of SOD and CAT and the content of GSH were significantly reduced after APAP administration, however, FA treatment up-regulated both anti-oxidative enzymes and restored the GSH level. These findings indicates FA attenuates APAP-induced liver injury though decreased oxidative stress which was associated to down-regulated CYP 2E1, thus suppressed the APAP protein adducts generation and further balanced the pro-oxidative system and anti-oxidative system.
Activation of PRRs by DMAPs triggers acute inflammatory responses that amplify the damage in APAP-induced hepatotoxicity [42,43]. As the first identified PRRs, TLRs play a predominant role to active the innate immune. Evidences from published researches revealed that TLR4 and TLR9 were mainly involved in APAP model, which were primarily activated by HMGB1 and DNA, respectively [6,10,30,44]. TLR4, a founding member of TLRs, is the only TLR initiating the innate immune by MyD88-and TRIF-dependent signaling pathways, thus exerts a vital role to evoke inflammatory responses [9,45]. TLR4 recognizes DMAPs and PAMPs, mainly depends on MyD88, phosphorylate p38-MAPK and IκB, then activate the transcriptional factors of AP-1 and NF-κB, respectively, ultimately lead to the release of pro-inflammatory cytokines such as IL-1β and TNF-α and the recruitment of inflammatory cells. However, the role of TLR4 in APAP poisoning is controversial. A previous study demonstrated deficient TLR4 signaling protected against APAP-induced hepatotoxicity while an investigation found that TLR4 signaling seem to not be involved in APAP model with TLR4 knockout mice [10,46]. To address whether TLR4 signaling is related to the protection against APAP-induced liver injury, we first measured the protein level of TLR4 in liver tissue by western blot. The result demonstrated that FA treatment reduced APAP-induced TLR4 expression. Next, the phosphorylated levels of primary downstream signaling molecules mediated by TLR4 including IRAK1, IκB and p38 were determined to further validated TLR4 signaling involves in APAP-induced liver injury. We found that FA treatment inhibited the phosphorylation of these molecules. The findings indicated FA could block TLR4-mediated MAPK and NF-κB pathways. Furthermore, the main pro-inflammatory cytokines detected by ELISA showed the increased secretions of IL-1β and TNF-α in serum following the activation TLR4 after APAP treatment were reversed by FA treatment. Together, these data suggested that FA attenuates APAP hepatotoxicity though the suppression of inflammation modulated by MyD88-dependent TLR4 signaling pathway.
In conclusion, the protection of FA pretreatment against APAP induced acute liver injury was demonstrated and the underlying mechanism was investigated in this study. The data indicated that a directly suppressed influence on APAP metabolism as well as anti-inflammatory responses in TLR4-dependent manner are associated to the protective effects of FA in APAP-challenged mice. Our study supported that FA may be a novel candidate for APAP induced acute liver failure.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (NO. 81373870).
Disclosure of conflict of interest
None.
References
- 1.Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: Summary of a workshop. Hepatology. 2008;47:1401–1415. doi: 10.1002/hep.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet. 2010;376:190–201. doi: 10.1016/S0140-6736(10)60274-7. [DOI] [PubMed] [Google Scholar]
- 3.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 2003;31:1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 4.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 5.Tujios S, Fontana RJ. Mechanisms of drug-induced liver injury: from bedside to bench. Nat Rev Gastroenterol Hepatol. 2011;8:202–211. doi: 10.1038/nrgastro.2011.22. [DOI] [PubMed] [Google Scholar]
- 6.Maher JJ. DAMPs ramp up drug toxicity. J Clin Invest. 2009;119:246–249. doi: 10.1172/JCI38178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology. 2004;127:1760–1774. doi: 10.1053/j.gastro.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 8.Ju C. Damage-associated molecular patterns: their impact on the liver and beyond during acetaminophen overdose. Hepatology. 2012;56:1599–1601. doi: 10.1002/hep.25920. [DOI] [PubMed] [Google Scholar]
- 9.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 10.Yohe HC, O’Hara KA, Hunt JA, Kitzmiller TJ, Wood SG, Bement JL, Bement WJ, Szakacs JG, Wrighton SA, Jacobs JM, Kostrubsky V, Sinclair PR, Sinclair JF. Involvement of Toll-like receptor 4 in acetaminophen hepatotoxicity. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1269–1279. doi: 10.1152/ajpgi.00239.2005. [DOI] [PubMed] [Google Scholar]
- 11.Shah N, Montes de Oca M, Jover-Cobos M, Tanamoto K, Muroi M, Sugiyama K, Davies NA, Mookerjee RP, Dhar DK, Jalan R. Role of toll-like receptor 4 in mediating multiorgan dysfunction in mice with acetaminophen induced acute liver failure. Liver Transpl. 2013;19:751–761. doi: 10.1002/lt.23655. [DOI] [PubMed] [Google Scholar]
- 12.Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med. 2008;359:285–292. doi: 10.1056/NEJMct0708278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park K, Antoine DJ, Pirmohamed M. Treatment of paracetamol overdose: room for improvement? Lancet. 2014;383:672–674. doi: 10.1016/S0140-6736(13)62303-X. [DOI] [PubMed] [Google Scholar]
- 14.Ferner RE, Dear JW, Bateman DN. Management of paracetamol poisoning. BMJ. 2011;342:d2218. doi: 10.1136/bmj.d2218. [DOI] [PubMed] [Google Scholar]
- 15.Ramar M, Manikandan B, Raman T, Priyadarsini A, Palanisamy S, Velayudam M, Munusamy A, Marimuthu Prabhu N, Vaseeharan B. Protective effect of ferulic acid and resveratrol against alloxan-induced diabetes in mice. Eur J Pharmacol. 2012;690:226–235. doi: 10.1016/j.ejphar.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 16.Roy S, Metya SK, Sannigrahi S, Rahaman N, Ahmed F. Treatment with ferulic acid to rats with streptozotocin-induced diabetes: effects on oxidative stress, pro-inflammatory cytokines, and apoptosis in the pancreatic beta cell. Endocrine. 2013;44:369–379. doi: 10.1007/s12020-012-9868-8. [DOI] [PubMed] [Google Scholar]
- 17.Cui L, Zhang Y, Cao H, Wang Y, Teng T, Ma G, Li Y, Li K, Zhang Y. Ferulic acid inhibits the transition of amyloid-beta42 monomers to oligomers but accelerates the transition from oligomers to fibrils. J Alzheimers Dis. 2013;37:19–28. doi: 10.3233/JAD-130164. [DOI] [PubMed] [Google Scholar]
- 18.Jayaprakasam B, Vanisree M, Zhang Y, Dewitt DL, Nair MG. Impact of alkyl esters of caffeic and ferulic acids on tumor cell proliferation, cyclooxygenase enzyme, and lipid peroxidation. J Agric Food Chem. 2006;54:5375–5381. doi: 10.1021/jf060899p. [DOI] [PubMed] [Google Scholar]
- 19.Karthikeyan S, Kanimozhi G, Prasad NR, Mahalakshmi R. Radiosensitizing effect of ferulic acid on human cervical carcinoma cells in vitro. Toxicol In Vitro. 2011;25:1366–1375. doi: 10.1016/j.tiv.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Alam MA, Sernia C, Brown L. Ferulic acid improves cardiovascular and kidney structure and function in hypertensive rats. J Cardiovasc Pharmacol. 2013;61:240–249. doi: 10.1097/FJC.0b013e31827cb600. [DOI] [PubMed] [Google Scholar]
- 21.Senaphan K, Kukongviriyapan U, Sangartit W, Pakdeechote P, Pannangpetch P, Prachaney P, Greenwald SE, Kukongviriyapan V. Ferulic Acid Alleviates Changes in a Rat Model of Metabolic Syndrome Induced by High-Carbohydrate, High-Fat Diet. Nutrients. 2015;7:6446–6464. doi: 10.3390/nu7085283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HY, Lee SM. Ferulic acid attenuates ischemia/reperfusion-induced hepatocyte apoptosis via inhibition of JNK activation. Eur J Pharm Sci. 2012;45:708–715. doi: 10.1016/j.ejps.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 23.Kim HY, Park J, Lee KH, Lee DU, Kwak JH, Kim YS, Lee SM. Ferulic acid protects against carbon tetrachloride-induced liver injury in mice. Toxicology. 2011;282:104–111. doi: 10.1016/j.tox.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Kim EO, Min KJ, Kwon TK, Um BH, Moreau RA, Choi SW. Anti-inflammatory activity of hydroxycinnamic acid derivatives isolated from corn bran in lipopolysaccharide-stimulated Raw 264.7 macrophages. Food Chem Toxicol. 2012;50:1309–1316. doi: 10.1016/j.fct.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Das U, Manna K, Sinha M, Datta S, Das DK, Chakraborty A, Ghosh M, Saha KD, Dey S. Role of ferulic acid in the amelioration of ionizing radiation induced inflammation: a murine model. PLoS One. 2014;9:e97599. doi: 10.1371/journal.pone.0097599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SS, Buters JT, Pineau T, Fernandez-Salguero P, Gonzalez FJ. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J Biol Chem. 1996;271:12063–12067. doi: 10.1074/jbc.271.20.12063. [DOI] [PubMed] [Google Scholar]
- 27.Vatansever F, de Melo WC, Avci P, Vecchio D, Sadasivam M, Gupta A, Chandran R, Karimi M, Parizotto NA, Yin R, Tegos GP, Hamblin MR. Antimicrobial strategies centered around reactive oxygen species--bactericidal antibiotics, photodynamic therapy, and beyond. FEMS Microbiol Rev. 2013;37:955–989. doi: 10.1111/1574-6976.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, Bao W, Pavlidis S, Barnes PJ, Kanerva J, Bittner A, Rao N, Murphy MP, Kirkham PA, Chung KF, Adcock IM COPDMAP. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;136:769–780. doi: 10.1016/j.jaci.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antoniades CG, Khamri W, Abeles RD, Taams LS, Triantafyllou E, Possamai LA, Bernsmeier C, Mitry RR, O’Brien A, Gilroy D, Goldin R, Heneghan M, Heaton N, Jassem W, Bernal W, Vergani D, Ma Y, Quaglia A, Wendon J, Thursz M. Secretory leukocyte protease inhibitor: a pivotal mediator of anti-inflammatory responses in acetaminophen-induced acute liver failure. Hepatology. 2014;59:1564–1576. doi: 10.1002/hep.26933. [DOI] [PubMed] [Google Scholar]
- 30.Adebayo D, Mookerjee RP, Jalan R. Mechanistic biomarkers in acute liver injury: are we there yet? J Hepatol. 2012;56:1003–1005. doi: 10.1016/j.jhep.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 31.Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, Loike JD, Jenkins RE, Antoine DJ, Schwabe RF. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2015;125:539–550. doi: 10.1172/JCI76887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122:1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulze-Osthoff K, Bantel H. Necrosis versus apoptosis in acetaminophen-induced hepatotoxicity. Hepatology. 2011;53:1070. doi: 10.1002/hep.24027. [DOI] [PubMed] [Google Scholar]
- 34.Jaeschke H, Cover C, Bajt ML. Role of caspases in acetaminophen-induced liver injury. Life Sci. 2006;78:1670–1676. doi: 10.1016/j.lfs.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 35.Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci. 2002;67:322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- 36.Gujral JS, Farhood A, Jaeschke H. Oncotic necrosis and caspase-dependent apoptosis during galactosamine-induced liver injury in rats. Toxicol Appl Pharmacol. 2003;190:37–46. doi: 10.1016/s0041-008x(03)00154-6. [DOI] [PubMed] [Google Scholar]
- 37.Antoine DJ, Williams DP, Kipar A, Jenkins RE, Regan SL, Sathish JG, Kitteringham NR, Park BK. High-mobility group box-1 protein and keratin-18, circulating serum proteins informative of acetaminophen-induced necrosis and apoptosis in vivo. Toxicol Sci. 2009;112:521–531. doi: 10.1093/toxsci/kfp235. [DOI] [PubMed] [Google Scholar]
- 38.Wolf KK, Wood SG, Allard JL, Hunt JA, Gorman N, Walton-Strong BW, Szakacs JG, Duan SX, Hao Q, Court MH, von Moltke LL, Greenblatt DJ, Kostrubsky V, Jeffery EH, Wrighton SA, Gonzalez FJ, Sinclair PR, Sinclair JF. Role of CYP3A and CYP2E1 in alcohol-mediated increases in acetaminophen hepatotoxicity: comparison of wild-type and Cyp2e1(-/-) mice. Drug Metab Dispos. 2007;35:1223–1231. doi: 10.1124/dmd.107.014738. [DOI] [PubMed] [Google Scholar]
- 39.Chen X, Sun CK, Han GZ, Peng JY, Li Y, Liu YX, Lv YY, Liu KX, Zhou Q, Sun HJ. Protective effect of tea polyphenols against paracetamol-induced hepatotoxicity in mice is significantly correlated with cytochrome P450 suppression. World J Gastroenterol. 2009;15:1829–1835. doi: 10.3748/wjg.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Copple IM, Goldring CE, Jenkins RE, Chia AJ, Randle LE, Hayes JD, Kitteringham NR, Park BK. The hepatotoxic metabolite of acetaminophen directly activates the Keap1-Nrf2 cell defense system. Hepatology. 2008;48:1292–1301. doi: 10.1002/hep.22472. [DOI] [PubMed] [Google Scholar]
- 41.Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehendale HM. Once initiated, how does toxic tissue injury expand? Trends Pharmacol Sci. 2012;33:200–206. doi: 10.1016/j.tips.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 44.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119:305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 46.Cai C, Huang H, Whelan S, Liu L, Kautza B, Luciano J, Wang G, Chen G, Stratimirovic S, Tsung A, Billiar TR, Zuckerbraun BS. Benzyl alcohol attenuates acetaminophen-induced acute liver injury in a Toll-like receptor-4-dependent pattern in mice. Hepatology. 2014;60:990–1002. doi: 10.1002/hep.27201. [DOI] [PubMed] [Google Scholar]