

Abstract
Aggregation of free alpha-hemoglobin proteins forms harmful reactive oxygen radicals during the development of normal erythroid cell, which can be prevented by a chaperone, alpha hemoglobin stabilizing protein (AHSP). Mutations at the AHSP gene may affect its interacting ability with other globin proteins. Various state-of-the-art tools have been extensively used to identify the most deleterious nsSNPs at the AHSP and their pathogenic effect during AHSP-globin interaction. Comprehensive analysis revealed that the V56G of the AHS protein is the most pathogenic amino acid substitution, agreed consistently and significantly (P=1.27E-13) by all the state-of-the-art tools (PROVEAN <-2.5, SIFT=0, SNAP2 >50, SNPs&GO >0.5, PolyPhen >0.5, FATHMM >0.6, PANTHER <-3, VEST P<0.05) and protein-protein interaction analysis. The V56G exists near the hot spot and was found to be the highly pathogenic and it forms an extra helix on mutation. The unchaperoned HBA2 and KLF1 proteins with the AHSP mutant (V56G) chains denote the non-interactive nature. Binding energies were significantly varied upon highly deleterious mutation at AHSP and/or HBA1 gene. The study endorses the mutated AHSP protein, p.val56Gly for detailed confirmatory wet lab analysis.
Keywords: α-Hemoglobin stabilizing protein (AHSP), nsSNPs, bioinformatics, protein-protein interaction, globin genes, mutation, molecular modeling, interface residues, interaction sites
Introduction
Aggregation of free alpha-globin proteins forms toxic reactive oxygen radicals, the phenomenon is prevented by a chaperone, alpha hemoglobin stabilizing protein (AHSP) during the development of normal erythroid cell [1,2]. In beta-thalassemia patents lack/reduced synthesis of β globin chains leaving free alpha globin protein, which can causes mature red cells hemolysis and premature death of erythroid precursors [2,3]. Alpha-globin/non-alpha globin protein imbalance reflects in the severity of thalassemia [2,4]. Comprehensive studies on the effect of the non-synonymous SNPs in the AHSP gene and their impact on the alpha globin-AHSP interaction is needed to device their direct and indirect impact. Wet lab experimental methods to identify the protein-protein interactions are expensive and tedious; it can be unraveled cost effectively by in-silico approaches. In the study we aimed to use various state-of-the-art tools extensively to understand the effect of nsSNPs on the structural and functional impacts to categorize the most deleterious nsSNPs at the AHSP and their pathogenic effect during AHSP-globin interaction.
Materials and methods
Datasets and SNP retrieval
AHSP, HBA1, HBA2, HBB, HBD, KLF1 and HBQ1 gene sequences were downloaded during December 2015 from NCBI [5,6]. The protein sequences of the HBA1, HBA2, HBB, HBD, KLF1, HBQ1 and AHSP proteins were retrieved by limiting our search only to human from RCSB Protein Data Bank (PDB ID: 1a00, 1hbb, 2w72, 5bnv, 2l2i.1.A, P09105 and Q9NZD4) [6]. The dbSNPs of AHSP gene was retrieved from NCBI. The non-synonymous SNPs (nsSNPs) of AHSP gene were screened to identify their damaging effects on AHSP protein.
Mutation effect prediction
The state-of-art-tools such as SIFT (Sorts intolerant from tolerant) [7], PolyPhen 2.0 (Polymorphism Phenotyping v2) [8], PROVEAN (Protein variation effect analyzer) [9], SNAP2 (Screening for Non-Acceptable Polymorphisms) [10], SNPs&GO (Single nucleotide polymorphisms and Gene Ontology) [11,12], PANTHER [13], FATHMM (Functional Analysis through Hidden Markov Models) [14] and VEST3 (Variant Effect Scoring Tool) [15] were used to predict the effect of the substitution mutations on the AHSP gene. The standard cutoff or the threshold values for PROVEAN (<-2.5, SIFT (=0), SNAP2 (>50), SNPs&GO (>0.5), PolyPhen (>0.5), FATHMM (>0.6), PANTHER (<-3), VEST (P<0.05) were maintained to predict the effect of change in amino acid sequence in the biological function, solvent accessibility, and structure of the ANSP protein [7-15].
Structure modeling and predicting residue positions
The 3D structures (resolution 2.80 Å) of the native and the mutated AHSP chains were designed based on template PDB ID: 1a00 to evaluate the stability of mutant using automated homology modeling tool such as SWISS MODEL [16-18]. The designed AHSP 3D structure was validated using PROCHECK [19]. The Swiss-Pdb Viewer software was used to generate mutated AHSP models using the validated AHSP 3D structure [20]. The native and mutated AHSP 3D models were subjected for energy minimization using the GROMACS program [21]. FASTA format of the AHSP protein sequence were provided to FlexPred to identify the solvent accessibility of AHSP protein to predict conformational switches and residue positions involved in kinetic energy and pathogenic disorders.
Visualize the nsSNPs location
Three-dimensional (3D) crystal structure of AHSP protein corresponding to the ID: 1Y01 was generated using the muPIT interactive [22]. The binding nature of the AHSP protein with Fe (II) alpha-hemoglobin was checked. All the 13 nsSNPs listed in the Table 1 was given as input to visualize the location of the substitutions at AHSP.
Table 1.
Possible pathogenic non-synonymous substitution mutations in the ANSP gene predicted using various state-of-art-tools
| S. No. | SNP | Coordinate | Amino Acid change | SIFT score | SIFT prediction | PolyPhen Score | Prediction | FATHMM Coding Score | PANTHER subPSEC | PROVEAN score | PREDICTION (cutoff=-2.5) | SNAP2 Score | Predicted Effect | SNPs&GO Effect | VEST p-value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs144861094 | 31539497 | A13P | 0.073 | TOLERATED | 0.028 | BENIGN | 0.06082 | -2.43754 | -2.04 | Neutral | -43 | Neutral | Neutral | 0.2587 |
| 2 | rs201919859 | 31539509 | E17K | 0.054 | TOLERATED | 0.936 | PROBABLY DAMAGING | 0.76085 | -2.48368 | -3.47 | Deleterious | 50 | Effect | Disease | 0.0043 |
| 3 | rs372264195 | 31539994 | H97Q | 0.113 | TOLERATED | 0 | UNKNOWN | 0.1351 | -1.15018 | -1.92 | Neutral | 12 | Effect | Neutral | 0.5607 |
| 4 | rs147251409 | 31539897 | K65M | 0.032 | DELETERIOUS | 0.329 | POSSIBLY DAMAGING | 0.03781 | -3.40556 | -4.07 | Deleterious | 30 | Effect | Neutral | 0.3437 |
| 5 | rs140200160 | 31539522 | L21P* | 0 | DELETERIOUS | 0.999 | PROBABLY DAMAGING | 0.8301 | -4.87126 | -6.24 | Deleterious | 83 | Effect | Disease | 0.0061 |
| 6 | rs10920 | 31539837 | M45K | 0.008 | DELETERIOUS | 0.001 | BENIGN | 0.8711 | -3.16676 | -3.72 | Deleterious | 73 | Effect | Disease | 0.0229 |
| 7 | rs75782426 | 31539927 | N75I | 0.05 | TOLERATED | 0.902 | PROBABLY DAMAGING | 0.05718 | -2.70452 | -6.02 | Deleterious | 6 | Effect | Neutral | 0.3279 |
| 8 | rs36018996 | 31540001 | P100T | 0 | DELETERIOUS | 0 | UNKNOWN | 0.15999 | -2.99152 | -4.03 | Deleterious | 76 | Effect | Neutral | 0.5319 |
| 9 | rs147349976 | 31539999 | P99L | 1 | TOLERATED | 0 | UNKNOWN | 0.00783 | -0.84752 | -0.33 | Neutral | 25 | Effect | Neutral | 0.6600 |
| 10 | rs142369727 | 31539929 | T76A | 0.667 | TOLERATED | 0.001 | BENIGN | 0.04407 | -1.23877 | -2.2 | Neutral | 26 | Effect | Neutral | 0.8676 |
| 11 | rs200722385 | 31539824 | V41L | 0.154 | TOLERATED | 0.002 | BENIGN | 0.46605 | -2.61048 | -2.46 | Neutral | 28 | Effect | Disease | 0.2642 |
| 12 | rs186590045 | 31539870 | V56G* | 0.01 | DELETERIOUS | 0.932 | PROBABLY DAMAGING | 0.68878 | -3.77106 | -5.07 | Deleterious | 69 | Effect | Disease | 0.0162 |
| 13 | rs372200025 | 31539957 | Y85C* | 0 | DELETERIOUS | 1 | PROBABLY DAMAGING | 0.87283 | -5.22604 | -8.84 | Deleterious | 76 | Effect | Disease | 0.0083 |
Highly pathogenic nsSNPs of ANSP gene agreed unanimously as deleterious by all the tools.
Shaded cells indicate the deleterious nsSNPs predicted by the respective state-of-art-tool. Threshold values: PROVEAN (<-2.5, SIFT (=0), SNAP2 (>50), SNPs&GO (>0.5), PolyPhen (>0.5), FATHMM (>0.6), PANTHER (<-3), VEST (P<0.05).
AHSP and globin interaction
We applied a comprehensive protein-protein interaction prediction structure and modeling assembly tool named PRISM for modeling the interactions between the AHSP and other proteins [23]. Two different input sets such as the template and the target sets were provided to the PRISM algorithm to obtain interaction predictions between them. HBA1, HBA2, HBB, HBD, KLF1 and HBQ1 proteins were included in the study; selection was based on the reported interaction in STRING 10 [4]. PRISM explores the hot spots of the interface between the target and template by analyzing the geometrical and evolutionary conservation of both the proteins using the 3D structures of the proteins [24].
Statistical analysis
The correlations between the predictions of various bioinformatics tools were carried out using SPSS. Significance between the predictions of various state-of-art-tools was tested using student T-test. Probabilities were maintained at P<0.0001 for most significant combinations.
Results
A total of 244 SNPs at AHSP gene were retrieved from the dbSNP including 53 nsSNPs. All the nsSNPs were subjected for the prediction analysis using various effective state of art bioinformatics tools. The prediction from PROVEAN (<-2.5, SIFT (=0), SNAP2 (>50), SNPs&GO (>0.5), PolyPhen (>0.5), FATHMM (>0.6), PANTHER (<-3), VEST (P<0.05) were found to be highly significant (P=1.27E-13 of single factor ANOVA test). Heatmap of AHSP protein generated using SNAP2 reveals most of the substitution with dark red corresponding to a strong signal for high level of pathogenicity (Figure 1). The correlations between the predictions of the tools were found to be varied (Figure 1). Prediction between two state-of-art-tools were significant at P<0.0001 (student T-test) for most the combinations (Figure 1). All the state-of-the-art tools (PROVEAN <-2.5, SIFT=0, SNAP2 >50, SNPs&GO >0.5, PolyPhen >0.5, FATHMM >0.6, PANTHER <-3, VEST P<0.05) have consistently and significantly agreed the pathogenicity of V56G, L21P and Y85C substitution of the AHS protein (P=1.27E-13) (Table 1).
Figure 1.
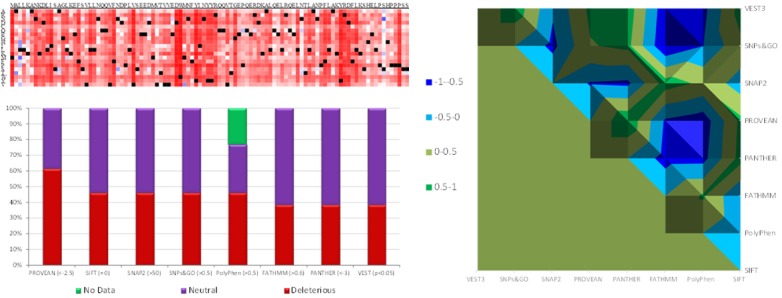
Heatmap of AHSP protein. Top Left: Heatmap of AHSP protein generated using SNAP2. Dark red: Strong signal for high level pathogenicity. Bottom Left: Pathogenicity of nsSNPs in the predicted using various state-of-art-tools. A value in the parenthesis indicates the threshold value for pathogenicity. Right: Surface chart of correlation between the predictions of pathogenicity of nsSNPs in AHSP gene by various state-of-art-tools.
Three-dimensional (3D) crystal structure of AHSP protein (ID: 1a00) was generated and its binding site with Fe(II) alpha-hemoglobin was checked using the muPIT interactive (Figure 2). Total of 80.39% of amino acids (R=rigid=82) in the AHSP protein are not flexible enough to participate in the processes of interaction as per the output from FlexPred. All the 13 nsSNPs were given as input to visualize the location of the substitutions at AHSP. Identification of location of various nsSNPs using muPIT interactive exposed the position of the V56G, which is near the physical contact site or hot spot between AHSP and HBA1 (Figure 2). The nsSNP, V56G exists near the interactive area and was found to be the highly pathogenic (Figure 3; Table 1). As an extra helix was formed due to the V56G mutation in the AHSP gene, it blocks some of the protein to be engaged physically (Figure 3). AHSP proteins always act with various type globin proteins (Figure 4). It was highly evident from the interaction of AHSP with HBA2 and KLF1 proteins, that the physical contact between AHSP with V56G and HBA2 or KLF1 was found to be completely absent (Figure 5). Interface energy and predicted hot spots interface residues of the template with pathogenic substitution at target-template protein complexes are enormously varied when compare with the wild template-wild target protein complex (Supplementary Table 1; Figure 6). Leucine at the position 21 is involved in the binding of HBA1 and AHSP. No involvement was observed for valine and tyrosine at position 56 and 85 respectively (Supplementary Table 1). Significant (>100%) variations in binding energies were observed in the interaction between wild HBA1 with mutated AHSP gene. Similar significant (>100%) variations were noted among the highly deleterious mutation of HBA1 (G60V and W15R) and wild type AHSP (Figure 6).
Figure 2.
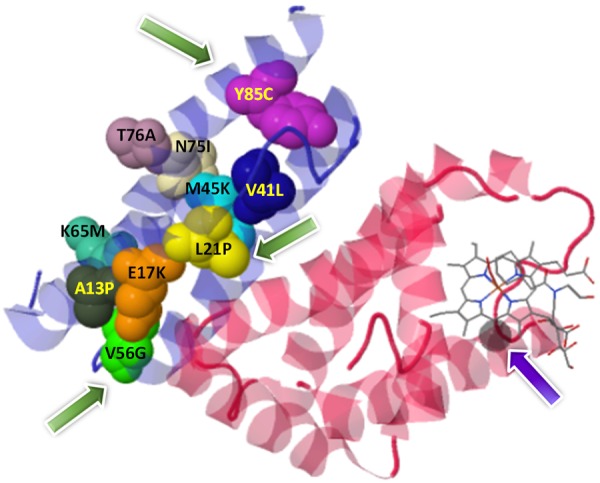
Three-dimensional (3D) protein structure of the α-Hemoglobin stabilizing protein (chain colored blue) complex with hemoglobin alpha chain (chain colored red). Green arrow: Most deleterious substitutions. Violet arrow: Iron (Fe) atom.
Figure 3.
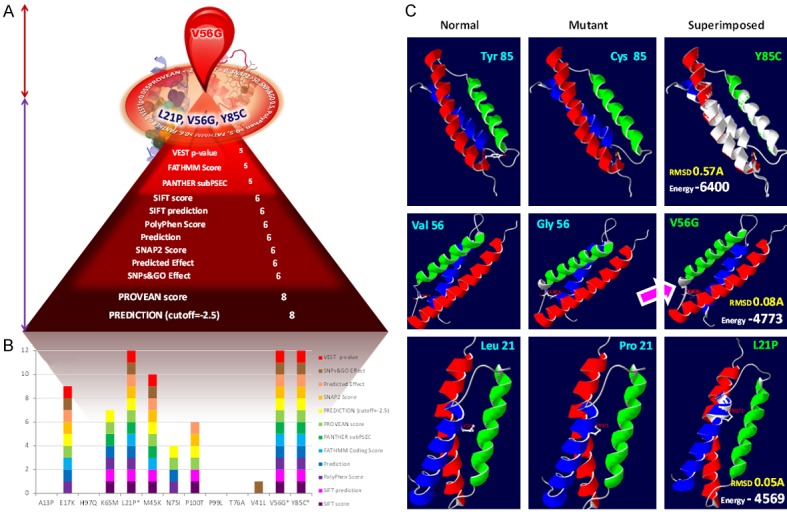
Graphic illustration of the state of art tools applied for the accurate detection of the highly pathogenic nsSNPs of AHSP gene. A: SNPs on the circle: The most deleterious nsSNPs of ANSP gene agreed consistently by all the state of art tools. Nut brown colored double headed arrow region: Region holds the substitution highly pathogenic as per the structural and protein-protein interaction analysis. Violet colored double headed arrow region: Number of deleterious SNPs by a particular tool. B: Deleterious effect of each substitution. Number of blocks corresponds to the number of tools agreed as deleterious. C: Superimposed models of AHSP normal and mutant. Arrow locates the extra helix due to the V56G mutation in the AHSP gene. Pink arrow indicates the resulted additional helix due to the glycine substitution at the 56th position.
Figure 4.
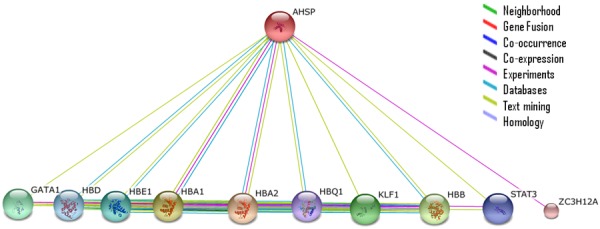
AHSP interacts with other proteins (using STRING 10).
Figure 5.
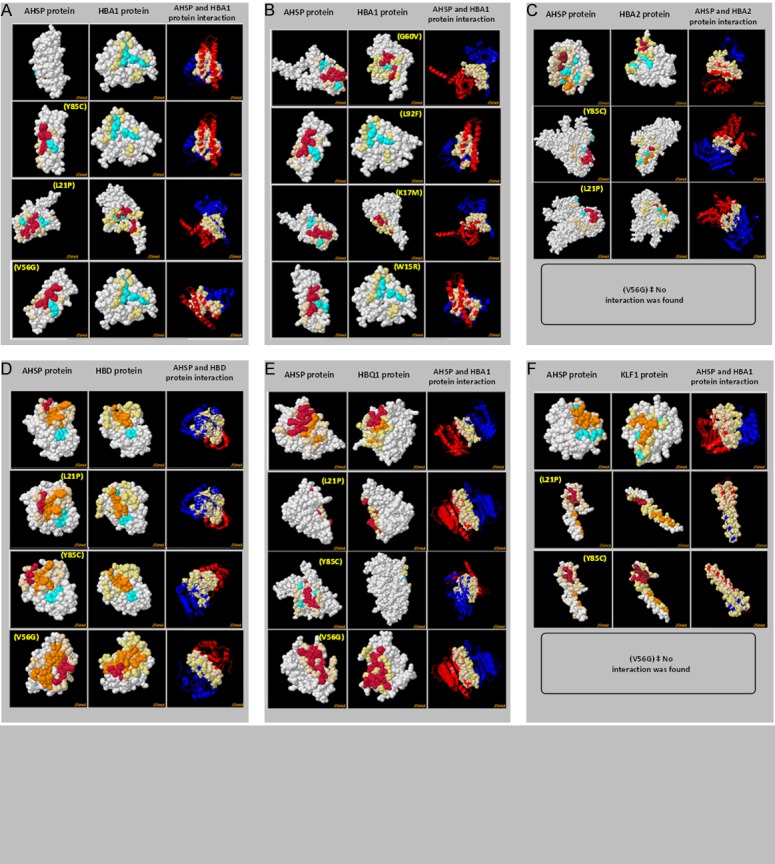
AHSP protein and globin protein interaction. Colum number 1 of each box indicates the template AHSP protein, column 2 denotes the target globin protein and text in parenthesis indicate the amino acid substitution. Column number 3 indicates the complex of the target protein-template protein interaction of the protein in the respective rows. A: Interaction between wild/mutated AHSP and native HBA1. B: Interaction between native AHSP and mutated HBA1. C: Interaction between mutated AHSP and native HBA2. D: Interaction between mutated AHSP and native HBD. E: Interaction between mutated AHSP and native HBQ1. F: Interaction between mutated AHSP and native KLF1.
Figure 6.
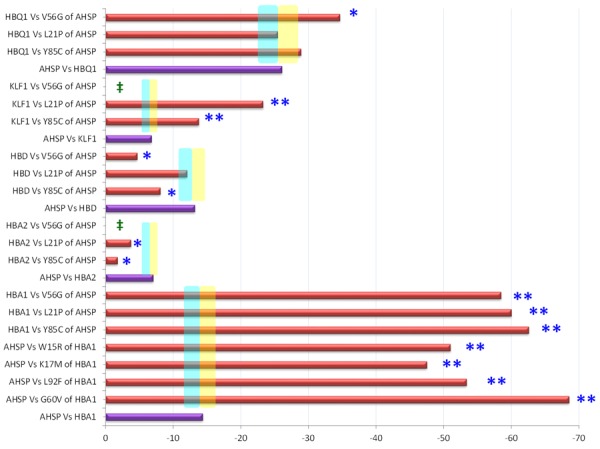
Binding energy of the template-target protein complex. ‡No interaction was found. *Less significantly at 10% varied binding energy compare to the wild type interaction. Shades colored sky blue and yellow indicates +10% and -10% variation compare to the wild type. **More Significant at >100% varied binding energy.
Discussion
The AHSP and the HBA1 genes are almost similar in size, but there were 201 non-coding synonymous (nsSNPs) retrieved in the HBA1 genes [25], while in the present study we have retrieved only 53 nsSNPs in the AHSP gene it indicates clearly and confirm that the AHSP gene is highly conserved during the evolution. As we shown in the previous study [25], the selected tools and their threshold values, PROVEAN (<-2.5), SIFT (=0), SNAP2 (>50), SNPs&GO (>0.5), PolyPhen (>0.5), FATHMM (>0.6), PANTHER (<-3), VEST (P<0.05) are adequate to calculate and predict the deleterious nature of the nsSNPs as they cover the needed attributes such as structural and sequence also the conservation analysis [7-15,25]. All the state-of-the-art tools were unanimously agreed (P=1.27E-13) the pathogenicity of V56G, L21P and Y85C amino acid substitutions at AHSP based on the calculations such as homology-based analysis, position-specific independent count score, sequence clustering and alignment-based scoring, Hidden Markov Models, Variant Effect Scoring and unfolding Gibbs free energy score [7-15].
A well-known fact is that the free alpha globin chains always unstable, it is stabilized by a chaperone AHSP, and prevents its precipitation, which is toxic to the body [18,26-28]. Sequence changes in the HBA1 and HBA2 genes are more common [29,30] their effect on the HBA1-AHSP or HBA2-AHSP interaction would disclose the adverse effects. Detailed study on 30 point mutants by Feng et al. [18] revealed the adverse effect of three amino acid substitutions (Lys99, His103 and Phe117) in alpha globin chain. Mutations such Hb Constant Spring, Hb Pakse, and cod 117 (GH5) in the alpha globin were reported to be affecting the AHSP-HBA1 interactions significantly [28,31,32]. We have taken a comprehensive computational step to identify the interaction difficulties of the most pathogenic HBA1 nsSNP mutations (W15R, K17M, L92F and G60V) [25] with the wild and mutated AHSP proteins. Non-interaction between the AHSP with V56G vs HBA2 and AHSP with V56G vs KLF1 indicates the negative impact of the mutation. V56G was tested in wet lab by Feng et al. [18] while L21P and Y85C were not. Here we report the pathogenicity of L21P and Y85C through a collective, comprehensive and computational approach.
The hot regions are tightly packed regions in protein-protein interfaces, have the most prominent cooperative behavioral property to interpret the protein-protein interface and their stability [24,33-35]. Binding energies were significantly varied upon highly deleterious mutation at AHSP and/or HBA1 gene. A threshold of 10% variation in the binding energy has been set as less significant by Chen et al. [36] to identify the very little influence and sensitivity of the binding energy on amino acid substitution. While we have set 100% variation as the threshold to predict the most influential effect of the amino acid substitution on the binding energy, which disclosed the most deleterious effect of the amino acid substitution among the 47.36% of tested combinations (Figure 6). Decrease in the binding energy is a prominent value that could justify the pathogenic effect of mutations as described earlier by Thorn and Bogan [37], which was reflected in 47.36% of tested combinations (Figure 6). Furthermore, the V56G substitution is not participating directly in the host spot binding region, while it affects the AHSP interaction badly with HBA1 and KLF1 proteins.
The ethical approval was obtained from the University of Dammam (IRB-2014-08-041) for the study to identify the prevalence of mutations at AHSP gene in Saudi population. Blood samples were collected from transfusion dependent Saudi beta thalassemia major patients (n=100) and healthy controls (n=100) after getting informed consent. All the samples were subjected for the sequencing of the AHSP gene. Interestingly we have identified the most pathogenic mutations AHSP:c.167T>G (V56G) in three (1♂ patient and 2♂ controls) subjects. Furthermore, three other mutations were also identified in the coding regions such as AHSP:c.231G>T (L77L), AHSP:c.45G>T (L15F), and AHSP:c.168G>T (V56V). All these samples were screened for the presence mutation in the KLF1, HBA1, HBA2 and HBB genes [29,38,39]. Subjects with the AHSP gene mutation were found to be free from KLF1 gene mutations, hence we couldn’t confirm the predictions. We have also identified the co-inheritance of AHSP:c.167T>G (V56G) and -α2 3.7/α1α2. Moreover, a large scale study on the subjects with AHSP:c.167T>G (V56G) may give the actual influence of the mutation. At the end of the story we could say that the limitation of the study is that the computational results needs to be taken for the wet lab analysis to confirm the results.
In conclusion we could say that the glycine substitution at the 56th position (V56G) is the most pathogenic. Replacing wet lab by web lab is not completely advisable, but can be considered to reduce the cost and time by selecting best options to proceed to wet lab. Furthermore, the study endorses the mutated AHSPV56G protein for detailed confirmatory wet lab analysis. We are in the stage of proposing possible ways to confirm the results in wet lab.
Acknowledgements
The authors would like to extend our gratitude to the King Abdulaziz City for Science and Technology and the Deanship of Scientific Research, University of Dammam (To JFB, 2014024) for the funding of this project. Authors would also like to thank Mr. Ranilo M. Tumbaga, Mr. Horace T. Pacifico, Ms. Jee E. Aquino, Mr. Edwardson G. Evangelista and Mr. Mohammed Al-Shmalan for their technical work.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Mahmoud HM, Shoeib AA, Abd El Ghany SM, Reda MM, Ragab IA. Study of alpha hemoglobin stabilizing protein expression in patients with beta thalassemia and sickle cell anemia and its impact on clinical severity. Blood Cells Mol Dis. 2015;55:358–362. doi: 10.1016/j.bcmd.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 2.Sagar CS, Kumar R, Sharma DC, Kishor P. Alpha hemoglobin stabilizing protein: its causal relationship with the severity of beta thalassemia. Blood Cells Mol Dis. 2015;55:104–107. doi: 10.1016/j.bcmd.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Schrier SL. Pathophysiology of thalassemia. Curr Opin Hematol. 2002;9:123–126. doi: 10.1097/00062752-200203000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodsell DS, Dutta S, Zardecki C, Voigt M, Berman HM, Burley SK. The RCSB PDB “molecule of the month”: inspiring a molecular view of biology. PLoS Biol. 2015;13:e1002140. doi: 10.1371/journal.pbio.1002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4:1073–1082. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 8.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hecht M, Bromberg Y, Rost B. Better prediction of functional effects for sequence variants. BMC Genomics. 2015;16(Suppl 8):S1. doi: 10.1186/1471-2164-16-S8-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat. 2009;30:1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 12.Capriotti E, Calabrese R, Fariselli P, Martelli PL, Altman RB, Casadio R. WS-SNPs&GO: a web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genomics. 2013;14(Suppl 3):S6. doi: 10.1186/1471-2164-14-S3-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, Gaunt TR. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter H, Douville C, Stenson PD, Cooper DN, Karchin R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics. 2013;14(Suppl 3):S3. doi: 10.1186/1471-2164-14-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng L, Gell DA, Zhou S, Gu L, Kong Y, Li J, Hu M, Yan N, Lee C, Rich AM, Armstrong RS, Lay PA, Gow AJ, Weiss MJ, Mackay JP, Shi Y. Molecular mechanism of AHSP-mediated stabilization of alpha-hemoglobin. Cell. 2004;119:629–640. doi: 10.1016/j.cell.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 19.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck-a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–291. [Google Scholar]
- 20.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 21.Lindahl E, Azuara C, Koehl P, Delarue M. NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 2006;34:W52–56. doi: 10.1093/nar/gkl082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niknafs N, Kim D, Kim R, Diekhans M, Ryan M, Stenson PD, Cooper DN, Karchin R. MuPIT interactive: webserver for mapping variant positions to annotated, interactive 3D structures. Hum Genet. 2013;132:1235–1243. doi: 10.1007/s00439-013-1325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baspinar A, Cukuroglu E, Nussinov R, Keskin O, Gursoy A. PRISM: a web server and repository for prediction of protein-protein interactions and modeling their 3D complexes. Nucleic Acids Res. 2014;42:W285–289. doi: 10.1093/nar/gku397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tuncbag N, Gursoy A, Nussinov R, Keskin O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat Protoc. 2011;6:1341–1354. doi: 10.1038/nprot.2011.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.AbdulAzeez S, Borgio JF. In-Silico computing of the most deleterious nsSNPs in HBA1 gene. PLoS One. 2016;11:e0147702. doi: 10.1371/journal.pone.0147702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khafizov K, Ivanov MV, Glazova OV, Kovalenko SP. Computational approaches to study the effects of small genomic variations. J Mol Model. 2015;21:251. doi: 10.1007/s00894-015-2794-y. [DOI] [PubMed] [Google Scholar]
- 27.Feng L, Zhou S, Gu L, Gell DA, Mackay JP, Weiss MJ, Gow AJ, Shi Y. Structure of oxidized alpha-haemoglobin bound to AHSP reveals a protective mechanism for haem. Nature. 2005;435:697–701. doi: 10.1038/nature03609. [DOI] [PubMed] [Google Scholar]
- 28.Lacerra G, Scarano C, Musollino G, Flagiello A, Pucci P, Carestia C. Hb Foggia or alpha 117(GH5)Phe ->Ser: a new alpha 2 globin allele affecting the alpha Hb-AHSP interaction. Haematologica. 2008;93:141–142. doi: 10.3324/haematol.11789. [DOI] [PubMed] [Google Scholar]
- 29.Borgio JF, AbdulAzeez S, Al-Nafie AN, Naserullah ZA, Al-Jarrash S, Al-Madan MS, Al-Muhanna F, Steinberg MH, Al-Ali AK. A novel HBA2 gene conversion in cis or trans: “alpha12 allele” in a Saudi population. Blood Cells Mol Dis. 2014;53:199–203. doi: 10.1016/j.bcmd.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Borgio JF. Molecular nature of alpha-globin genes in the Saudi population. Saudi Med J. 2015;36:1271–1276. doi: 10.15537/smj.2015.11.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vasseur-Godbillon C, Marden MC, Giordano P, Wajcman H, Baudin-Creuza V. Impaired binding of AHSP to alpha chain variants: Hb groene hart illustrates a mechanism leading to unstable hemoglobins with alpha thalassemic like syndrome. Blood Cells Mol Dis. 2006;37:173–179. doi: 10.1016/j.bcmd.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Turbpaiboon C, Limjindaporn T, Wongwiwat W, U-Pratya Y, Siritanaratkul N, Yenchitsomanus PT, Jitrapakdee S, Wilairat P. Impaired interaction of alpha-haemoglobin-stabilising protein with alpha-globin termination mutant in a yeast two-hybrid system. Br J Haematol. 2006;132:370–373. doi: 10.1111/j.1365-2141.2005.05865.x. [DOI] [PubMed] [Google Scholar]
- 33.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 34.Cukuroglu E, Gursoy A, Keskin O. Analysis of hot region organization in hub proteins. Ann Biomed Eng. 2010;38:2068–2078. doi: 10.1007/s10439-010-0048-9. [DOI] [PubMed] [Google Scholar]
- 35.Liu T, Whitten ST, Hilser VJ. Functional residues serve a dominant role in mediating the cooperativity of the protein ensemble. Proc Natl Acad Sci U S A. 2007;104:4347–4352. doi: 10.1073/pnas.0607132104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen DE, Willick DL, Ruckel JB, Floriano WB. Principal component analysis of binding energies for single-point mutants of hT2R16 bound to an agonist correlate with experimental mutant cell response. J Comput Biol. 2015;22:37–53. doi: 10.1089/cmb.2014.0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thorn KS, Bogan AA. ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics. 2001;17:284–285. doi: 10.1093/bioinformatics/17.3.284. [DOI] [PubMed] [Google Scholar]
- 38.Borgio JF, AbdulAzeez S, Naserullah ZA, Al-Jarrash S, Al-Ali RA, Al-Madan MS, Al-Muhanna F, Al-Suliman AM, Al-Nafie A, Steinberg MH, Al-Ali AK. Mutations in the beta-globin gene from a Saudi population: an update. Int J Lab Hematol. 2016;38:e38–40. doi: 10.1111/ijlh.12463. [DOI] [PubMed] [Google Scholar]
- 39.Singleton BK, Burton NM, Green C, Brady RL, Anstee DJ. Mutations in EKLF/KLF1 form the molecular basis of the rare blood group In(Lu) phenotype. Blood. 2008;112:2081–2088. doi: 10.1182/blood-2008-03-145672. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.