

Abstract
Background: Previous findings indicate that testosterone level is negatively correlated with the incidence and mortality of cardiovascular diseases in men. Endothelial progenitor cells (EPCs) play a critical role in endothelial healing and vascular integrity. This study aimed to examine the effects of dihydrotestosterone (DHT), an active metabolite of testosterone, on human EPC function and investigate the underlying mechanism. Methods: EPCs were isolated from peripheral blood of healthy adult males and incubated with a series of concentrations (1, 10, and 100 nmol/L in dimethyl sulfoxide) of DHT for 24 h or with 10 nmol/L DHT for different periods (6, 12, 24, 36, and 48 h). EPC proliferation, migration, and adhesion were determined by MTT assay, modified Boyden chamber assay, and cell counting, respectively. Furthermore, vascular endothelial growth factor (VEGF) production was examined by ELISA, RhoA activity was determined through pull-down assay. The protein level of RhoA was quantified by Western blot analysis. Results: DHT significantly increased the proliferative, migratory, and adhesive abilities of EPCs in a dose- and time-dependent manner and upregulated the levels of VEGF and activated RhoA. However, RhoA inhibitor C3 exoenzyme or ROCK inhibitor Y-27632 significantly inhibited DHT-induced proliferation, migration, and adhesion, as well as VEGF production. Moreover, C3 exoenzyme inhibited the activation of RhoA stimulated by DHT. Conclusions: DHT promotes EPC proliferation, migration, and adhesion activities via RhoA/ROCK pathway.
Keywords: Endothelial progenitor cells, dihydrotestosterone, cardiovascular disease, RhoA/ROCK pathway
Introduction
Reconstitution and maintenance of an intact endothelial layer play a fundamental role in the prevention of cardiovascular diseases (CVDs) [1-4]. Risk factors such as hypertension, hyperlipidemia, insulin resistance, obesity, metabolic syndrome, excessive drinking and smoking, and diabetes are thought to significantly contribute to endothelial dysfunction and atherosclerosis [5-11]. Recent studies have demonstrated that an injured endothelial monolayer can be restored not only by resident cells within the wounded vascular wall but also by circulating endothelial progenitor cells (EPCs) [12-15].
As a circulating pool of cells, EPCs originally reside in the bone marrow and other putative niches and migrate into peripheral circulation and home into the sites of endothelial injury in response to many stimuli of endothelial repair and neovascularization [16]. In addition, EPCs can differentiate into mature endothelial cells [17,18]. Although the effects of androgens on EPCs are controversial, numerous studies have demonstrated that androgen level is associated with EPC function in men, and immunohistochemical analysis showed that EPCs express a testosterone receptor. Furthermore, a synthetic androgen called methyltrienolone (R1881) can augment the proliferation, migration, and colony formation of EPCs, which could be abolished by pretreatment with flutamide [19]. EPC proliferation and adhesion are regulated by several pathways such as androgen receptor-mediated pathway and phosphatidyl-inositol-3-kinase (PI3K) signaling [20]. However, the molecular mechanisms by which androgen modulates EPCs remain unclear. Therefore, in this study we aimed to determine the signaling pathway underlying the effects of male hormones on EPCs.
Rho family is an important member of small G proteins that regulate a diverse array of cellular processes, including cytoskeletal dynamics, cell polarity, membrane transport, and gene expression [21]. RhoA and Rho-associated kinases (ROCKs) are widely studied as prototypical members of Rho family [22]. RhoA/ROCK signaling pathway plays an important role in angiogenesis and may be a promising target for antiangiogenic treatment [23]. RhoA/ROCK has been indicated as an upstream regulator of mitogen-activated protein kinase (MAPK) family members, including extracellular signal-regulated protein kinase, c-Jun-NH2 kinase, and p38 MAPK (p38) [24-26]. Recently, Sandra et al. demonstrated that EPCs proliferated through MEK-dependent p42 MAPK signaling pathway [27]. Thus we speculated that the activation of RhoA/ROCK pathway might play a significant role in the imodulation of EPC function by androgens. We investigated the effects of dihydrotestosterone (DHT), an active metabolite of testosterone, on human EPCs and explored the role of RhoA/ROCK signaling in this process.
Methods
EPC isolation and culture
This study conforms to the principles outlined in the Declaration of Helsinki for use of human tissue and was approved by the Institutional Review Board of the Affiliated Hospital of Jiangsu University. Detailed informed consent for the procedures was obtained from all subjects. EPC was isolated and cultured as previously described [5,20]. Briefly, peripheral blood mononuclear cells (PB-MNCs) were fractionated from other blood components through density gradient centrifugation using Ficoll solution according to the manufacturer’s instructions. PB-MNCs were plated on fibronectin-coated 24-well dish at a density of 1×106 cells/well and maintained in EGM endothelial cell growth medium (Clonetics, Lonza, MD, USA) at 37°C in a humidified 5% CO2 incubator. EMG medium consisted of endothelial basal medium-2 (EBM-2), gentamicin, vascular endothelial growth factor (VEGF), 5% fetal bovine serum, fibroblast growth factor-2, insulin-like growth factor-1, epidermal growth factor, and ascorbic acid. Culture medium was first changed on day 4 and then every 2 days. Starting from day 7, culture medium was supplemented with 1, 10, or 100 nmol/L DHT to examine the effects of on cultured EPCs. The adherent cells were selected for the experiment after 48 h.
Characterization of cultured EPCs
Fluorescent detection of EPCs was performed on the attached MNCs after 7 days of culture. Those adherent cells were washed three times with PBS, incubated with 2.4 μg/mL 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI)-labeled acetylated low-density lipoprotein (acLDL; Molecular Probe, Invitrogen, USA) at 37°C for 1 h and then fixed with 2% paraformaldehyde for 10 min. The cells were then incubated with 10 μg/mL fluorescein isothiocyanate (FITC)-conjugated Ulex europaeus agglutinin (UEA)-I (Sigma-Aldrich, St. Louis, MO, USA) for 1 h. After staining, the samples were observed under a laser scanning confocal microscope (×200). Those attached cells with DiI-AcLDL and FITC-UEA-I double-positive were identified as early EPCs.
Proliferation assay
EPC proliferation was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described [20,22,27]. Briefly, cells were digested with 0.25% trypsin (Keyi, Hangzhou, China) and then diluted to 2×105/mL. Cell suspension was seeded into 96-well plates (100 ml/well) and cultured in serum-free EGM-2 medium for 24 h. EPCs in each well were treated with DHT at different concentrations (0, 1, 10, and 100 nmol/L) and cultured for 48 h. Next 20 ml of MTT (5 g/L; Sigma-Aldrich) were added into each well and incubated for another 4 h. The supernatant was removed, and 150 ml dimethyl sulfoxide were added into each well and the plates were shaken for 10 min before the optical density (OD) was measured at 490 nm. RhoA inhibitor C3 exoenzyme (30 mg/mL; Alexis Biochemicals, Carlsbad, CA, USA) and ROCK inhibitor Y-27632 (10 mM; Sigma, St. Louis, MO, USA) were added 1 h before DHT treatment.
EPC migration assay
EPC migration was evaluated using a Transwell chamber (Greiner; Monroe, NC, USA) with 8 mm-pore filters as previously described [22]. The cultured cells were digested with 0.25% trypsin and diluted to 2×105/mL. EPCs were placed in the upper chamber of a Transwell chamber (100 μl/well). VEGF in serum-free EBM-2 medium was placed in the lower compartment of the chamber and EPCs were incubated at 37°C in 5% humidified CO2 for 24 h. The lower face of the filter was washed with PBS, and cells remaining on the upper side were removed using a cotton wool swab. EPCs were then fixed with 2% paraformaldehyde for 10 min and stained with hematoxylin for 30 min. Migratory cells in the lower chamber were counted in five random microscopic fields (×200). RhoA inhibitor C3 exoenzyme or ROCK inhibitor Y-27632 was added 1 h before DHT treatment.
EPC adhesion assay
EPC adhesion was evaluated as previously described [20,22]. Briefly, EPCs were washed with PBS and gently detached with 0.25% trypsin after treatment overnight with DHT at different concentrations (0, 1, 10, and 100 nmol/L) alone or in combination with RhoA inhibitor C3 exoenzyme or ROCK inhibitor Y-27632. An equal number of EPCs were seeded into fibronectin-coated 96-well plates and incubated for 30 min at 37°C in 5% humidified CO2. The adherent cells were independently counted by blinded investigators in five randomly selected microscopic fields (×200).
ELISA
VEGF protein level in conditioned medium was detected by ELISA. EPCs were treated as described above. The conditioned medium was collected, and VEGF level in the conditioned medium was measured using a high-sensitivity ELISA kit (Bender, Vienna, Austria) according to the manufacturer’s instructions.
RhoA activation assay
RhoA activation assay was performed as previously described with some modifications [22,28]. RhoA activity was determined by pull-down assay using a Rho activation assay kit (Cytoskeleton, USA) [29]. After culture in serum-free EBM-2 medium for 24 h, EPCs were treated with C3 exoenzyme for 48 h before treatment with 10 nmol/L DHT and then lyzed in lysis buffer. Cell lysates were incubated with agarose-conjugated Rhotekin-RBD at 4°C for 1 h, and then Agarose beads were boiled to release active Rho. Precipitation with Rhotekin GTP-Rho was performed thereafter. Finally, Rho protein was detected by specific anti-RhoA antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). 20 μg of total cell lysate per sample was used to detect the total amount of RhoA.
Western blot analysis
EPCs were collected and lysed in lysis buffer. Total protein content was quantified using the BCA Protein Assay Kit (Boster, Wuhan, Hubei, China). Equal amounts of proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto PVDF membranes (Millipore, Bedford, MA, USA), which were blocked with 5% BSA for 1 h and then incubated with antibodies against RhoA (1:1000). The membranes were washed in TBST and subsequently incubated with HRP-conjugated goat anti-rabbit IgG antibody (1:2500; MultiSciences, Hangzhou, Zhejiang, China). Antigen-antibody complex was quantified using an ECL Plus system (Amersham Bioscience, Piscataway, NJ, USA).
Statistical analysis
All data are presented as mean ± SD. Statistical significance was analyzed using unpaired Student’s t-test or ANOVA for comparisons between two groups and multiple comparisons, respectively. SPSS version 17.0 was used for the analysis, and P < 0.05 was considered statistically significant.
Results
EPC characterization
MNCs were isolated from peripheral blood and cultured for 7 days, resulting in a spindle-shaped morphology and formed cell colonies as previously reported [17,30,31] (Figure 1A). EPCs were characterized as spindle-shaped adherent cells double positive for DiI-AcLDL (DiI-labeled AcLDL) uptake and FITC-UEA-I (FITC-conjugated Bandeiraea simplicifolia lectin I) binding using LSCM (87.2% ± 6.5%) (Figure 1D). Flow cytometry confirmed the expression of CD133 (82.2% ± 5.6%), VEGFR-2 (93.6% ± 5.7%) and CD34 (85.3% ± 6.2%) in EPCs (Figure 1C).
Figure 1.
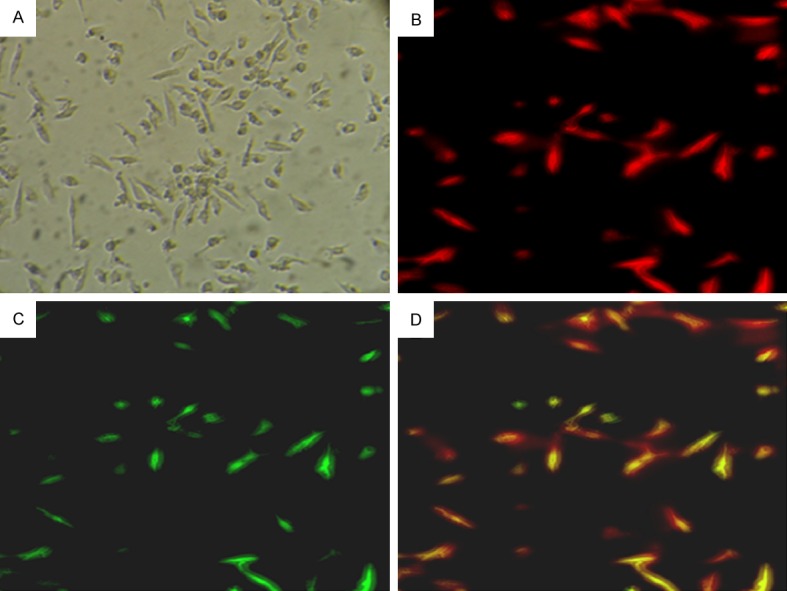
Characterization of EPCs. (A) The attached cells exhibited an oval or spindle morphology after 7 days of culture. Adherent cells stained with DiI-AcLDL (B. Red; excitation wave length at 543 nm) and FITC-UEA-I (C. Green; excitation wave length at 477 nm) were detected under a fluorescent microscope. Double positive cells (D. Yellow) were identified as differentiating early EPCs. Magnification at ×200.
DHT promotes EPC proliferation in vitro
EPC proliferation was examined using MTT assay. We observed that DHT dose-dependently promoted the proliferation of EPCs, with maximal effect at 10 nmol/L DHT (10 nM DHT group vs. control group, 0.216 ± 0.204 vs. 0.167 ± 0.056, P < 0.001; Figure 2A). Time-course experiments showed that DHT improved EPC proliferation time-dependently, reaching a peak value at 24 h (P < 0.01; Figure 2B). However, after pretreatment with C3 exoenzyme or Y-27632, the proliferation activity of EPCs was significantly inhibited even in the presence of DHT (C3 exoenzyme + DHT vs. DHT, 0.204 ± 0.026 vs. 0.226 ± 0.022, P = 0.018; Y-27632 + DHT vs. DHT, 0.196 ± 0.018 vs. 0.226 ± 0.022, P = 0.002) (n = 10, Figure 2C).
Figure 2.
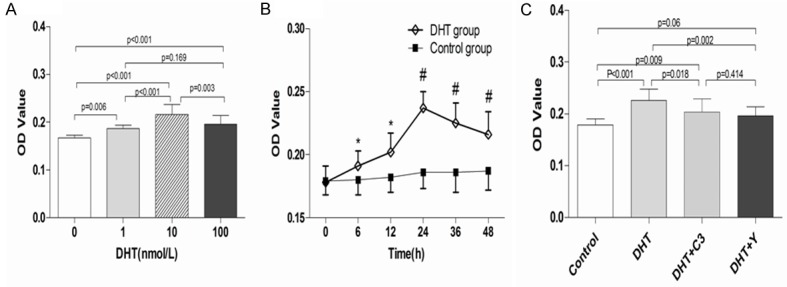
Effects of DHT, C3 exoenzyme, and Y-27632 on EPC proliferation. A. DHT dose-dependently promoted the proliferation of EPCs, with a maximal effect achieved at 10 nmol/L DHT. B. 10 nmol/L DHT increased EPC proliferation time-dependently. C. C3 exoenzyme and Y-27632 attenuated the stimulatory effects of DHT on EPC proliferation. The data are expressed as the mean ± SD; n = 10 separate experiments. *P < 0.05; #P < 0.01 versus control; DHT: dihydrotestosterone; C3: C3 exoenzyme; Y: Y-27632.
DHT promotes EPC adhesion in vitro
The adhesive capacity of EPCs was promoted by DHT in a dose-dependent manner, with maximal effect at 10 nmol/L DHT (10 nM DHT group vs. control group, 46.8 ± 5.67 vs. 30.6 ± 2.63, P < 0.001; Figure 3A). Time-course experiments showed that DHT promoted the adhesive capacity of EPCs in a time-dependent manner, with maximum effect at 24 h and then gradually decreased (P < 0.01; Figure 3B). However, C3 exoenzyme or Y-27632 reversed the enhanced EPC adhesion ability induced by DHT (C3 exoenzyme + DHT vs. DHT, 39.7 ± 4.74 vs. 45.5 ± 7.06, P = 0.037; Y-27632 + DHT vs. DHT, 35.6 ± 7.31 vs. 45.5 ± 7.06, P = 0.001; Figure 3C).
Figure 3.
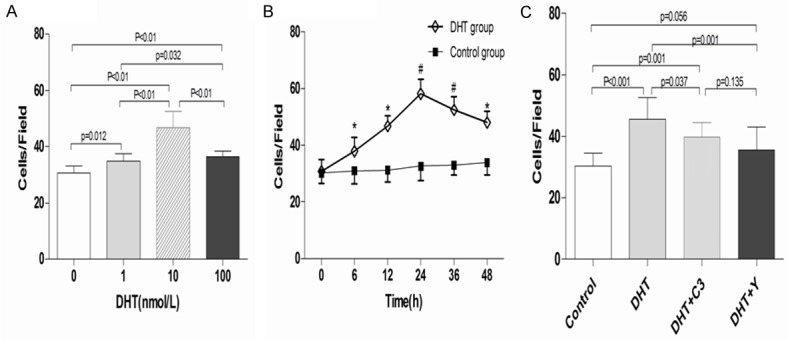
Effects of DHT, C3 exoenzyme, and Y-27632 on EPC adhesion. Cell counting assay was used to measure the adhesive activity of EPCs (×200). A. EPCs were incubated with DHT of different concentrations for 24 h. DHT concentration-dependently enhanced EPC adhesion, with a maximal effect achieved at 10 nmol/L. B. DHT time-dependently enhanced EPC adhesion. C. C3 exoenzyme and Y-27632 significantly decreased the adhesion of EPCs enhanced by DHT. *P < 0.05; #P < 0.01 versus control; data are presented as the mean ± SD; n =10 separate experiments. DHT dihydrotestosterone; C3: C3 exoenzyme; Y: Y-27632.
DHT promotes EPC migration in vitro
Transwell chamber assay was performed to determine the effect of DHT on EPC migration. The results revealed that DHT dose-dependently increased the migratory activity of EPCs, with the maximum migration rate at 10 nmol/L DHT (10 nM DHT group vs. control group, 39.4 ± 5.97 vs. 25.4 ± 3.13, P < 0.001; Figure 4A). The migration capacity of EPCs reached a maximum at 24 h and then progressively declined in time-course experiments with 10 nmol/L DHT (Figure 4B). However, the enhanced effect of DHT on EPC migration was attenuated with the administration of C3 exoenzyme or Y-27632 (C3 exoenzyme + DHT vs. DHT, 33.6 ± 6.28 vs. 42.4 ± 5.76, P = 0.004; Y-27632 + DHT vs. DHT, 31.1 ± 5.7 vs. 42.4 ± 5.76, P < 0.001; Figure 4C).
Figure 4.
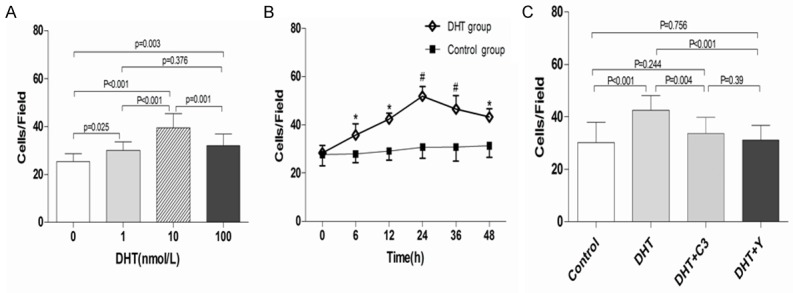
Effects of DHT, C3 exoenzyme, and Y-27632 on EPC migration. Transwell chamber assay was used to evaluate EPC migration (×200). A. EPCs were incubated with DHT at a series of concentrations for 24 h. DHT concentration-dependently promoted EPC migration. B. DHT time-dependently enhanced the migration capacity of EPCs. C. C3 exoenzyme and Y-27632 attenuated EPC migration enhanced by DHT. EPCs without treatment served as the control group. *P < 0.05; #P < 0.01 versus control; data are presented as the mean ± SD; n = 10 separate experiments. DHT dihydrotestosterone; C3: C3 exoenzyme; Y: Y-27632.
DHT upregulates VEGF secretion by EPCs
The protein concentration of VEGF in conditioned medium of EPCs was examined by using an ELISA kit. The results showed that DTH increased the secretion of VEGF by EPCs in a concentration-dependent manner, with the maximal effect at 10 nmol/L (DHT vs. control, 227.25 ± 28.17 vs. 128.82 ± 19.51 pg/mL; P < 0.001; Figure 5A). However, RhoA/ROCK inhibitor C3 exoenzyme and Y-27632 attenuated DHT-induced EPC secretion of VEGF (C3 exoenzyme + DHT vs. DHT, 156.60 ± 23.73 vs. 221. 04 ± 28.24, P < 0.001; Y-27632 + DHT vs. DHT, 161.63 ± 29.69 vs. 221. 04 ± 28.24, P < 0.001) (n = 10, Figure 5B).
Figure 5.
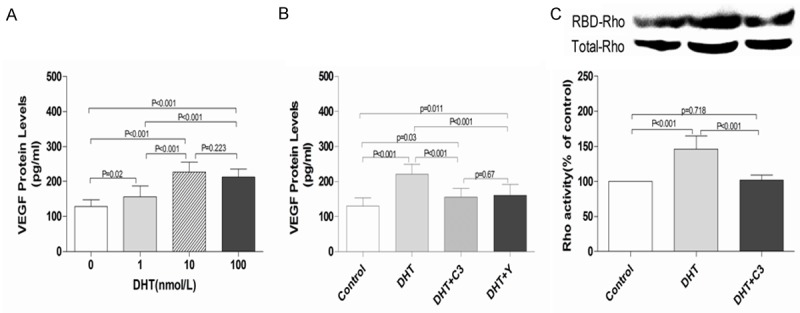
Effects of DHT, C3 exoenzyme, and Y-27632 on VEGF secretion and RhoA activation in EPCs. A. EPCs were incubated with DHT at different concentrations (0, 1, 10, and 100 nmol/L) for 24 h, VEGF protein levels in culture medium were determined by ELISA. DHT dose-dependently increased VEGF secretion with the maximal effect achieved at 10 nmol/L. B. C3 exoenzyme or Y-27632 markedly attenuated VEGF protein secretion induced by DHT. EPCs without treatment served as the control group. Data are presented as the mean ± SD; n = 10 separate experiments. DHT dihydrotestosterone; C3: C3 exoenzyme; Y: Y-27632. C. Serum-starved EPCs were pretreated with or without C3 exoenzyme (30 ng/mL) before incubation with 10 nmol/L DHT for 15 min. RBD-RhoA proteins and the total amount of RhoA were examined by Western blot analysis. The ratio of active RhoA vs. total RhoA was calculated to indicate the activation of RhoA. The results are representative of three independent experiments. EPCs without treatment served as the control group. Data are presented as the mean ± SD; n = 10 separate experiments. DHT: dihydrotestosterone; C3: C3 exoenzyme.
Effects of DHT and C3 exoenzyme on RhoA activation in EPC in vitro
RhoA activity was determined through pull-down assay. The results showed that the active form of RhoA (GTP-bound) was elevated to 146.00% ± 18.67% after DHT treatment compared with control (Figure 5C). Pretreatment with C3 exoenzyme significantly inhibited RhoA activation induced by DHT (C3 exoenzyme + DHT vs. DHT, 101.90% ± 7.52% vs. 146.00% ± 18.67%, P < 0.01; Figure 5C).
Discussion
In this study, we demonstrated that DHT significantly enhanced the proliferation, adhesion, and migration of human EPCs and upregulated VEGF secretion in vitro in a dose-dependent manner within a specific concentration range. Furthermore, we revealed that the stimulatory effects of DHT on EPC activity are dependent on RhoA/ROCK pathway. These results support the potential role of DHT in upregulating EPC function and may provide novel strategy for the prevention and treatment of early atherosclerosis associated with EPCs and endothelial damage.
The role of androgens in the cardiovascular system is controversial. In clinical studies, androgen excess and deficiency have been reported to exert negative effects on cardiovascular parameters and endothelial function [32,33]. Moreover, a low level of androgen is closely correlated to increased male CVD morbidity and mortality [34-36]. Integrated vascular endothelium is associated with the protection of cardiovascular system. Numerous environmental factors, especially cardiovascular risk factors, would damage the structure and function of the endothelium, resulting in the development of various diseases [37]. Endothelial dysfunction may induce a series of pathogenic events, including atherosclerosis, hypertension, diabetes, and thrombosis [38,39]. Therefore, the repair of endothelial dysfunction is essential to the prevention of the initiation and development of atherosclerosis [40]. Accumulating evidence has suggested that EPCs are involved in multiple aspects of this repair process and play a significant role in the protection of cardiovascular function [41-43]. Accelerated re-endothelialization by EPCs may effectively abrogate smooth muscle cell proliferation, migration, and neointima formation, thereby preventing the initiation and development of atherosclerosis and restenosis following vascular injury [41,44,45]. Therefore, great efforts have been taken to improve EPCs function through different strategies ranging from available medication to the frontiers of cell therapy [18].
Recent studies have indicated that androgens or their derivatives have conspicuous protective effects on vascular function [46,47]. Androgens can stimulate endothelial progenitor cells through an androgen receptor-mediated pathway [19]. Moreover, DHT can modulate EPC proliferation and adhesion via PI3K/Akt pathway [20]. These findings collectively suggest that androgens play important role in regulating EPCs in human. However, the molecular mechanisms remain unclear. In the present study, we explored the effects of DHT, an active metabolite of testosterone, on EPCs. Our results revealed that DHT could promote proliferation, migration, and adhesion of EPCs in a dose-dependent manner, with the peak value at 10 nmol/L DHT. These findings are partly consistent with the results of Liu et al. [20] but in contrast to other studies. Several reasons may explain the discrepancy, including the use of different androgens, androgen concentrations, various experimental conditions, as well as EPC culture methods.
We further demonstrated that DHT promoted the proliferative, migratory, and adhesive capacities of EPCs via RhoA/ROCK pathway. A growing body of experimental evidence revealed that RhoA/ROCK pathway is crucial to many cell physiological and pathological processes, including cytoskeletal dynamics, cell polarity, cell proliferation, survival and migration. A recent study showed that zoledronate attenuated the angiogenic effects of angiotensin II-stimulated endothelial progenitor cells through RhoA and MAPK signaling [22]. Bryan et al. revealed that RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis [23]. In this study, RhoA/ROCK signaling pathway was blocked by C3 exoenzyme or Y-27632, and the proliferative, migratory, and adhesive ability of EPCs were significantly attenuated. In addition, the effect of DHT on EPC secretion of VEGF was investigated in the present study. The results indicate that DHT enhanced the secretion of VEGF by EPCs, an effect that was effectively abrogated by C3 exoenzyme or Y-27632. These data suggest that the role of DHT in the promotion of EPC function may be due to the stimulation of paracrine effects of EPC through the secretion of VEGF.
Conclusion
This study demonstrates that DHT enhances the proliferative, migratory, and adhesive abilities of EPCs and promotes the secretion of VEGF by EPCs. Furthermore, these effects of DHT are mostly likely mediated by RhoA/ROCK signaling. Our data suggest that DHT may play a protective role in vascular repair, especially for patients with low levels of androgens.
Acknowledgements
This study was supported by grants from Zhen Jiang Social Development Fund (No. SH2014033), Jiangsu Provincial Key Research and Development Program (BE2016721), and Jiangsu Province TCM Project (No. YB2015184).
Disclosure of conflict of interest
None.
Authors’ contribution
HZ and LS carried out the studies and were co-first author. GR and WS participated in the design of the study. YW, YC and JY helped to finish the studies. BW drafted the manuscript. All authors read and approved the final manuscript.
References
- 1.Brugaletta S, Martin-Yuste V, Padro T, Alvarez-Contreras L, Gomez-Lara J, Garcia-Garcia HM, Cola C, Liuzzo G, Masotti M, Crea F, Badimon L, Serruys PW, Sabate M. Endothelial and smooth muscle cells dysfunction distal to recanalized chronic total coronary occlusions and the relationship with the collateral connection grade. JACC Cardiovasc Interv. 2012;5:170–178. doi: 10.1016/j.jcin.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 2.Fadini GP, Agostini C, Sartore S, Avogaro A. Endothelial progenitor cells in the natural history of atherosclerosis. Atherosclerosis. 2007;194:46–54. doi: 10.1016/j.atherosclerosis.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 3.Hirase T, Node K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am J Physiol Heart Circ Physiol. 2012;302:H499–505. doi: 10.1152/ajpheart.00325.2011. [DOI] [PubMed] [Google Scholar]
- 4.Terashima M, Kaneda H, Nasu K, Matsuo H, Habara M, Ito T, Tanaka N, Rathore S, Kinoshita Y, Kimura M, Ehara M, Suzuki Y, Suzuki T. Protective effect of telmisartan against endothelial dysfunction after coronary drug-eluting stent implantation in hypertensive patients. JACC Cardiovasc Interv. 2012;5:182–190. doi: 10.1016/j.jcin.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 5.Huang C, Zhang L, Wang Z, Pan H, Zhu J. Endothelial progenitor cells are associated with plasma homocysteine in coronary artery disease. Acta Cardiol. 2011;66:773–777. doi: 10.1080/ac.66.6.2136962. [DOI] [PubMed] [Google Scholar]
- 6.Foesta C, Caretta N, Lana A, De Toni L, Biagioli A, Ferlin A, Garolla A. Reduced number of circulating endothelial progenitor cells in hypogonadal men. J Clin Endocrinol Metab. 2006;91:4599–4602. doi: 10.1210/jc.2006-0763. [DOI] [PubMed] [Google Scholar]
- 7.Davel AP, Wenceslau CF, Akamine EH, Xavier FE, Couto GK, Oliveira HT, Rossoni LV. Endothelial dysfunction in cardiovascular and endocrine-metabolic diseases: an update. Braz J Med Biol Res. 2011;44:920–932. doi: 10.1590/s0100-879x2011007500104. [DOI] [PubMed] [Google Scholar]
- 8.Ciftci O, Topcu S, Caliskan M, Gullu H, Erdogan D, Yildirim E, Yildirir A, Muderrisoglu H. Smoking mentholated cigarettes impairs coronary microvascular function as severely as does smoking regular cigarettes. Acta Cardiol. 2008;63:135–140. doi: 10.2143/AC.63.2.2029518. [DOI] [PubMed] [Google Scholar]
- 9.Hamed S, Brenner B, Roguin A. Nitric oxide: a key factor behind the dysfunctionality of endothelial progenitor cells in diabetes mellitus type-2. Cardiovasc Res. 2011;91:9–15. doi: 10.1093/cvr/cvq412. [DOI] [PubMed] [Google Scholar]
- 10.Tomada N, Tomada I, Botelho F, Pacheco-Figueiredo L, Lopes T, Negrao R, Pestana M, Cruz F. Endothelial function in patients with metabolic syndrome and erectile dysfunction: a question of angiopoietin imbalance? Andrology. 2013;1:541–548. doi: 10.1111/j.2047-2927.2013.00102.x. [DOI] [PubMed] [Google Scholar]
- 11.Pelliccione F, D’Angeli A, Filipponi S, Falone S, Necozione S, Barbonetti A, Francavilla F, Francavilla S. Serum from patients with erectile dysfunction inhibits circulating angiogenic cells from healthy men: relationship with cardiovascular risk, endothelial damage and circulating angiogenic modulators. Int J Androl. 2012;35:645–652. doi: 10.1111/j.1365-2605.2012.01253.x. [DOI] [PubMed] [Google Scholar]
- 12.Capobianco S, Chennamaneni V, Mittal M, Zhang N, Zhang C. Endothelial progenitor cells as factors in neovascularization and endothelial repair. World J Cardiol. 2010;2:411–420. doi: 10.4330/wjc.v2.i12.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grisar JC, Haddad F, Gomari FA, Wu JC. Endothelial progenitor cells in cardiovascular disease and chronic inflammation: from biomarker to therapeutic agent. Biomark Med. 2011;5:731–744. doi: 10.2217/bmm.11.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sen S, McDonald SP, Coates PT, Bonder CS. Endothelial progenitor cells: novel biomarker and promising cell therapy for cardiovascular disease. Clin Sci (Lond) 2011;120:263–283. doi: 10.1042/CS20100429. [DOI] [PubMed] [Google Scholar]
- 15.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 16.Aicher A, Rentsch M, Sasaki K, Ellwart JW, Fandrich F, Siebert R, Cooke JP, Dimmeler S, Heeschen C. Nonbone marrow-derived cir culating progenitor cells contribute to postnatal neovascularization following tissue ischemia. Circ Res. 2007;100:581–589. doi: 10.1161/01.RES.0000259562.63718.35. [DOI] [PubMed] [Google Scholar]
- 17.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 18.Fadini GP, Albiero M, Cignarella A, Bolego C, Pinna C, Boscaro E, Pagnin E, De Toni R, de Kreutzenberg S, Agostini C, Avogaro A. Effects of androgens on endothelial progenitor cells in vitro and in vivo. Clin Sci (Lond) 2009;117:355–364. doi: 10.1042/CS20090077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foresta C, Zuccarello D, De Toni L, Garolla A, Caretta N, Ferlin A. Androgens stimulate endothelial progenitor cells through an androgen receptor-mediated pathway. Clin Endocrinol (Oxf) 2008;68:284–289. doi: 10.1111/j.1365-2265.2007.03036.x. [DOI] [PubMed] [Google Scholar]
- 20.Liu R, Ding L, Yu MH, Wang HQ, Li WC, Cao Z, Zhang P, Yao BC, Tang J, Ke Q, Huang TZ. Effects of dihydrotestosterone on adhesion and proliferation via PI3-K/Akt signaling in endothelial progenitor cells. Endocrine. 2014;46:634–643. doi: 10.1007/s12020-013-0081-1. [DOI] [PubMed] [Google Scholar]
- 21.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 22.Yang JX, Chen B, Pan YY, Han J, Chen F, Hu SJ. Zoledronate attenuates angiogenic effects of angiotensin II-stimulated endothelial progenitor cells via RhoA and MAPK signaling. PLoS One. 2012;7:e46511. doi: 10.1371/journal.pone.0046511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryan BA, Dennstedt E, Mitchell DC, Walshe TE, Noma K, Loureiro R, Saint-Geniez M, Campaigniac JP, Liao JK, D’Amore PA. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. Faseb J. 2010;24:3186–3195. doi: 10.1096/fj.09-145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marinissen MJ, Chiariello M, Tanos T, Bernard O, Narumiya S, Gutkind JS. The small GTP-binding protein RhoA regulates c-jun by a ROCK-JNK signaling axis. Mol Cell. 2004;14:29–41. doi: 10.1016/s1097-2765(04)00153-4. [DOI] [PubMed] [Google Scholar]
- 25.Shatanawi A, Romero MJ, Iddings JA, Chandra S, Umapathy NS, Verin AD, Caldwell RB, Caldwell RW. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am J Physiol Cell Physiol. 2011;300:C1181–1192. doi: 10.1152/ajpcell.00328.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong SY, Jeon YM, Lee HJ, Kim JG, Baek JA, Lee JC. Activation of RhoA and FAK induces ERK-mediated osteopontin expression in mechanical force-subjected periodontal ligament fibroblasts. Mol Cell Biochem. 2010;335:263–272. doi: 10.1007/s11010-009-0276-1. [DOI] [PubMed] [Google Scholar]
- 27.Sandra F, Oktaviono YH, Widodo MA, Dirgantara Y, Chouw A, Sargowo D. Endothelial progenitor cells proliferated via MEK-dependent p42 MAPK signaling pathway. Mol Cell Biochem. 2015;400:201–206. doi: 10.1007/s11010-014-2276-z. [DOI] [PubMed] [Google Scholar]
- 28.Bryan BA, Li D, Wu X, Liu M. The Rho family of small GTPases: crucial regulators of skeletal myogenesis. Cell Mol Life Sci. 2005;62:1547–1555. doi: 10.1007/s00018-005-5029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye Y, Hu SJ, Li L. Inhibition of farnesylpyrophosphate synthase prevents angiotensin II-induced hypertrophic responses in rat neonatal cardiomyocytes: involvement of the RhoA/Rho kinase pathway. FEBS Lett. 2009;583:2997–3003. doi: 10.1016/j.febslet.2009.08.034. [DOI] [PubMed] [Google Scholar]
- 30.Hur J, Yoon CH, Kim HS, Choi JH, Kang HJ, Hwang KK, Oh BH, Lee MM, Park YB. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. 2004;24:288–293. doi: 10.1161/01.ATV.0000114236.77009.06. [DOI] [PubMed] [Google Scholar]
- 31.Ruifrok WP, de Boer RA, Iwakura A, Silver M, Kusano K, Tio RA, Losordo DW. Estradiol-induced, endothelial progenitor cell-mediated neovascularization in male mice with hind-limb ischemia. Vasc Med. 2009;14:29–36. doi: 10.1177/1358863X08096666. [DOI] [PubMed] [Google Scholar]
- 32.Rosano GM, Sheiban I, Massaro R, Pagnotta P, Marazzi G, Vitale C, Mercuro G, Volterrani M, Aversa A, Fini M. Low testosterone levels are associated with coronary artery disease in male patients with angina. Int J Impot Res. 2007;19:176–182. doi: 10.1038/sj.ijir.3901504. [DOI] [PubMed] [Google Scholar]
- 33.Fukui M, Kitagawa Y, Ose H, Hasegawa G, Yoshikawa T, Nakamura N. Role of endogenous androgen against insulin resistance and athero- sclerosis in men with type 2 diabetes. Curr Diabetes Rev. 2007;3:25–31. doi: 10.2174/157339907779802094. [DOI] [PubMed] [Google Scholar]
- 34.Wehr E, Pilz S, Boehm BO, Marz W, Grammer T, Obermayer-Pietsch B. Low free testosterone is associated with heart failure mortality in older men referred for coronary angiography. Eur J Heart Fail. 2011;13:482–488. doi: 10.1093/eurjhf/hfr007. [DOI] [PubMed] [Google Scholar]
- 35.Patel SM, Ratcliffe SJ, Reilly MP, Weinstein R, Bhasin S, Blackman MR, Cauley JA, Sutton-Tyrrell K, Robbins J, Fried LP, Cappola AR. Higher serum testosterone concentration in older women is associated with insulin resistance, metabolic syndrome, and cardiovascular disease. J Clin Endocrinol Metab. 2009;94:4776–4784. doi: 10.1210/jc.2009-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikeda Y, Aihara K, Yoshida S, Akaike M, Matsumoto T. Effects of androgens on cardiovascular remodeling. J Endocrinol. 2012;214:1–10. doi: 10.1530/JOE-12-0126. [DOI] [PubMed] [Google Scholar]
- 37.Ding M, Ye TX, Zhao GR, Yuan YJ, Guo ZX. Aqueous extract of Salvia miltiorrhiza attenuates increased endothelial permeability induced by tumor necrosis factor-alpha. Int Immunopharmacol. 2005;5:1641–1651. doi: 10.1016/j.intimp.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Ross R. Atherosclerosis--an inflammatory disease. New Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 39.Yang GD, Zhang H, Lin R, Wang WR, Shi XL, Liu Y, Ji QL. Down-regulation of CD40 gene expression and inhibition of apoptosis with Danshensu in endothelial cells. Basic Clin Pharmacol Toxicol. 2009;104:87–92. doi: 10.1111/j.1742-7843.2008.00342.x. [DOI] [PubMed] [Google Scholar]
- 40.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 41.Werner N, Junk S, Laufs U, Link A, Walenta K, Bohm M, Nickenig G. Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res. 2003;93:e17–24. doi: 10.1161/01.RES.0000083812.30141.74. [DOI] [PubMed] [Google Scholar]
- 42.Dimitrova KR, Leitman IM. Intramyocardial transplantation of endothelial progenitor cells and erythropoietin: a new scope for the treatment of cardiovascular disease. J Surg Res. 2013;183:550–552. doi: 10.1016/j.jss.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 43.Bakogiannis C, Tousoulis D, Androulakis E, Briasoulis A, Papageorgiou N, Vogiatzi G, Kampoli AM, Charakida M, Siasos G, Latsios G, Antoniades C, Stefanadis C. Circulating endothelial progenitor cells as biomarkers for prediction of cardiovascular outcomes. Curr Med Chem. 2012;19:2597–2604. doi: 10.2174/092986712800492995. [DOI] [PubMed] [Google Scholar]
- 44.Griese DP, Ehsan A, Melo LG, Kong D, Zhang L, Mann MJ, Pratt RE, Mulligan RC, Dzau VJ. Isolation and transplantation of autologous circulating endothelial cells into denuded vessels and prosthetic grafts: implications for cell-based vascular therapy. Circulation. 2003;108:2710–2715. doi: 10.1161/01.CIR.0000096490.16596.A6. [DOI] [PubMed] [Google Scholar]
- 45.Ji KT, Chai JD, Xing C, Nan JL, Yang PL, Tang JF. Danshen protects endothelial progenitor cells from oxidized low-density lipoprotein induced impairment. J Zhejiang Univ Sci B. 2010;11:618–626. doi: 10.1631/jzus.B1001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campelo AE, Cutini PH, Massheimer VL. Cellular actions of testosterone in vascular cells: mechanism independent of aromatization to estradiol. Steroids. 2012;77:1033–1040. doi: 10.1016/j.steroids.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 47.Xu ZR, Hu L, Cheng LF, Qian Y, Yang YM. Dihydrotestosterone protects human vascular endothelial cells from H(2)O(2)-induced apoptosis through inhibition of caspase-3, caspase-9 and p38 MAPK. Eur J Pharmacol. 2010;643:254–259. doi: 10.1016/j.ejphar.2010.06.039. [DOI] [PubMed] [Google Scholar]