

Abstract
Autophagy has been shown to antagonize the development of Lumbar disc degeneration (LDD), whereas the molecular regulation of autophagy is unknown. We recently reported a potential role of Insulin-like growth factor 1 receptor (IGF1R)/phosphatidylinositol-3 kinase (PI3k)/Akt signaling in the initiation and progression of LDD. Here, we studied the effects of IGF1R signaling on disc cell autophagy. We showed a correction of activation of IGF1R and disc cell autophagy in the resected discs in LDD patients. In vitro, activation of IGF1R signaling antagonized the decreases in cell viability of human disc cells, HNPSV, through suppression of apoptosis and enhancement of autophagy. Suppression of IGF1R signaling or inhibition of autophagy abolished the effects of activation of IGF1R signaling on disc cell survival upon compression. Together, our data suggest that activation of IGF1R may antagonize LDD, at least partially through enhanced autophagy.
Keywords: Lumbar disc degeneration (LDD), insulin-like growth factor 1 receptor (IGF1R), apoptosis, autophagy
Introduction
More than half the population will experience significant low back pain during their life, largely due to the occurrence of lumbar disc degeneration (LDD), which frequently results from a degenerated disc in the spine [1-3]. The lumbar disc consists of nucleus pulposus (NP), annulus fibrosus and cartilage end plates. Previous studies have highlighted excessive apoptosis of NP cells which are capable of producing cartilage-specific extracellular matrix components as one of the most evident cellular and biochemical changes in LDD. The decreased NP cells caused by excessive apoptosis, leads to that the decreases in extracellular matrix synthesis, and then LDD [4]. Therefore, excessive apoptosis of NP cells has been believed to contribute to the degradation of ECM and plays an important role in the process of LDD degeneration [5-9]. Disc damage caused by mechanical injury, inflammation, or aging may modulate the structure of the discs through regulating disc homeostasis [10]. Consequently, inhibition of apoptosis of NP cells may decrease the degradation of extracellular matrix and postpone the progression of the IVD degeneration.
Autophagy was initially characterized based on its ultrastructural features, in particular, double-membraned structures that surrounded cytoplasm and organelles in cells, known as autophagosomes [11-14]. Autophagy is a catabolic program, which is activated in response to starvation or alteration in nutrient conditions [11-14]. Previous studies have identified a number of proteins, and elucidated several biochemical pathways, that are essential for autophagy. Microtubule-associated protein 1A/1B-light chain 3 (LC3) is a soluble cellular protein and a marker for autophagy. During autophagy, autophagosomes engulf cytoplasmic components, resulting in changes of the cytosolic LC3-I into LC3-II. Thus, the ratio of LC3-II to LC3-I has been widely used as a marker of the autophagic activity [11-14]. Among all these autophagy-associated proteins, autophagy-related protein 6 (ATG6, or Beclin-1) appears to be a critical one, which is a component of a class III PI3-K-containing complex [15]. Recent studies have demonstrated a role of autophagy in antagonizing apoptosis in LDD [10,16-19]. However, the regulation of autophagy in LDD remains largely unknown.
Matrix metalloproteinase 3 (MMP3) is involved in the disease initiation and progression of LDD [20,21]. However, only recently, we reported that MMP3 is suppressed by insulin-like growth factor 1 receptor (IGF1R) signaling in disc cells to prevent the development of LDD [22]. The IGF1R signaling pathway initiates with binding of Insulin-like growth factor 1 (IGFI) to its cell-surface receptor IGF1R to activate phosphatidylinositol-3 kinase (PI3K)/Akt signaling pathway, to stimulate cell growth and proliferation, and to inhibit programmed cell death [23-25]. In our previous study, we showed that and IGF1 induced phosphorylation of IGF1R, and then phosphorylation of Akt in the human disc cells to inhibit MMP3 through nuclear retention of forkhead box protein O1 (FoxO1) [22]. Nevertheless, the role of activation of IGF1R signaling on disc cell autophagy against apoptosis has not been studied before. Here, we showed a correction of activation of IGF1R and disc cell autophagy in the resected discs in LDD patients. In vitro, activation of IGF1R signaling antagonized the decreases in cell viability of human disc cells, HNPSV, through suppression of apoptosis and enhancement of autophagy. Suppression of IGF1R signaling or inhibition of autophagy abolished the effects of activation of IGF1R signaling on disc cell survival upon compression.
Materials and methods
Specimens from patients
A total of 39 subjects (17 with resected LDD discs and 22 healthy subjects with fractured discs as controls) were included in this study. All the subjects had no accompanying diseases (e.g. diabetes, Crohn’s disease and Parkinson’s disease) that may affect the IGF1 metabolism. All specimens had been histologically and clinically diagnosed at Qingpu Branch of Zhongshan Hospital Affiliated to Fudan University from 2009 to 2015. For the use of these clinical materials for research purposes, prior patient’s consents and approval from the Institutional Research Ethics Committee were obtained. Phosphorylated IGF1R (pIGF1R) levels were measured by Western blot and normalized to total IGF1R levels. LC3 levels were measured by Western blot and the ratio of LC3 II vs I was used as an autophagy marker.
Human disc cell line, reagents, and treatment
A human disc cell line HNPSV has been described before [22]. The HNPSV cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Gibco; Life Technologies, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS, Sigma-Aldrich, St Louis, MO, USA), penicillin (100 μg/ml) and streptomycin (250 ng/ml) at 37°C, in a 5% CO2 atmosphere. Recombinant human IGF1 (100 ng/ml) and Akt inhibitor LY294002 (20 µmol/l) were also purchased from Sigma-Aldrich. 3-Methyladenine (3-MA, Sigma-Aldrich) was prepared in DMSO and given to cultured HNPSV cells at a dose of 5 mmol/l. For application of a compression apparatus on HNPSV cells, the cells were cultured in a stainless steel pressure vessel to mimic in vivo conditions. The pressure apparatus (capacity, 8.2 L) was constructed to withstand up to 1×106 pa pressure. HNPSV cells were placed on cell culture plates or culture dishes, and subjected to a 1×106 pa mechanical stress.
Western blot
Protein was extracted from the resected disc cells or cultured cells by RIPA buffer (Sigma-Aldrich) for Western Blot. The supernatants were collected after centrifugation at 12000×g at 4°C for 20 min. Protein concentration was determined using BCA protein assay, and whole lysates were mixed with 4×SDS loading buffer (125 mmol/l Tris-HCl, 4% SDS, 20% glycerol, 100 mmol/l DTT, and 0.2% bromophenol blue) at a ratio of 1:3. Samples were heated at 100°C for 5 min and were separated on SDS-polyacrylamide gels. The separated proteins were then transferred to a PVDF membrane. The membrane blots were first probed with a primary antibody. After incubation with horseradish peroxidase-conjugated second antibody, autoradiograms were prepared using the enhanced chemiluminescent system to visualize the protein antigen. The signals were recorded using X-ray film. Primary antibodies for Western Blot are anti-IGF1R, anti-phosphorylated-IGF1R (pIGF1R), anti-Caspase 3, anti-LC3, anti-Beclin-1 and anti-α-tubulin (all purchased from Cell Signaling, San Jose, CA, USA). Secondary antibody is HRP-conjugated anti-rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). Images shown in the figure were representative from 5 repeats. Densitometry of Western blots was quantified with NIH ImageJ software. The phosphorylated protein levels were first normalized to total protein, and then normalized to controls. MMP3 were first normalized to α-tubulin, and then normalized to controls.
Cell counting kit-8 (CCK-8) assay
The CCK-8 detection kit (Sigma-Aldrich) was used to measure cell viability according to the manufacturer’s instructions. Briefly, cells were seeded in a 96-well microplate at a density of 5×104/ml. After 24 h, cells were treated with resveratrol. Subsequently, CCK-8 solution (20 ml/well) was added and the plate was incubated at 37°C for 2 h. The viable cells were counted by absorbance measurements with a monochromator microplate reader at a wavelength of 450 nm. The optical density value was reported as the percentage of cell viability in relation to the control group (set as 100%).
Apoptosis assay by flow cytometry
The cultured cells were re-suspended at a density of 106 cells/ml in PBS. After double staining with FITC-Annexin V and propidium iodide (PI) from a FITC Annexin V Apoptosis Detection Kit I (Becton-Dickinson Biosciences, San Jose, CA, USA), cells were analyzed using FACScan flow cytometer (Becton-Dickinson Biosciences) for determination of Annexin V+ PI- apoptotic cells, with Flowjo software (Flowjo LLC, Ashland, OR, USA).
Statistical analysis
All statistical analyses were carried out using GraphPad prism 6.0 (GraphPad Software Inc. La Jolla, CA, USA). All values are depicted as mean ± SD and are considered significant if p < 0.05. All data were statistically analyzed using one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test for comparison of two groups. Bivariate correlations were calculated by Spearman’s Rank Correlation Coefficients.
Results
Activation of IGF1R signaling correlates with autophagy levels in LDD specimens
We examined the IGF1R, phosphorylated IGF1R (pIGF1R) and LC3 levels in the 17 LDD disc specimens by Western blot (Figure 1A). The levels of pIGF1R vs IGF1R were used as an indicator for IGF1R signaling activation, while the levels of LC3 II vs I were used as a maker for autophagy. We found a strong correlation between pIGF1R/IGF1R and LC3 II/I in LDD specimens (Figure 1B, r=0.90; p < 0.0001), suggesting a causal relationship between activation of IGF1R signaling and autophagy levels in LDD specimens.
Figure 1.
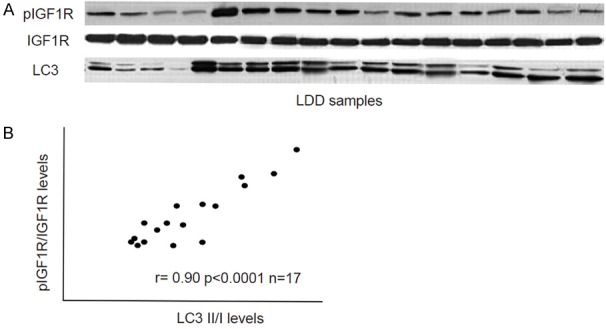
Activation of IGF1R signaling correlates with autophagy levels in LDD specimens. A. IGF1R, phosphorylated IGF1R (pIGF1R) and LC3 levels in the 17 LDD disc specimens by Western blot. B. A strong correlation was detected between pIGF1R/IGF1R and LC3II/I in LDD specimens (r=0.90; p < 0.0001). N=17.
Activation of IGF1R signaling suppresses compression-induced disc cell death
We further analyzed the activation of IGF1R signaling on disc cell death. Thus, a human disc cell line HNPSV was subjected to compression. Then, IGF1 was given to the cultured cells to activate IGF1R signaling. Moreover, a specific Akt inhibitor LY294002 was also used to some IGF1-treated HNPSV cells for inhibiting the activation of IGF1R signaling by IGF1. We found that activation of IGF1R signaling significantly decreased the cell death by compression, and suppression of the phosphorylation of IGF1R signaling downstream target Akt abolished the effects of activation of IGF1R signaling on cell survival, in a CCK-8 assay (Figure 2). Thus, activation of IGF1R signaling suppresses compression-induced disc cell death in an Akt-dependent manner.
Figure 2.
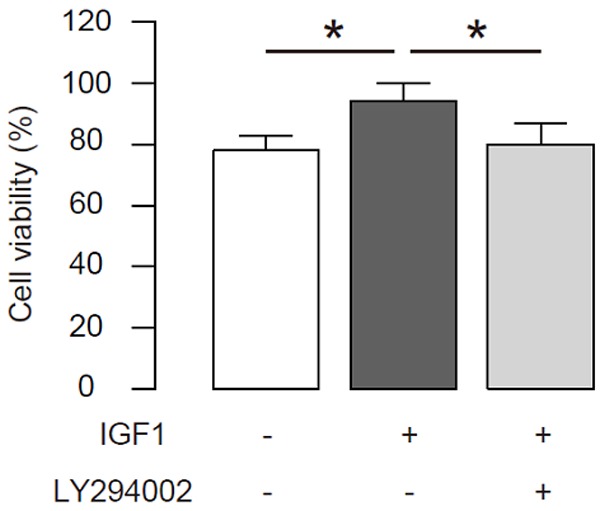
Activation of IGF1R signaling suppresses compression-induced disc cell death. A human disc cell line HNPSV was subjected to compression. Then, IGF1 was given to the cultured cells to activate IGF1R signaling. Moreover, a specific Akt inhibitor LY294002 was also used to some IGF1-treated HNPSV cells for inhibiting the activation of IGF1R signaling by IGF1. The cell viability was analyzed in a CCK-8 assay. *p < 0.05. N=5.
Activation of IGF1R signaling suppresses compression-induced disc cell apoptosis
Next, we examined the effects of activation of IGF1R signaling on disc cell apoptosis. We found that activation of IGF1R signaling significantly decreased the cell death by compression, and suppression of the phosphorylation of IGF1R signaling downstream target Akt abolished the inhibitory effects of activation of IGF1R signaling on cell apoptosis, by representative flow charts (Figure 3A), and by quantification (Figure 3B). Thus, activation of IGF1R signaling suppresses compression-induced disc cell apoptosis in an Akt-dependent manner.
Figure 3.
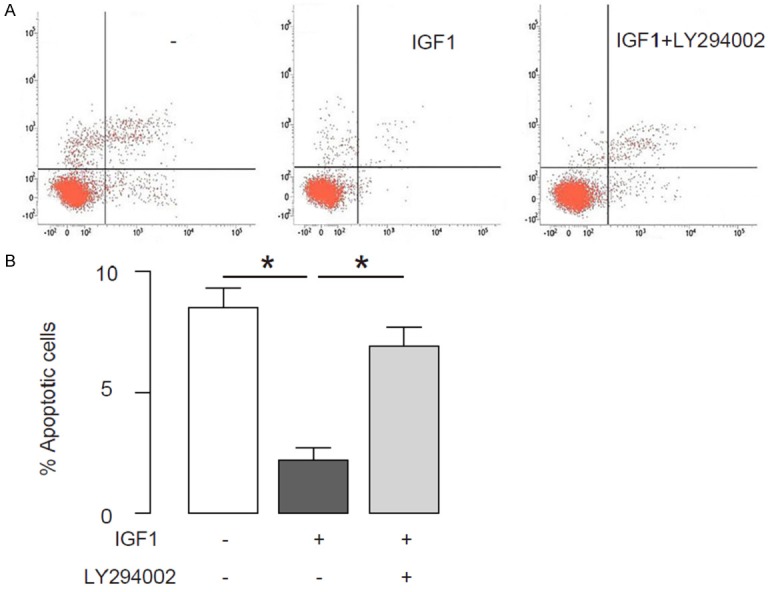
Activation of IGF1R signaling suppresses compression-induced disc cell apoptosis. A human disc cell line HNPSV was subjected to compression. Then, IGF1 was given to the cultured cells to activate IGF1R signaling. Moreover, a specific Akt inhibitor LY294002 was also used to some IGF1-treated HNPSV cells for inhibiting the activation of IGF1R signaling by IGF1. (A, B) Disc cell apoptosis was examined in a FITC Annexin V Apoptosis Detection assay, shown by representative flow charts (A), and by quantification (B). *p < 0.05. N=5.
Activation of IGF1R signaling enhances disc cell autophagy upon compression treatment
Then we examined the effects of activation of IGF1R signaling on cell autophagy of disc cells upon compression treatment. First, the activation of IGF1R signaling was confirmed by analyses of pIGF1R/IGF1R, shown by representative images (Figure 4A), and by quantification (Figure 4B). Activation of IGF1R signaling also significantly decreased the levels of Caspase 3, an apoptotic marker, shown by representative images (Figure 4A), and by quantification (Figure 4C). Moreover, the activation of IGF1R signaling increased the ratio of LC3 II/I, shown by representative images (Figure 4A), and by quantification (Figure 4D), possibly through its effects on Beclin-1, an autophagy-related protein, by representative images (Figure 4A), and by quantification (Figure 4E). Furthermore, a specific Akt inhibitor LY294002 completely abolished the effects of IGF1 on Caspase 3, LC3 and Beclin-1 (Figure 4A-E), without affecting IGF1R phosphorylation (Figure 4A, 4B). These data suggest that activation of IGF1R signaling enhances disc cell autophagy upon compression treatment.
Figure 4.
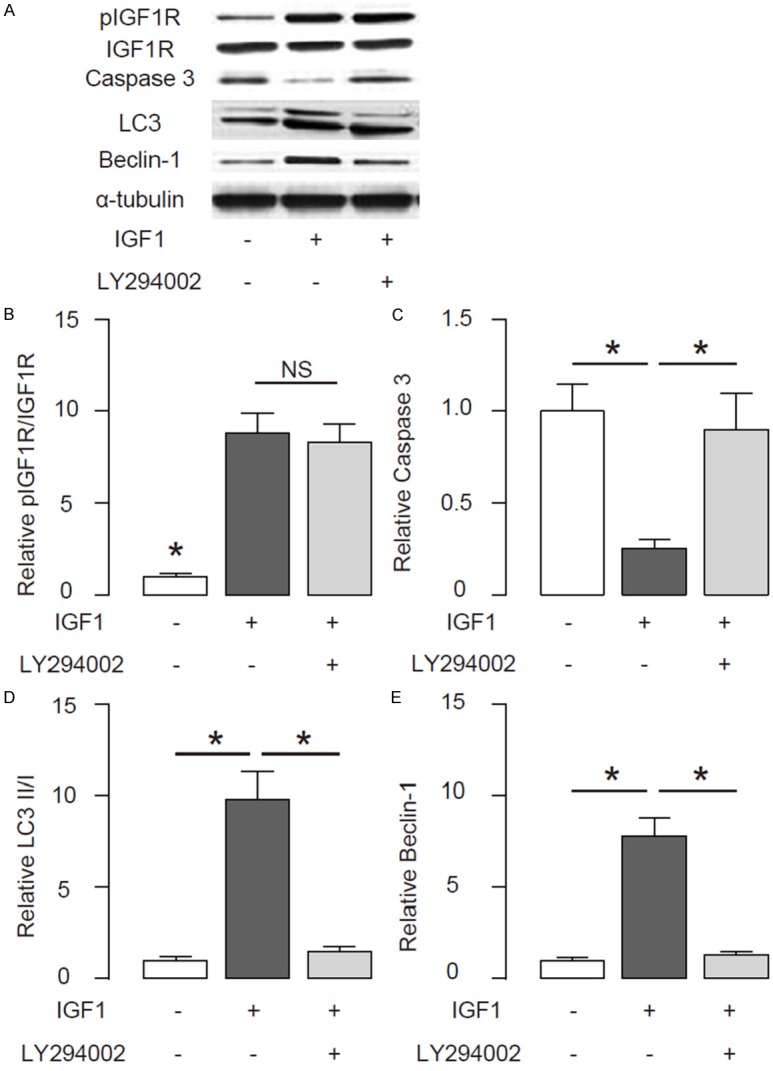
Activation of IGF1R signaling enhances disc cell autophagy upon compression treatment. A human disc cell line HNPSV was subjected to compression. Then, IGF1 was given to the cultured cells to activate IGF1R signaling. Moreover, a specific Akt inhibitor LY294002 was also used to some IGF1-treated HNPSV cells for inhibiting the activation of IGF1R signaling by IGF1. (A) Representative Western blot for IGF1R, Caspase 3, LC3 and Beclin-1 in disc cells. (B-E) Quantification for levels of pIGF1R/IGF1R (B), Caspase 3 (C), LC3 II/I (D) and Beclin-1 (E) in disc cells. *p < 0.05. NS: non-significant. N=5.
Autophagy is needed for the effects of activation of IGF1R signaling on disc cell survival upon compression treatment
In order to determine whether autophagy is critical for the protective effects of activation of IGF1R signaling on disc cell survival upon compression treatment, we gave the IGF1-treated cells with an autophagy inhibitor, 3-Methyladenine (3-MA) at a dose of 5 mmol/l. We found that blocking of cell autophagy by 3-MA did not alter activation of IGF1R signaling, shown by representative images (Figure 5A), and by quantification (Figure 5B). However, blocking of cell autophagy by 3-MA significantly increased the levels of Caspase 3, shown by representative images (Figure 5A), and by quantification (Figure 5C). Moreover, blocking of cell autophagy by 3-MA significantly decreased the ratio of LC3 II/I, shown by representative images (Figure 5A), and by quantification (Figure 5D), possibly through its effects on Beclin-1, shown by representative images (Figure 5A), and by quantification (Figure 5E). Furthermore, blocking of cell autophagy by 3-MA resulted in abolishment of the protective effects of activation of IGF1R signaling on disc cell survival upon compression treatment (Figure 5F).
Figure 5.
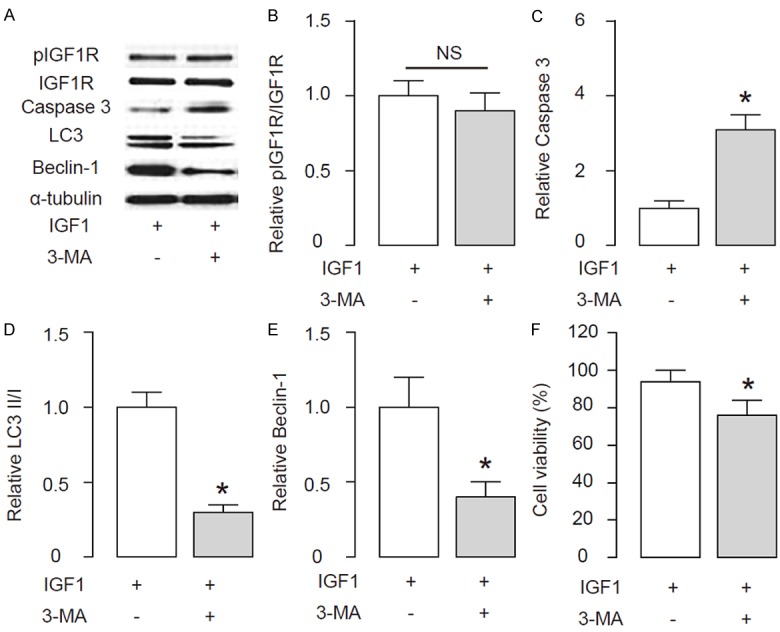
Autophagy is needed for the effects of activation of IGF1R signaling on disc cell survival upon compression treatment. The IGF1-treated cells were given an autophagy inhibitor, 3-Methyladenine (3-MA) at a dose of 5 mmol/l. (A) Representative Western blot for IGF1R, Caspase 3, LC3 and Beclin-1 in disc cells. (B-E) Quantification for levels of pIGF1R/IGF1R (B), Caspase 3 (C), LC3 II/I (D) and Beclin-1 (E) in disc cells. (F) The cell viability was analyzed in a CCK-8 assay. *p < 0.05. NS: non-significant. N=5.
Discussion
LDD affects the health of many people worldwide, whereas the molecular mechanisms underlying the development of LDD have not been elucidated [1-3]. Very recently, we reported that LDD patients had significantly lower levels of serum IGF1, and LDD discs had significantly lower levels of activated IGF1R. These clinical data provide a clinical relevance of analyzing IGF1R signaling on the development of LDD. In that study, we also showed that IGF1 induced phosphorylation of IGF1R and then phosphorylation of its downstream factor Akt, which further suppressed MMP3 in the human disc cells. Moreover, FoxO1 nuclear retention in human disc cells completely abolished the effects of IGF1 on MMP3, suggesting that Akt-induced phosphorylation and nuclear exclusion FoxO1 is responsible for the IGF1-induced MMP3 suppression.
In the current study, we tried to connect the disc cell survival upon compression treatment, since excessive or inappropriate compressive force stimulus is a demonstrative contributing factor in LDD occurrence. Compression directly affects the synthesis of collagen and proteoglycan in disc cells, and triggers necrosis and programmed cell death in disc cells. Thus, understanding the mechanisms of programmed cell death and survival are crucial for treating IDD. We showed a correction of activation of IGF1R and disc cell autophagy in the resected discs in LDD patients. These data provide clinical relevance of an association between activation of IGF1R and the levels of disc cell autophagy in the development of LDD. Since apoptosis and autophagy are coupled molecular events that regulate cell death and survival, it is expected that two pathways affect each other. We analyzed the molecular mechanisms in vitro using a compression model in human disc cells. We found that activation of IGF1R signaling antagonized the decreases in cell viability of human disc cells through suppression of apoptosis and enhancement of autophagy. Suppression of IGF1R signaling or inhibition of autophagy abolished the effects of activation of IGF1R signaling on disc cell survival upon compression.
Combined with results from our two studies on IGF1R/PI3k/Akt signaling in MMP3 and autophagy related development of LDD, here we proposed a model in which IGF1R/Akt controls LDD through FoxO1/MMP3 axis and through regulation of autophagy vs apoptosis. IGF1R signaling thus appears as a novel therapeutic target for inhibiting the development of LDD. This pathway at least has three critical control points, one is the initial stimulator, IGF1, as a trigger of activation of the system, and it functions mainly through induction of receptor phosphorylation. The second control point is FoxO1, since its cellular localization decides the activation MMP3-associated disc cell degeneration. The third control is autophagy, and it is regulated by IGF1R signaling and controls the survival of disc cells through crosstalk with apoptosis. The effects of IGF1R signaling on autophagy may through modulation of apoptosis-associated protein, e.g. Caspase 3, and autophagy-associate protein, e.g. Belcin-1. Further studies may address the exact molecular regulation of these proteins that control apoptosis and autophagy by IGF1R signaling. Together, our data suggest that activation of IGF1R may antagonize LDD, at least partially through enhanced autophagy.
Acknowledgements
The study was supported by Shanghai Municipal Science and Technology Commission, No: 1441197300.
Disclosure of conflict of interest
None.
References
- 1.Wang SZ, Rui YF, Lu J, Wang C. Cell and molecular biology of intervertebral disc degeneration: current understanding and implications for potential therapeutic strategies. Cell Prolif. 2014;47:381–390. doi: 10.1111/cpr.12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang HQ, Samartzis D. Clarifying the nomenclature of intervertebral disc degeneration and displacement: from bench to bedside. Int J Clin Exp Pathol. 2014;7:1293–1298. [PMC free article] [PubMed] [Google Scholar]
- 3.Risbud MV, Shapiro IM. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat Rev Rheumatol. 2014;10:44–56. doi: 10.1038/nrrheum.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng C, Liu H, Yang Y, Huang B, Zhou Y. Growth and differentiation factor-5 contributes to the structural and functional maintenance of the intervertebral disc. Cell Physiol Biochem. 2015;35:1–16. doi: 10.1159/000369670. [DOI] [PubMed] [Google Scholar]
- 5.Gruber HE, Ingram JA, Norton HJ, Hanley EN Jr. Senescence in cells of the aging and degenerating intervertebral disc: immunolocalization of senescence-associated beta-galactosidase in human and sand rat discs. Spine (Phila Pa 1976) 2007;32:321–327. doi: 10.1097/01.brs.0000253960.57051.de. [DOI] [PubMed] [Google Scholar]
- 6.Gruber HE, Norton HJ, Hanley EN Jr. Anti-apoptotic effects of IGF-1 and PDGF on human intervertebral disc cells in vitro. Spine (Phila Pa 1976) 2000;25:2153–2157. doi: 10.1097/00007632-200009010-00002. [DOI] [PubMed] [Google Scholar]
- 7.Gruber HE, Hanley EN Jr. Analysis of aging and degeneration of the human intervertebral disc. Comparison of surgical specimens with normal controls. Spine (Phila Pa 1976) 1998;23:751–757. doi: 10.1097/00007632-199804010-00001. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Yang JK, Kabaleeswaran V, Rice AJ, Cruz AC, Park AY, Yin Q, Damko E, Jang SB, Raunser S, Robinson CV, Siegel RM, Walz T, Wu H. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol. 2010;17:1324–1329. doi: 10.1038/nsmb.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JB, Chang H, Kim KW. Expression of Fas ligand and apoptosis of disc cells in herniated lumbar disc tissue. Spine (Phila Pa 1976) 2001;26:618–621. doi: 10.1097/00007632-200103150-00011. [DOI] [PubMed] [Google Scholar]
- 10.Chen JW, Ni BB, Li B, Yang YH, Jiang SD, Jiang LS. The responses of autophagy and apoptosis to oxidative stress in nucleus pulposus cells: implications for disc degeneration. Cell Physiol Biochem. 2014;34:1175–1189. doi: 10.1159/000366330. [DOI] [PubMed] [Google Scholar]
- 11.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Liu H, Guan Y, Wang Q, Zhou F, Jie L, Ju J, Pu L, Du H, Wang X. The altered autophagy mediated by TFEB in animal and cell models of amyotrophic lateral sclerosis. Am J Transl Res. 2015;7:1574–1587. [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Wang Q, Yin FQ, Zhang W, Yan LH, Li L. MTRR silencing inhibits growth and cisplatin resistance of ovarian carcinoma via inducing apoptosis and reducing autophagy. Am J Transl Res. 2015;7:1510–1527. [PMC free article] [PubMed] [Google Scholar]
- 14.Nandi SS, Duryee MJ, Shahshahan HR, Thiele GM, Anderson DR, Mishra PK. Induction of autophagy markers is associated with attenuation of miR-133a in diabetic heart failure patients undergoing mechanical unloading. Am J Transl Res. 2015;7:683–696. [PMC free article] [PubMed] [Google Scholar]
- 15.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gruber HE, Hoelscher GL, Ingram JA, Bethea S, Hanley EN Jr. Autophagy in the Degenerating Human Intervertebral Disc: In Vivo Molecular and Morphological Evidence, and Induction of Autophagy in Cultured Annulus Cells Exposed to Proinflammatory Cytokines-Implications for Disc Degeneration. Spine (Phila Pa 1976) 2015;40:773–782. doi: 10.1097/BRS.0000000000000865. [DOI] [PubMed] [Google Scholar]
- 17.Ding F, Shao ZW, Xiong LM. Cell death in intervertebral disc degeneration. Apoptosis. 2013;18:777–785. doi: 10.1007/s10495-013-0839-1. [DOI] [PubMed] [Google Scholar]
- 18.Jiang L, Zhang X, Zheng X, Ru A, Ni X, Wu Y, Tian N, Huang Y, Xue E, Wang X, Xu H. Apoptosis, senescence, and autophagy in rat nucleus pulposus cells: Implications for diabetic intervertebral disc degeneration. J Orthop Res. 2013;31:692–702. doi: 10.1002/jor.22289. [DOI] [PubMed] [Google Scholar]
- 19.Ye W, Zhu W, Xu K, Liang A, Peng Y, Huang D, Li C. Increased macroautophagy in the pathological process of intervertebral disc degeneration in rats. Connect Tissue Res. 2013;54:22–28. doi: 10.3109/03008207.2012.715702. [DOI] [PubMed] [Google Scholar]
- 20.Shen B, Melrose J, Ghosh P, Taylor F. Induction of matrix metalloproteinase-2 and -3 activity in ovine nucleus pulposus cells grown in three-dimensional agarose gel culture by interleukin-1beta: a potential pathway of disc degeneration. Eur Spine J. 2003;12:66–75. doi: 10.1007/s00586-002-0454-2. [DOI] [PubMed] [Google Scholar]
- 21.Yurube T, Nishida K, Suzuki T, Kaneyama S, Zhang Z, Kakutani K, Maeno K, Takada T, Fujii M, Kurosaka M, Doita M. Matrix metalloproteinase (MMP)-3 gene up-regulation in a rat tail compression loading-induced disc degeneration model. J Orthop Res. 2010;28:1026–1032. doi: 10.1002/jor.21116. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Zhou K, Fu W, Zhang H. Insulin-Like Growth Factor 1 Activates PI3k/Akt Signaling to Antagonize Lumbar Disc Degeneration. Cell Physiol Biochem. 2015;37:225–232. doi: 10.1159/000430347. [DOI] [PubMed] [Google Scholar]
- 23.Tao Y, Pinzi V, Bourhis J, Deutsch E. Mechanisms of disease: signaling of the insulin-like growth factor 1 receptor pathway--therapeutic perspectives in cancer. Nat Clin Pract Oncol. 2007;4:591–602. doi: 10.1038/ncponc0934. [DOI] [PubMed] [Google Scholar]
- 24.Ewing GP, Goff LW. The insulin-like growth factor signaling pathway as a target for treatment of colorectal carcinoma. Clin Colorectal Cancer. 2010;9:219–223. doi: 10.3816/CCC.2010.n.032. [DOI] [PubMed] [Google Scholar]
- 25.Tognon CE, Sorensen PH. Targeting the insulin-like growth factor 1 receptor (IGF1R) signaling pathway for cancer therapy. Expert Opin Ther Targets. 2012;16:33–48. doi: 10.1517/14728222.2011.638626. [DOI] [PubMed] [Google Scholar]